
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffective electro-oxidation of hydroxymethylfurfural using the electrografted immobilized aminoxyl radical†
Jiaxun
Guo
a,
Maryam
Abdinejad
 b,
Amirhossein
Farzi
a,
Mahdi
Salehi
a and
Ali
Seifitokaldani
*a
b,
Amirhossein
Farzi
a,
Mahdi
Salehi
a and
Ali
Seifitokaldani
*a
aDepartment of Chemical Engineering, McGill University, Montreal, QC H3A 0C5, Canada. E-mail: ali.seifitokaldani@mcgill.ca
bDepartment of Mechanical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
First published on 25th April 2023
Abstract
2,5-Furandicarboxylic acid (FDCA) is an eco-friendly substitute of fossil fuel-derived polymers that can be produced from biowaste. One of the methods for its production is electrochemical oxidation, through which hydroxymethylfurfural (HMF)–a readily available lignocellulosic compound from biorefineries as well as the pulp and paper industry–can be converted into FDCA. The primary challenge of this process is catalyst design, which encompasses characteristics such as reaction rate, conditions, and product yield in determining the overall performance. Herein, a reactive analogue of an aminoxyl radical (TEMPO) was prepared through organic synthesis and immobilized through the electrografting method as an anodic catalyst, with the cathode available for pairing with desirable reduction reactions. Through facile electrochemical methods, the immobilization procedure allows the traditionally homogeneous TEMPO catalyst to function heterogeneously, eliminating the need for catalyst separation and recycling. The attained catalyst demonstrated excellent performance: the synthesized electrografted TEMPO showed a low onset potential (1.45 V vs. RHE) and a high faradaic efficiency (>90%) and was catalytically active even in a mildly alkaline environment–a distinct advantage over metal-based catalysts as HMF decomposes in high pH electrolytes. Furthermore, due to the versatility of aminoxyl radicals, this catalyst can be adapted to other oxidative organic electrochemical reactions in electrolyzer anodes.
Introduction
In the quest to limit global carbon dioxide emissions and shift away from the reliance on fossilized resources, concerted efforts have been made to develop processes utilizing biomass as platform chemicals.1–5 In particular, biomass sources from agriculture as well as the pulp and paper industry are interesting due to the availability of lignocellulosic biomass as waste. Through catalytic reactions, lignocellulosic biomass can be upgraded to value-added chemicals of commercial value.6–95-Hydroxymethylfurfural (HMF) is a platform chemical obtained from lignocellulosic biomass via sugar dehydration,10 which has been extensively studied as the basis for biomass upgrading into many furan-derived chemicals. The product garnering the most interest is 2,5-furandicarboxylic acid (FDCA), which has been identified as a top chemical for the green chemistry industry of the future. 3 FDCA's prominent status as a renewable platform chemical is due to its potential to supplant petrochemicals such as terephthalic acid in the production of polyethylene terephthalate (PET) polyester, one of the most widely produced plastics in the world.3,11–14 FDCA can be produced from HMF via complete oxidation of its alcohol and aldehyde functional groups to carboxyl groups. As shown in Fig. 1, there are two possible pathways: (i) oxidation of the HMF alcohol group to form diformylfuran (DFF), and (ii) oxidation of the HMF aldehyde group to form 5-hydroxymethyl-2-furancarboxylic acid (HMFCA). Both pathways converge upon the formation of 5-formyl-2-furancarboxylic acid (FFCA), before the final oxidation step forming FDCA.
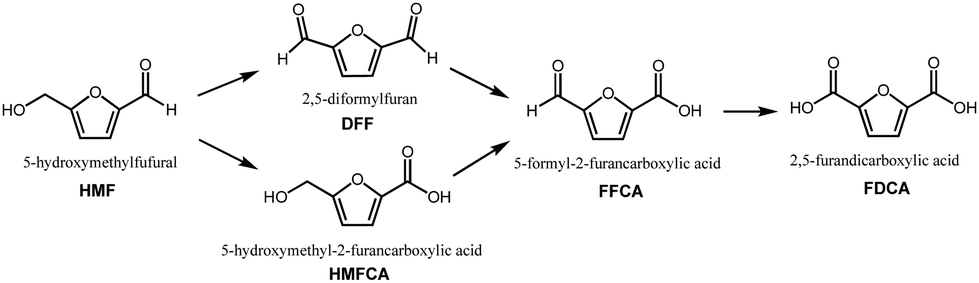 | ||
| Fig. 1 5-Hydroxymethylfurfural (HMF) oxidation pathways. | ||
There have been extensive efforts in studying heterogeneous catalysts for HMF oxidation, which mostly utilizes noble metal catalysts requiring harsh reaction conditions.15–18 A promising alternative to aerobic oxidation is electrochemical oxidation, where oxidation is driven by an applied potential. Electrochemical processes have the distinct advantage of high viability at mild temperature and pressure. Furthermore, the oxidation reaction can be paired with suitable reduction reactions that produce valuable products (such as H2 with water reduction) that augment the productivity of electrochemical systems.19 Many heterogeneous electrochemical catalysts based on transitional metals (especially nickel) have been previously reported to feature high faradaic efficiencies (FE). However, these catalysts are limited by their high pH requirement, a condition under which HMF readily decomposes into humins. Additionally, high alkalinity also increases separation costs when dissolved carboxylate products need to be acidified.
As shown in Table 1, although most of the reported heterogeneous catalysts are nickel-based with high faradaic efficiency and current densities, they require highly alkaline electrolytes such as potassium hydroxide, which decomposes HMF. Therefore, to explore alternatives, we looked towards heterogeneous aminoxyl radicals as potential candidates as catalysts/mediators in an aqueous environment.
| Electrode | HMF (mM) | V (RHE) | j (mA cm−2) | FDCA FE |
|---|---|---|---|---|
| Ni2P NPA/NF20 | 10 | 1.423 | >200 | 98 |
| NixB/NF21 | 10 | 1.45 | 100 | ∼100 |
| NiFe LDH22 | 10 | 1.33 | 36.9 | 98.6 |
| NiCo2O423 | 5 | 1.53 | 14.83 | 87.5 |
| Ni3N@C24 | 10 | 1.38 | 50 | 99 |
| NiCoFe25 | 5 | 1.52 | 10 | ∼90 |
| Co9S8-NiS2@NSOC/NF26 | 10 | 1.40 | 32 | ∼98 |
| NiO-CMK-127 | 20 | 1.75 | 15 | ∼80 |
| NiP@NiC13 | 15 | 1.37 | 10 | 87 |
| Cr-Ni(OH)228 | 10 | 1.47 | 60 | 96 |
| TEMPO29 | 100 | 1.60 | 200> | 91.5 |
Aminoxyl radicals have long been known as an effective oxidizing agent for organic oxidative reactions.30,31 For example, 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) has been shown to be effective for oxidation of alcohols to aldehydes. TEMPO is a homogeneous mediator that is dissolved in an electrolyte (pH 9∼12) and can be oxidized by carbon electrodes into oxoammonium cations, which act as an oxidizing agent for the HMF oxidation reaction (HMFOR).29 The use of homogeneous TEMPO has been widely explored, with innovative works such as combined HMF hydrogenation (BHMF) and HMF oxidation, achieving encouraging performance of over 97% FE for FDCA and a current density of 10 mA cm−2.32 Although the use of TEMPO allows for milder pH (9–11) conditions, it is disadvantageous due to the need to separate and replenish TEMPO in the electrolyte of the homogeneous media. There have been forays into heterogenization of TEMPO through several immobilization techniques, but most are done through polymer immobilization methods31 that are limited by low TEMPO stability or added complexity.33 In addition to TEMPO, Taitt et al. investigated AZADO, another aminoxyl radical similar in mechanism to TEMPO but sterically less hindered, and reported lower onset potential for AZADO oxidation and AZADO-mediated alcohol oxidation compared to TEMPO, along with higher current densities.34 While the reported performance is promising, the present work uses TEMPO for immobilization due to the abundance of previous work studying homogeneous TEMPO, as well as ease of access of the relevant chemical reagents.
Abdinejad et al.35,36 previously reported a promising technique for functionalizing carbon or silver nanoparticle electrodes with amino-compounds, and successfully implemented this technique on cathodic catalysts for carbon dioxide reduction reactions.37 The present study takes inspiration from their method and aims to develop a facile and one-pot aminoxyl immobilization process on carbon electrodes for the HMFOR. We synthesized 4-amino-TEMPO, a TEMPO derivative suitable for immobilization, and then adapted the electrografting methodology for facile immobilization. The functionalized electrode was characterized, and its electrochemical properties were thoroughly investigated. Through electrochemical experiments such as cyclic voltammetry (CV), chronoamperometry (CA), electrochemical impedance spectroscopy (EIS), and X-ray photoelectron spectroscopy (XPS), we confirm the successful immobilization of TEMPO through electrografting and report the HMFOR performance of ∼95% FE at ∼1.6 V vs. RHE (reversible hydrogen electrode), and that the aminoxyl radicals are stable during prolonged reactions. To the best of our knowledge, this is the first report of direct covalent immobilization of TEMPO used for the HMFOR. Its promising performance and potential for modifications render it a notable catalyst for the HMFOR and other organic oxidative reactions.
Results and discussion
Immobilization
The necessity of synthesizing 4-amino-TEMPO arises from the fact that an amine group is necessary for immobilization through electrografting. The synthesis steps are summarized in Fig. 2A. Primary amines (as present on 4-amino-TEMPO) are known to be selectively oxidized by nitrosonium ions into diazonium ions. Under the appropriate reduction potential, diazonium ions can be reduced to a piperidine radical intermediate, likely to be followed by covalent bonding to the carbon substrate with the leaving of molecular nitrogen (Fig. 2A).38,39 This scheme can be realized through a procedure similar to the Sandmeyer reaction, where NaNO2 and HCl are the reagents. The reaction is described in the ESI.† A CV program between 0.5 V and −0.7 V vs. Ag/AgCl was carried out at a scan rate of 20 mV s−1 to perform the reaction, as shown in Fig. 2B. The choice of using a CV program to apply reductive potential may appear counter-intuitive at first, but CV carries several advantages over constant potential experiment that aid in the understanding of this process.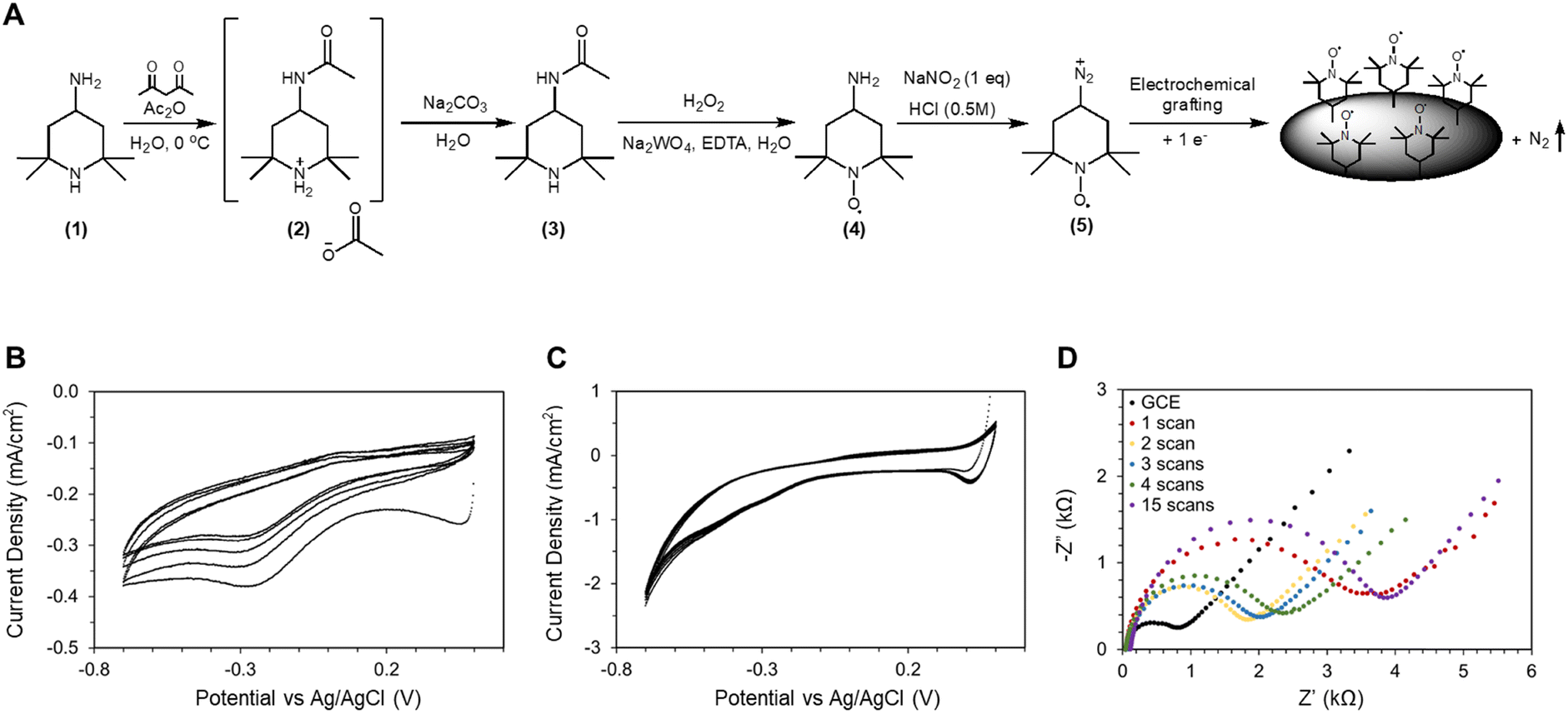 | ||
| Fig. 2 (A) Synthesis steps for 4-amino-TEMPO and reaction pathway for amine electrografting onto carbon electrode surfaces. (B) CV of the electrografting on a glassy carbon electrode at 20 mV s−1 scan rate in 5 mM 4-amino-TEMPO, 5 mM sodium nitrite, and 0.5 M HCl; each subsequent scan has lower current.‡ (C) Electrografting experiment in the absence of 4-amino-TEMPO. (D) EIS data of the GCE electrode after various cycles of electrografting scans. | ||
Electrografting and electrochemical characterization
Firstly, CV reveals the exact onset potential of the electrografting reaction. As shown in Fig. 2B, the reduction peak is around −0.25 V vs. Ag/AgCl. A noteworthy observation is the general trend of this reduction peak shifting towards more negative potential in subsequent CV scans. This suggests that as the reaction progressed, the overpotential increased as the immobilization became ever more difficult. This is indirect evidence that the electrochemical surface area (ECSA) is decreasing over time, potentially due to the coverage of TEMPO. In a similar vein, another observation is that the overall current densities decrease with subsequent scans. There are two possible explanations: either electrografting covered the glassy carbon electrode (GCE) surface with TEMPO to reduce ECSA, or the CV scans were altering the GCE surface itself. Through a control experiment shown in Fig. 2C, it was determined that in the absence of TEMPO, none of the two characteristics (reduction peaks and diminishing current density) occurs, hence confirming that these features are the result of TEMPO immobilization. Earlier, we stipulated that TEMPO immobilization occurs through covalent bond formation under reductive potentials; hence a logical question is whether this process is electrochemically reversible. We note that during the oxidative portion of the CV scans, minor oxidation peaks around 0 V vs. Ag/AgCl were present. These oxidation peaks are comparably small compared to the reduction peaks, suggesting that overall, oxidation potentials should not pose a threat to immobilization, but it nevertheless raises some concerns over catalyst stability.From the literature36 and from our preliminary experiments, we learned that CV parameters play a role in the electrochemical performance of the immobilized catalyst. Specifically, the number of electrografting CV cycles applied influenced the current density during the HMFOR, where prolonged electrografting produced electrodes with low current density. We hypothesized that there exists an optimal degree of electrografting, beyond which further addition of TEMPO hampers the performance of TEMPO catalysts already present. Experimentally, we used EIS to determine the optimal electrografting scan cycle, and the result and the equivalent circuit model can be found in Fig. S8 (ESI†). Fig. 2D shows that after the initial CV cycle, the charge transfer resistance (Rct) increased from ∼800 Ω to 3.4 kΩ. But after the 2nd cycle, Rct decreased to ∼1.8 kΩ. However as further electrografting scans were applied, Rct increased to 2 kΩ, 2.3 kΩ (3rd and 4th scans), and 3.9 kΩ with an excessive 15 electrografting scans. The charge transfer resistance is the reaction resistance at the high frequency and small amplitude perturbations during EIS measurements, and a high charge transfer resistance signifies a large barrier that needs to be overcome for electron transfer. The EIS models are presented alongside the data; as part of this dataset, the constant phase element (CPE) was also monitored. The impedance of a CPE is defined by the following equation:
| Z = (1/Y0)/(jw)α | (1) |
In addition to the number of electrografting CV scans, the scan rate is also a significant factor, but its effects are convoluted. In theory, slower scan rates lead to more charges passed per CV cycle; hence slower scan rates effectively equate to more CV scans. However, it is not known whether electrografting procedures of different scan rate, but equal amount of charges passed would yield the same electrode as a result. This question is especially delicate as it has not been established whether the electrografting reaction accounts for 100% of the faradaic current. Hence even if a charge-controlled comparison study was performed, the results would not have been conclusive. To avoid confusion over this topic, we chose 20 mV s−1 as the electrografting scan rate through heuristic experience (unless otherwise specified).
Confirmation of immobilization – XPS
XPS is used to directly observe the presence of TEMPO on the electrografted graphite electrode. Fig. 3 shows the XPS spectra of electrografted graphite in comparison with those of the bare graphite. Fig. 3A shows the N1s spectrum of bare graphite, where no identifiable peaks were found. In Fig. 3B, the electrografted sample's raw data as well as fitted peaks are shown. By comparison with the published XPS tables, the peaks were identified as shown in Table S7 (ESI†). Re-calling the structure of the TEMPO molecule, the peaks identified match closely with the expected peaks based on the structure, with the slight deviations possibly due to the presence of the radical oxygen atom, and the fact that these two peaks are due to the same nitrogen atom. The nitro-peak should be associated with 1′ position N atom's bond with the oxygen atom as part of the aminoxyl group, whereas the amine peak is associated with this N atom's single bonds with the 2′ and 6′ position carbon atoms, forming a secondary amine. It is also worth noting that the approximate atomic % also displays the expected correlation: we expect the amine peak with two N1s bonds to be around two-thirds of the counts, and it was shown to be responsible for 65.3% of atomic %, with the nitro-peak constituting the remaining percentage, as expected. The overall elemental analysis is shown in Table S8 (ESI†), where the sample was shown to comprise 87.9% carbon, 11.2% oxygen and only 0.9% nitrogen. In Table S9 (ESI†) the same results are shown for bare graphite. As expected, neither of the two N1s peaks was observed, with further diminished oxygen composition.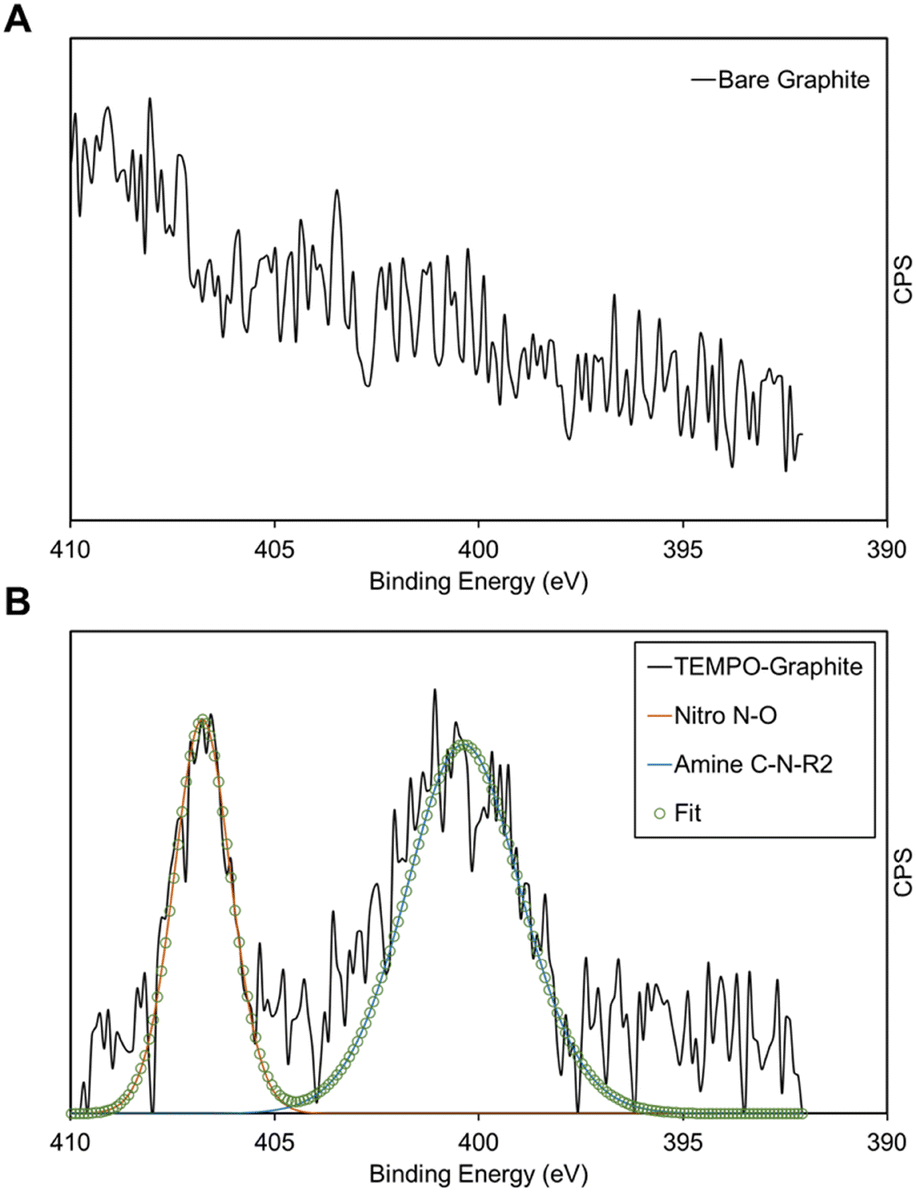 | ||
| Fig. 3 (A) X-ray photoelectron spectroscopy (XPS) N 1s spectra for the graphite electrode; (B) XPS N 1s spectra for the electrografted TEMPO-graphite electrode. | ||
Confirmation of immobilization – electrochemical method
In addition to spectroscopic techniques like XPS, the immobilized electrode can also be tested electrochemically to ascertain its heterogeneous nature. Here, we once again utilize CV to understand the underlying correlation between the peak current density and the scan rate. In Fig. 4A, the CV plots of the TEMPO-immobilized graphite electrode at various scan rates are shown, revealing oxidation potentials between 0.6 and 0.7 V vs. Ag/AgCl. As the scan rate increases, the redox peak currents also increase. For dissolved redox species, the correlation between the scan rate and the peak current is described by the Randles–Sevcik equation: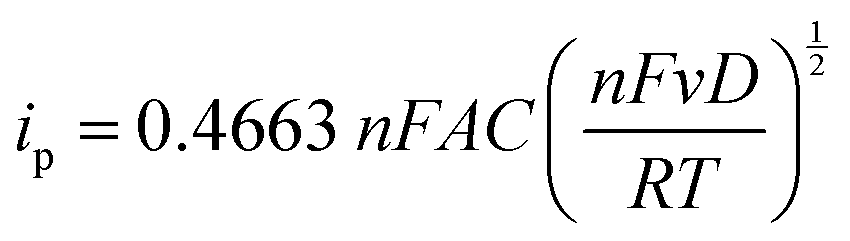 | (2) |
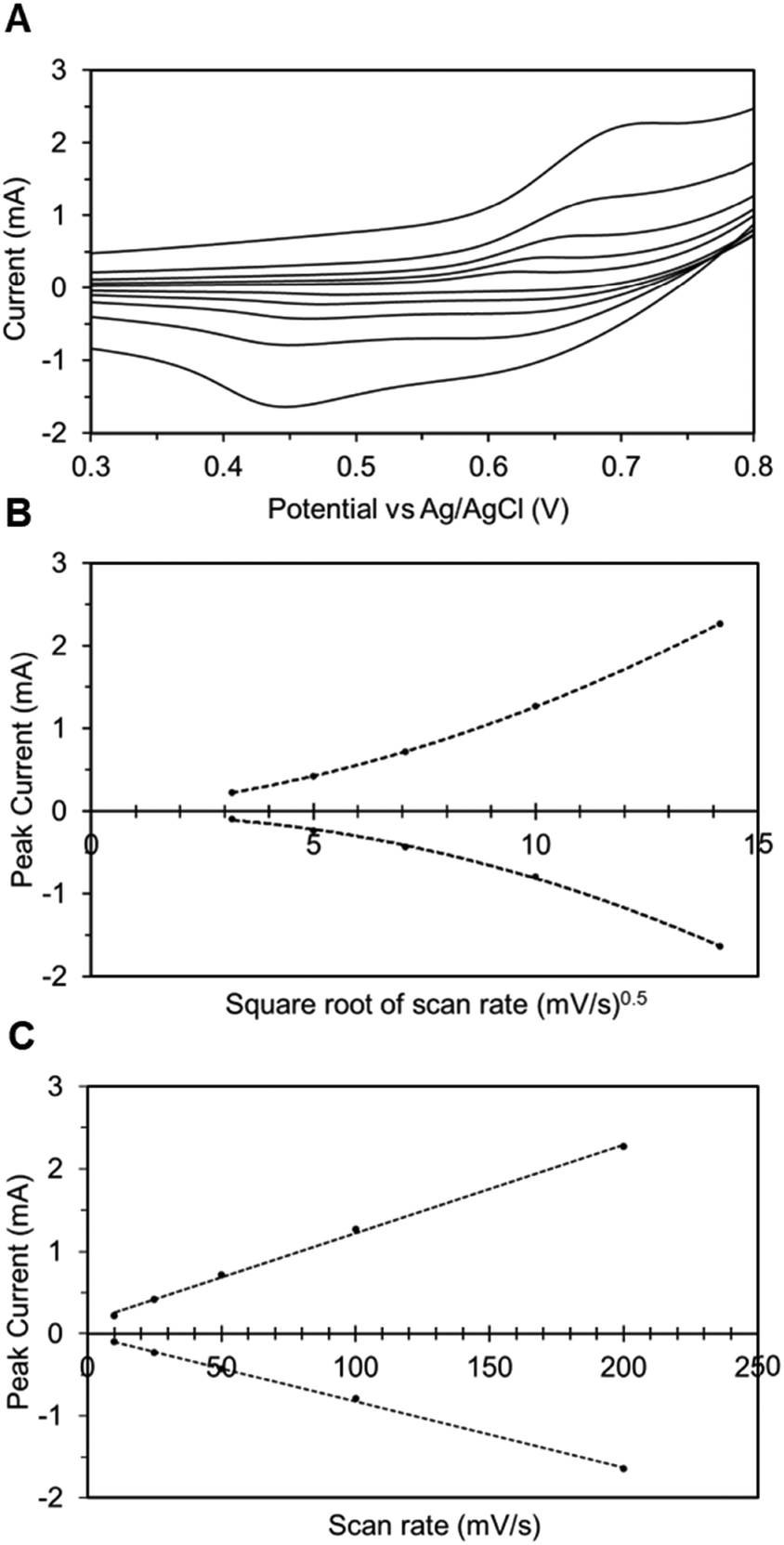 | ||
| Fig. 4 (A) CV of the TEMPO-immobilized glassy carbon electrode at scan rates 10, 25, 50, 100, and 200 mV s−1, and in a 0.2 M NaHCO3/Na2CO3 electrolyte. The peak current increases with the scan rate. (B) Correlation between scan rates and peak current, shown as peak current vs. square root of the scan rate. (C) Correlation between scan rates and the peak current shown as peak current vs. linear scan rate. | ||
In eqn (2), ip is the peak current in amps, n is the number of electrons, F is Faraday's constant (96485.3 C mol−1), A is the electrode area in cm2, C is the concentration in mol cm−3, D is the diffusion coefficient in cm2 s−1, v is the scan rate in V s−1, R is the gas constant (8.314 J K−1 mol−1), and T is the temperature in K. In this relationship, the square root of the scan rate is linearly correlated to the peak current ip. However, Fig. 4B clearly contradicts this: the relationship between the square root of scan rates and the peak currents is not linear. On the other hand, if the peak current is plotted against linear scan rates, the relationship is shown to be linear (Fig. 4C). This correlation reveals the heterogeneous nature of the electrode.40 Hence, the peak current and scan rate correlation corroborates the other supporting evidence previously presented, and it is clear that the TEMPO mediator acted heterogeneously, meaning it had been electrografted onto carbon substrates while remaining electrochemically active.
Electrochemical performance
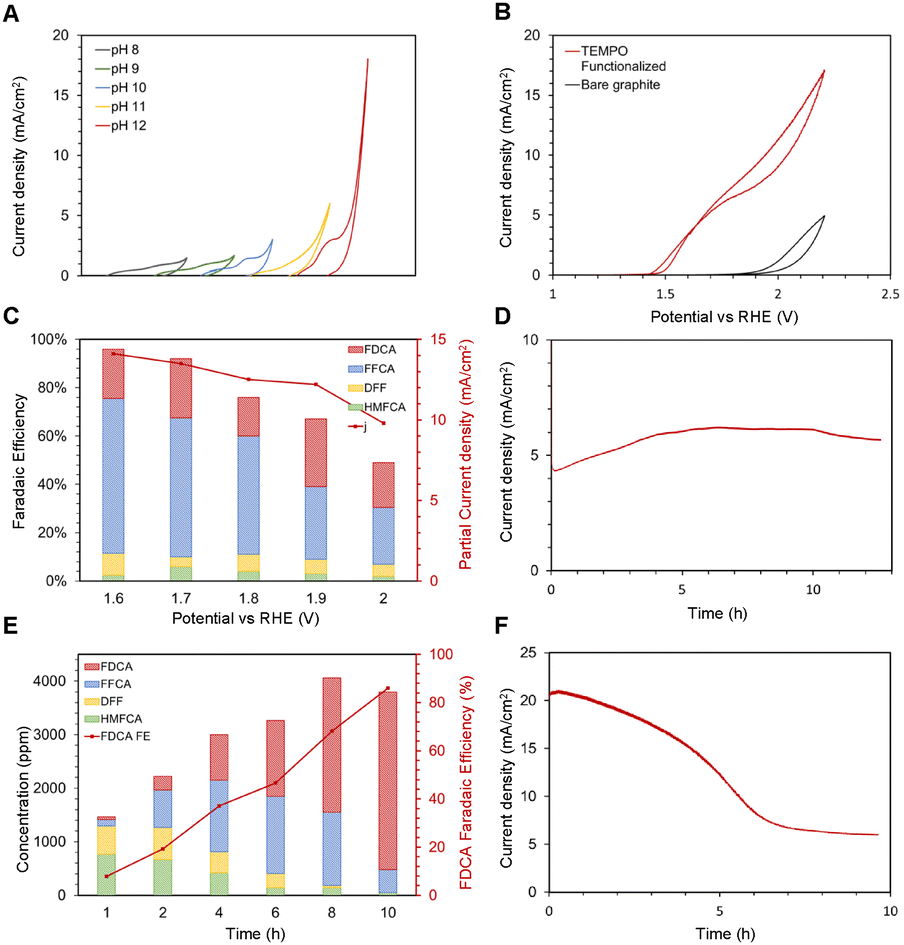 | ||
| Fig. 5 (A) Cyclic voltammetry (CV) of TEMPO functionalized graphite in different pH electrolytes from pH 8 to pH 12, shown from left to right.§ (B) Cyclic voltammetry of the TEMPO-immobilized graphite electrode (red) and the naked graphite electrode (black). Electrolyte conditions are 10 mM HMF and pH 12 electrolyte. (C) Faradaic efficiencies for each HMFOR product of immobilized TEMPO at various applied potentials. The partial current density is shown on the secondary y-axis, and balance FE is assumed to be the OER. (D) Chronoamperometry of TEMPO-GCE in 10 mM HMF and pH 12 electrolyte, at applied potential 1.7 V vs. RHE. (E) Product distribution over time of complete oxidation of 30 mM HMF, 1.7V vs. RHE.¶ (F) Chronoamperometry of the complete oxidation experiment. | ||
To observe the current stability of the system, a TEMPO-immobilized glassy carbon electrode (GCE) was prepared for the CA experiment. The GCE was selected over graphite due to its smaller ECSA, which leads to a lower current density. In this case, lower current density is preferred to ensure that the reactant concentration would not significantly shift over the course of the experiment. The CA experiment was conducted for 10 hours, and the system perfectly maintained the level of current for the duration of the experiment (Fig. 5D). This suggests that immobilization is relatively stable even under applied oxidative potentials for long periods. Curiously, the current density initially increased before it decreased. We suspect that this was caused by immobilized-TEMPO's preference towards alcohol oxidation instead of aldehyde oxidation. The initial increase in current density can be attributed to HMF forming HMFCA, which, once formed, can be easily oxidized to FFCA. But as FFCA concentration builds up over the course of the reaction, the reaction's focus shifted towards the FFCA to FDCA step, which is more difficult, reflected through the decreasing current. This is evident from the high FFCA concentration in Fig. 5C.
In order to further clarify the evolution of FE towards each species over time, the chronoamperometry experiment was performed, and the charge required for HMF to FDCA conversion was calculated. By controlling the charge passed, the faradaic efficiency of FDCA was assessed accurately. As shown in Fig. 5E, HMFCA and DFF were initially detected as the primary products. Over time, FFCA concentration stayed steady and became the dominant intermediate product between hours 4 and 6. This result explains why the reported FDCA faradaic efficiencies were low in Fig. 5C. The FDCA FE is a function of reaction time, and only reaches maximum towards the end of the reaction when HMF and all previous intermediates are converted. The overall FDCA FE of this reaction was 83%, signifying that electrografted TEMPO is capable of competitive FDCA FE if the batch reaction is allowed to reach its completion. Fig. 5F shows the CA result of the complete oxidation experiment. The current density gradually decreases over time, correlating to decreasing concentration of available oxidation reactants (excluding OER).
| Atom | Immobilized (TEMPO) | Homogeneous (4-amino-TEMPO) |
|---|---|---|
| N | 5.203 | 4.993 |
| O | 6.406 | 6.327 |
| C | 3.978 | 4.030 |
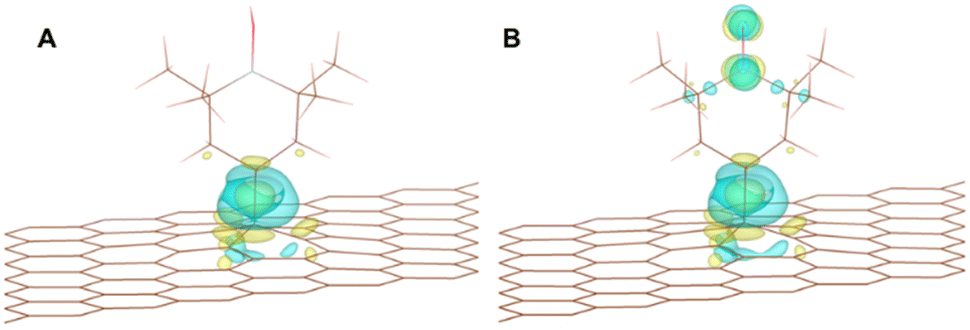 | ||
| Fig. 6 Charge delocalization in the (A) TEMPO radical and (B) oxoammonium cation with an iso-surface level of 0.004 e per Bohr3 in all cases. | ||
Conclusion
In conclusion, we have demonstrated a facile approach to immobilizing the TEMPO catalyst for the HMFOR through electrografting amine groups onto commercially available graphite and supported the validity of this approach through characterization and analysis of electrochemical characteristics. The catalyst showed promising performance with high total FE towards the HMFOR products, as well as decent current densities. In addition, it negates the need for a highly alkaline environment, as well as the homogeneous nature of aminoxyl radical catalysts, which renders this approach more suitable for industrial applications. Moreover, the versatility of aminoxyl radicals means that this approach can be applied to other organic electrochemical reactions. For future works, the tuning of electrografted TEMPO can be examined, both computationally and experimentally, through the addition of functional groups between TEMPO and the carbon electrode, as well as by immobilizing directly on metallic electrodes, with the aim of decreasing the overpotential and increasing the current density.Experimental
Materials
All chemicals except those synthesized were purchased as reagent grade from Sigma-Aldrich. 5-Hydroxymethyfurfural (HMF), acetic anhydride (Ac2O), diethyl ether (Et2O), ethylenediaminetetraacetic acid (EDTA), hydrochloric acid (HCl), potassium chloride (KCl), potassium hydroxide (KOH), potassium nitrate (KNO3), potassium nitrite (KNO2), sodium carbonate (Na2CO3), sodium hydroxide (NaOH), TEMPO, sodium nitrite (NaNO2) and sodium tungstate (Na2WO4) were purchased from Sigma-Aldrich; 4-amino-2,2,6,6-tetramethylpiperidine was purchased from Oakwood Chemical; and 37% hydrochloric acid (HCl) was purchased from Fisher Scientific. All solutions were prepared using RO water (resistivity >1.8 MΩ). Carbon felt and the anion exchange membrane (Fumasep FAA-3-PK-75) were purchased from the Fuel Cell Store. Graphite electrodes were purchased from Corrtest Instruments.Synthesis of 4-amino-TEMPO
In homogeneous systems, TEMPO can be directly dissolved in the electrolyte. However, in heterogeneous systems, TEMPO is unsuitable for immobilization due to the lack of reactive functional groups for covalent attachment. Hence, we proposed the use of 4-amino-TEMPO instead, a molecule analogous to TEMPO in function but contains primary amine at the 4′ position. The presence of an amine group enables immobilization via electrografting technique upon conversion to a diazonium salt. The compound was prepared through an organic synthesis procedure adopted in part from that published by Lu et al.44 The reaction steps are outlined in Figure 2: 4-amino-2,2,6,6-tetramethylpiperidine (Compound 1) is first reacted with acetic anhydride to render an acetamide acetate (Compound 2). In the presence of oxidant, Na2WO4, and EDTA45 the compound was converted into its corresponding aminoxyl radical (Compound 3) while the acetamide is converted into an amine, forming 4-amino-TEMPO (Compound 4). See the ESI† for details (Fig. S1 and S2) and characterization (Fig. S3–S6).Cyclic voltammetry and chromoamperometry
Cyclic voltammetry (CV) and chronoamperometry (CA) were performed in an H-cell (50 or 80 mL) divided by a Fumasep anion exchange membrane (AEM) with a three-electrode setup using a Metrohm Autolab PGSTAT204. The appropriate carbon electrode (graphite or glassy carbon electrode) was used as the working electrode with an Ag/AgCl (3 M KCl) reference electrode and a Pt foil counter electrode. A glassy carbon electrode (GCE) was used for electrochemical characterization such as EIS, showcase of electrografting CV, etc. Its benefit is that a GCE is less susceptible to impurities and changing surface roughness. Graphite was used when larger current density was desired, such as for product generation during CA. Between experiments, the surface was sanded with 600 grit sandpaper, and smoothed with a rotary tool using different grit wax compounds. GCE surface areas were calculated from measurements taken using a digital caliper, whereas graphite rods were masked with Kapton tape to restrict the exposed surface area to desired values. Unless otherwise specified, a pH 12 KCl/NaOH electrolyte solution of approximately 0.2 M concentration was used as the electrolyte. The solution was stirred at ∼200 rpm using a hotplate-stirrer at room temperature. For CA experiments, 10 mM HMF was added to the electrolyte unless otherwise specified. Reaction potentials are reported in Ag/AgCl for those that are pH independent, and in RHE for the HMFOR which are pH dependent. The potential was not corrected for the ohmic loss (iR drop). EIS was performed in a H-cell similar to that used in CV experiments, with the electrolyte solution consisting of 0.2 M potassium nitrate and 2.5 mM potassium hexacyanoferrate/ferrate probe. The experiments were done using the same Autolab PGSTAT204 but with an added FRA32M EIS module.Product quantification
The oxidation products are split between the HMFOR and the oxygen evolution reaction (OER); the former consists of dissolved organic compounds, whereas the latter produces oxygen. To quantitatively analyze the anolyte, the Thermo Ultimate 3000 high-performance liquid chromatography (HPLC) system with the internal UV-vis detector was used. Over the course of chronoamperometry experiments, dissolved oxidation products permeate through the cell membrane, and hence the catholyte was also analyzed. Product identification and calibration were done using respective standards prepared to known concentrations. 10 μL was injected into a Thermo Scientific™ Acclaim™ 300 C18 column with a guard column, and the temperature was maintained at a constant 25 °C. The mobile phase was 90% 5 mM ammonium formate and 10% methanol, pumped at a rate of 5 mL min−1. The respective retention times (in min) for HMF, HMFCA, DFF, FFCA and FDCA were 8.71, 4.41, 9.66–9.97, 4.88 and 3.89, respectively. The detection wavelength λmax was 265 nm. See Fig. S7 (ESI†) for the sample HPLC spectrum. Faradaic efficiency (FE) was calculated as follows: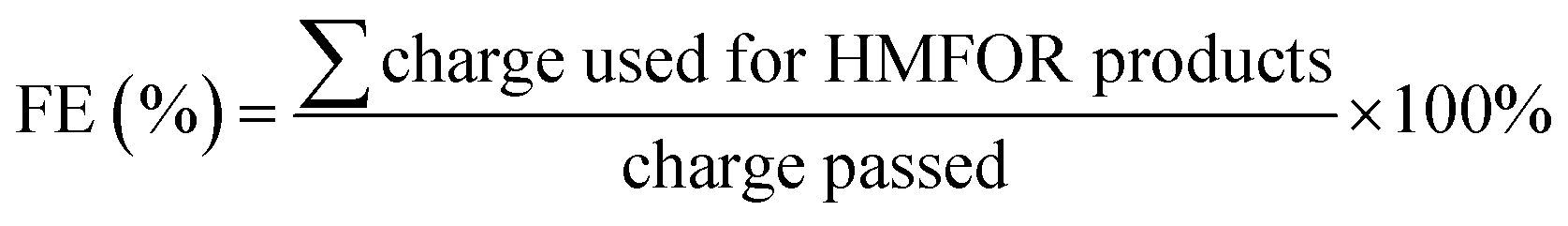 | (3) |
As the various HMFOR products require different number of electrons, their FE is not directly equivalent to their selectivity. Specifically, HMFCA and DFF require two electron transfers, FFCA four electrons, and FDCA six electrons. In this work, unless otherwise specified, the term FE is taken to mean the sum of all HMFOR products, as the intermediates represent various degrees of reaction towards FDCA.
DFT computational details
DFT computations were performed using the Vienna Ab initio simulation package (VASP),46 on Compute Canada clusters. In all computations, we used the projected augmented wave (PAW) pseudopotentials and the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) to describe the exchange–correlation functionals.47 A cut-off energy of 450 eV for the plane-wave basis sets and a 2 × 2 × 1 Γ-centered Monkhorst–Pack mesh for the k-point sampling in the first Brillouin zone, with a first order Methfessel–Paxton smearing parameter σ of 0.05 eV, ensured that the energy convergence criteria are better than 1 meV for a vacuum of 20 Å or greater. The self-consistent field (SCF) convergence criterion was set to 10−4 eV for electronic iteration, and the ionic relaxation continued till the maximum force was less than 0.02 eV Å−1 that was updated by the conjugate gradient approach. Dipole corrections and spin polarization were implemented. The DFT-D3 method with the Becke–Jonson damping was performed for the van der Waals correction. The electrostatic charge density around each ion was calculated by the Bader charge analysis method.42 VESTA software was used for visualization.48 Structure optimization, electron density and delocalization calculations followed the standard procedures described in the literature.49Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval for the final version of the manuscript. J. G. performed the synthesis, electrochemical tests, chemical analysis, and most of the characterization and prepared the first draft of the manuscript. A. F. performed the DFT computations. M. S. performed the XPS characterization and analysis. M. A. provided guidance on chemical synthesis, characterization, and troubleshooting the immobilization method. A. S. supervised the whole project, designed the experiments, and finalized the manuscript.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was financially supported by the McGill Sustainability Systems Initiation (MSSI) Ideas Fund, Fonds de recherche du Québec – Nature et technologies (FRQNT) New Researchers Fund (2021-NC-283234), NSERC Discovery Grant (RGPIN-2020-04960), and Canada Research Chair (950-23288). The DFT computations carried out in this study were supported by Calcul Quebec, Compute Canada, and the Digital Research Alliance of Canada.References
- S. Dutta, S. De and B. Saha, ChemPlusChem, 2012, 77, 259–272 CrossRef CAS.
- J. Gravitis, N. Vedernikov, J. Zandersons and A. Kokorevics, ACS Symp. Ser., 2001, 110–122 CrossRef CAS.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539 RSC.
- P. Okoye and B. Hameed, Renewable Sustainable Energy Rev., 2016, 53, 558–574 CrossRef CAS.
- C. Delhomme, D. Weuster-Botz and F. E. Kühn, Green Chem., 2009, 11, 13–26 RSC.
- X. Chen, Y. Liu and J. Wang, Ind. Eng. Chem. Res., 2020, 59, 17008–17025 CrossRef CAS.
- K. S. Kanakikodi, S. R. Churipard, R. Bai and S. P. Maradur, Mol. Catal., 2021, 510, 111716 CrossRef CAS.
- J. S. Luterbacher, D. Martin Alonso and J. A. Dumesic, Green Chem., 2014, 16, 4816–4838 RSC.
- R. S. Almeida Ribeiro, L. E. Monteiro Ferreira, V. Rossa, C. G. S. Lima, M. W. Paixão, R. S. Varma and T. Melo Lima, ChemSusChem, 2020, 13, 3992–4004 CrossRef PubMed.
- X. Tong, Y. Ma and Y. Li, Appl. Catal., A, 2010, 385, 1 CrossRef CAS.
- A. Gandini, A. J. D. Silvestre, C. P. Neto, A. F. Sousa and M. Gomes, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 295–298 CrossRef CAS.
- R.-J. Van Putten, J. C. Van Der Waal, E. De Jong, C. B. Rasrendra, H. J. Heeres and J. G. De Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- R. Lin, M. Salehi, J. Guo and A. Seifitokaldani, iScience, 2022, 25, 104744 CrossRef CAS PubMed.
- P. Patel, D. Schwartz, X. Wang, R. Lin, O. Ajao and A. Seifitokaldani, ACS Sustainable Chem. Eng., 2022, 10, 4206–4217 CrossRef CAS.
- J. Zhang, et al. , Carbohydr. Polym., 2015, 130, 420 CrossRef CAS.
- N. K. Gupta, S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2011, 13, 824 RSC.
- X. Wan, C. Zhou, J. Chen, W. Deng, Q. Zhang, Y. Yang and Y. Wang, ACS Catal., 2014, 4, 2175–2185 CrossRef CAS.
- S. E. Davis, B. N. Zope and R. J. Davis, Green Chem., 2012, 14, 143–147 RSC.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- B. You, N. Jiang, X. Liu and Y. Sun, Angew. Chem., Int. Ed., 2016, 55, 9913–9917 CrossRef CAS PubMed.
- S. Barwe, J. Weidner, S. Cychy, D. M. Morales, S. Dieckhöfer, D. Hiltrop, J. Masa, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2018, 57, 11460–11464 CrossRef CAS PubMed.
- W.-J. Liu, L. Dang, Z. Xu, H.-Q. Yu, S. Jin and G. W. Huber, ACS Catal., 2018, 8, 5533–5541 CrossRef CAS.
- M. J. Kang, H. Park, J. Jegal, S. Y. Hwang, Y. S. Kang and H. G. Cha, Appl. Catal., B, 2019, 242, 85–91 CrossRef CAS.
- N. Zhang, Y. Zou, L. Tao, W. Chen, L. Zhou, Z. Liu, B. Zhou, G. Huang, H. Lin and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 15895–15903 CrossRef CAS.
- M. Zhang, Y. Liu, B. Liu, Z. Chen, H. Xu and K. Yan, ACS Catal., 2020, 10, 5179–5189 CrossRef CAS.
- Y. Zhang, Z. Xue, X. Zhao, B. Zhang and T. Mu, Green Chem., 2022, 24, 1721–1731 RSC.
- F. J. Holzhäuser, T. Janke, F. Öztas, C. Broicher and R. Palkovits, Adv. Sustainable Syst., 2020, 4, 1900151 CrossRef.
- Z. Yang, B. Zhang, C. Yan, Z. Xue and T. Mu, Appl. Catal., B, 2023, 330, 122590 CrossRef CAS.
- A. C. Cardiel, B. J. Taitt and K.-S. Choi, ACS Sustainable Chem. Eng., 2019, 7, 11138–11149 CrossRef CAS.
- J. M. Bobbitt and N. Merbouh, Org. Synth., 2003, 82, 80–86 Search PubMed.
- Y. Kashiwagi, F. Kurashima, S. Chiba, J.-I. Anzai, T. Osa and J. M. Bobbitt, Chem. Commun., 2003, 114–115, 10.1039/b209871g.
- H. Liu, T.-H. Lee, Y. Chen, E. W. Cochran and W. Li, Green Chem., 2021, 23, 5056–5063 RSC.
- A. Das and S. S. Stahl, Angew. Chem., Int. Ed., 2017, 56, 8892–8897 CrossRef CAS.
- B. J. Taitt, M. T. Bender and K.-S. Choi, ACS Catal., 2020, 10, 265–275 CrossRef CAS.
- M. Abdinejad, I. Santos Da Silva and H. B. Kraatz, J. Mater. Chem. A, 2021, 9, 9791–9797 RSC.
- M. Abdinejad, E. Irtem, A. Farzi, M. Sassenburg, S. Subramanian, H.-P. Iglesias van Montfort, D. Ripepi, M. Li, J. Middelkoop, A. Seifitokaldani and T. Burdyny, ACS Catal., 2022, 12, 7862–7876 CrossRef CAS PubMed.
- M. Abdinejad, T. Yuan, K. Tang, S. Duangdangchote, A. Farzi, H. P. Iglesias Van Montfort, M. Li, J. Middelkoop, M. Wolff, A. Seifitokaldani, O. Voznyy and T. Burdyny, Chem. – Eur. J., 2023, 29, e202203977 CrossRef CAS PubMed.
- S. Baranton and D. Bélanger, J. Phys. Chem. B, 2005, 109, 24401–24410 CrossRef CAS PubMed.
- A. Adenier, M.-C. Bernard, M. M. Chehimi, E. Cabet-Deliry, B. Desbat, O. Fagebaume, J. Pinson and F. Podvorica, J. Am. Chem. Soc., 2001, 123, 4541–4549 CrossRef CAS PubMed.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical methods: fundamentals and applications, John Wiley & Sons, 2022 Search PubMed.
- J. M. Bobbitt, C. Brückner and N. Merbouh, Org. React., 2009, 103–424, DOI:10.1002/0471264180.or074.02.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- P. Kisszekelyi, R. Hardian, H. Vovusha, B. Chen, X. Zeng, U. Schwingenschlögl, J. Kupai and G. Szekely, ChemSusChem, 2020, 13, 3127–3136 CrossRef CAS PubMed.
- Y. Lu and S. Yamago, Angew. Chem., Int. Ed., 2019, 58, 3952 CrossRef CAS PubMed.
- J. Lind and G. Merényi, J. Phys. Chem. A, 2006, 110, 192–197 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653–658 CrossRef CAS.
- H. Al-Mahayni, X. Wang, J.-P. Harvey, G. S. Patience and A. Seifitokaldani, Can. J. Chem. Eng., 2021, 99, 1885–1911 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ya00108c |
| ‡ We make a point to report electrografting CVs vs. Ag/AgCl reference electrode to illustrate that the reaction is pH independent. |
| § For Fig. 5A, the x-axis represents applied potential, but the axis is not continuous, hence the lack of markers. Each CV is offset by +0.5 V to show them side by side and not overlap. |
| ¶ The decrease in total product concentration from 8 h to 10 h is likely due to the migration of HMF-OR products across the anion exchange membrane, as the 10 h sample was taken some time later, after the CA programme stopped. |
| This journal is © The Royal Society of Chemistry 2023 |