
SpecLipIDA: a pseudotargeted lipidomics approach for polyunsaturated fatty acids in milk†
Jingnan
Lei
a,
Yuan
He
a,
Shuang
Zhu
a,
Jiachen
Shi
a,
Chin-Ping
Tan
b,
Yuanfa
Liu
 a and
Yong-Jiang
Xu
*a
a and
Yong-Jiang
Xu
*a
aState Key Laboratory of Food Science and Resources, School of Food Science and Technology, Jiangnan University, No. 1800 Lihu Road, Binhu District, Wuxi, Jiangsu 214122, People's Republic of China. E-mail: yjxutju@gmail.com; Tel: +086-510-853262
bDepartment of Food Technology, Faculty of Food Science and Technology, University Putra Malaysia, Selangor 410500, Malaysia
First published on 9th January 2024
Abstract
Polyunsaturated fatty acids (PUFAs), such as arachidonic acid (ARA), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA), play an important role in the nutritional value of milk lipids. However, a comprehensive analysis of PUFAs and their esters in milk is still scarce. In this study, we developed a novel pseudotargeted lipidomics approach, named SpecLipIDA, for determining PUFA lipids in milk. Triglycerides (TGs) and phospholipids (PLs) were separated using NH2 cartridges, and mass spectrometry data in the information-dependent acquisition (IDA) mode were preprocessed by MS-DIAL, leading to improved identification in subsequent targeted analysis. The target matching algorithm, based on specific lipid cleavage patterns, demonstrated enhanced identification of PUFA lipids compared to the lipid annotations provided by MS-DIAL and GNPS. The approach was applied to identify PUFA lipids in various milk samples, resulting in the detection of a total of 115 PUFA lipids. The results revealed distinct differences in PUFA lipids among different samples, with 44 PUFA lipids significantly contributing to these differences. Our study indicated that SpecLipIDA is an efficient method for rapidly and specifically screening PUFA lipids.
1. Introduction
Milk is recognised for its significant nutritional research value as an excellent nourishment for newborns and young children.1,2 Milk's lipid content, constituting 3–5% (w/v) of its composition, primarily consists of milk fat globules composed of triglycerides (TGs) and phospholipids (PLs) found in hydrophobic nuclei and amphiphilic membranes.3 Milk lipids not only serve as a major energy source, but also provide abundant physiologically active components.4 TGs and PLs consist of a backbone structure and fatty acyl chains. Polyunsaturated fatty acids (PUFAs) such as arachidonic acid (ARA), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA) exhibit essential physiological functions, including the modulation of lipid metabolism, anti-inflammatory properties, neuroprotection, and support for brain growth and development.5–8 Since the body's natural production of these vital PUFAs is limited, their inclusion in the diet is necessary for supply.9 Notably, studies involving 2474 women indicated an average ARA of 0.47% in breast milk (with a range of 0.24% to 1.0%), while the average DHA concentration was 0.32% (with a range of 0.06% to 1.4%).5 In contrast, human milk's EPA content is minimal, measuring at 0.12%.10 The substantial and stable presence of ARA in human milk is of biological significance as it ensures a continuous supply during brain growth and development. Therefore, these crucial PUFAs are introduced to formulas to mimic breast milk's composition and structure.11 Accurate characterization of PUFA lipids in human milk is vital for the development of infant nutrition.Given the complexity of milk lipids, their compositions vary extensively, and lipids enriched with specific PUFAs exhibit diverse profiles. However, the concentration of various lipid types in milk varies significantly.12 Notably, milk lipids contain lower levels of total polar lipids compared to other biological samples, with TGs as primary lipids affecting polar lipid detection substantially.13 Previous studies often required purification to prevent TG interference, especially for precious milk samples, resulting in reduced sample consumption for improved identification of polar lipids.13,14 In addition, the health effects of different lipid types and PUFA molecules in milk remain unclear. Establishing a comprehensive analytical approach to characterize PUFA lipids in milk is pivotal for assessing their nutritional contribution.
Various analytical techniques, including thin-layer chromatography (TLC), nuclear magnetic resonance (NMR), liquid chromatography, and liquid chromatography-mass spectrometry (LC-MS), have been employed to examine milk lipids.15 LC-MS, known for its high sensitivity and precision, can collect multidimensional data suitable for complex lipid investigation.16 However, the analysis of specific fatty acid lipids in milk using traditional untargeted lipidomics presents challenges due to incomplete coverage and structural characterization of lipids in a given sample.17 Current milk lipid analyses often focus on major lipid classes, neglecting specific molecular species. Due to the lipid class diversity, untargeted data acquisition struggles to provide required insights, and lipid annotation relies heavily on manual work.18 In recent years, numerous data processing software tools have emerged, many relying on two algorithms: rule-based matching based for lipid class features (e.g., LDA, LipidMatch) and database matching using scoring algorithms (e.g., MS-DIAL, LipidSearch).19–22 For example, Zhang et al. used ultra-high performance liquid chromatography-quadrupole time-of-flight mass spectrometry (UHPLC-Q-TOF-MS) to analyze lipids in breast milk and infant formula, identifying six PUFA PLs using an in-house lipid analyzer.10 Another study identified PUFA TG and PUFA PL in donkey and human milk using UHPLC-Q-Exactive orbitrap mass spectrometry with Lipidsearch software.16 While utilizing existing data processing software is recommended, database variability and limitations persist, and non-targeted lipidomics is still susceptible to errors, requiring additional measures for identification credibility.23 Increasingly, researchers are focusing on identifying specific biologically active lipids. Yu et al. developed a precursor ion scanning mass spectrometry method to identify EPA/DHA PLs in fish oils, achieving targeted data acquisition and molecular-level PL identification through chromatogram comparison with the Lipid Maps Structure Database (LMSD).17 However, this method's reliance on manual lipid annotation demands substantial effort when identifying multiple lipids. There is a lack of methods for rapid identification of lipids containing specific fatty acids.
This study introduces a novel pseudotargeted method, combining quadrupole time-of-flight mass spectrometry with a specific lipid matching algorithm, to analyze PUFA lipids in milk. Unlike general lipid composition changes, this method provides detailed insights into specific lipids. Optimized lipid separation conditions and mass spectrometry data acquisition modes were followed by MS-DIAL preprocessing. Identification relied on a self-constructed theoretical information library of PUFA lipids algorithmically matched with preprocessed data. This approach enabled the identification of numerous PUFA lipids from small milk samples, confirming accuracy, precision and sensitivity. The study contributes to an improved understanding of the molecular composition of PUFA lipids in milk samples.
2. Experimental
2.1. Materials and methods
Matured milk from Sanhe cattle (SH), Saanen goat (SN) and DairyMeade sheep (DR) from different Inner Mongolia farms was selected. Human milk (HM) samples were obtained from healthy volunteer mothers of similar age at Wuxi Children's Hospital, classified as mature milk (>30 days). All volunteer mothers were provided thorough information about the study, and the protocol received approval from Wuxi Children's Hospital (WXCH-2021-09-012). All samples were freeze-dried and stored at −80 °C. Two infant formula brands (IFA, IFB) were purchased from an official Chinese online store.Triarachidonoyl glycerol (>98%), trieicosapentaenoyl glycerol (>98%) and tridocosahexaenoyl glycerol (>98%) standards were obtained from Macklin Biochemical Co., Ltd (Shanghai, China). Standards including 1,2-diarachidonoyl-sn-glycero-3-phosphatidylcholine (>90%), 1,2-diarachidonoyl-sn-glycero-3-phosphoethanolamine (>90%), 1-octadecanoyl-2-Eiocosapentaenoyl-sn-glycero-3-phosphocholine (>90%), 1-stearoyl-2-eicosapentaenoyl-sn-glycero-3-phosphoethanolamine (>98%), 1-palmitoyl-2-docosahexaenoyl-sn-glycero-3-phosphocholine (>95%) and 1-palmitoyl-2-docosahexaenoyl-sn-glycero-3-phosphoethanolamine (>95%) were purchased from Cayman Chemicals (Ann Arbor, MI, USA). A bonded phase amino (NH2) cartridge (500 mg, 3 mL) and silica (Si) cartridge (1 g, 6 mL) were obtained from ANPEL Laboratory Technologies (Shanghai, China). Chloroform, methanol, n-hexane, diethyl ether, ethyl acetate, acetic acid, and acetone were supplied by Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Isopropanol, methanol and acetonitrile of HPLC-grade were obtained from Thermo Fisher Scientific (Auckland, New Zealand). Ultrapure water was obtained using a Milli-Q system (Millipore, Bedford, MA, USA).
2.2. Lipid extraction
Lipid extraction was performed using a modified Folch method.14,24 Briefly, 60 mg of milk powder was placed in a centrifuge tube, followed by addition of 0.35 mL of ultrapure water for complete dissolution, and then 1.05 mL of Folch reagent (chloroform/methanol 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) was added to extract the lipids. After vortex mixing, ultrasonication-assisted dissolution was carried out for 10 min, followed by centrifugation at 4 °C and 8000 rpm for 10 min to separate the phases. The lower chloroform phase was moved to a new tube for vacuum concentration, while the upper aqueous phase was extracted using 0.45 mL of Folch reagent, repeating the process twice more. The chloroform phase was combined and vacuum concentrated to yield the total lipid extraction, which was stored at −80 °C.
:1 v/v) was added to extract the lipids. After vortex mixing, ultrasonication-assisted dissolution was carried out for 10 min, followed by centrifugation at 4 °C and 8000 rpm for 10 min to separate the phases. The lower chloroform phase was moved to a new tube for vacuum concentration, while the upper aqueous phase was extracted using 0.45 mL of Folch reagent, repeating the process twice more. The chloroform phase was combined and vacuum concentrated to yield the total lipid extraction, which was stored at −80 °C.
2.3. Lipid separation
Following total lipid extraction, TGs and PLs were separated using two solid-phase extraction methods and an acetone precipitation method.:1 v/v) and transferring it. Elution of TGs involved the sequential use of 5 mL hexane/ether (50:1, v/v), 3 mL hexane/ether (6:1, v/v), and 1 mL hexane/ether (1:1, v/v). Subsequent elution of PLs employed 6 mL of methanol and 3 mL of chloroform/methanol/water (3:5:2, v/v/v).13
:1 v/v) and transferring it. Elution of TGs was carried out with 4 mL of hexane/ether (85:15, v/v), 4 mL of hexane/ethyl acetate (15:85, v/v), and 4 mL of acetic acid/ether solution (2:98, v/v). Subsequent elution of PLs involved 3 mL of methanol.
000 rpm at 2 °C for 10 min, the supernatant was transferred to a new centrifugal tube. The process was repeated twice, and the supernatants were combined to obtain TGs while the precipitates represented PLs.
The eluate was dried using a gentle nitrogen stream, and the obtained TGs and PLs were stored at −80 °C for subsequent analysis. Prior to analysis, the samples were reconstituted with chromatographic reagents.
2.4. LC-MS/MS analysis
LC-MS/MS analysis was performed using ultra performance liquid chromatography (UPLC) coupled with an AB SCIEX 5600 (QTOF) system. The chromatographic column used was Phenomenex Kinetex C18 (2.1 × 100 mm, 2.6 μm, 100 Å). Mobile phase A was composed of 60% acetonitrile (ACN) and 40% ultrapure water (6:4), while mobile phase B was composed of 90% isopropanol (IPA) and 10% ACN, and both spiked with 10 mM ammonium acetate, at a flow rate of 0.3 mL min−1. Samples were injected at 40 °C with a volume of 1 μL. The mobile phase gradient started at 40% B, increased to 100% B from 0 to 12 min, held for 0.5 min, decreased to 40% B from 13.5 to 13.7 min, and maintained for 4.3 min, resulting in a total run time of 18 min.28
Primary mass spectra were acquired in full scan acquisition mode, covering a mass range of 100–1200 Da. Secondary mass spectra were acquired in both information dependent acquisition (IDA) mode and sequential window acquisition of all theoretical mass spectra (SWATH) mode, with a mass range of 50–1200 Da. The ESI source parameters are as follows: ion source temperature at 450 °C; air curtain gas, ion source gas 1, and ion source gas 2 set at 40 psi, 50 psi, and 50 psi, respectively. Spray voltages in positive and negative ion modes were 5500 V and −4500 V, respectively. The collision energy used was 5 V for primary mass spectra and 40 ± 20 eV for secondary mass spectra. The IDA acquisition mode selected the 10 most abundant peaks for fragmentation, with each IDA experiment involving 1 MS1 scan and 10 product ion scans in a cycle time of 1.1 s. The SWATH acquisition mode featured 10 mass windows (110 Da/SWATH window), with each SWATH experiment comprising 1 MS1 scan and 10 mass windows in a cycle time of 1.1 s. Quality control samples were injected after every six samples, and three quality control sample tests were performed prior to sample analysis.
2.5. SpecLipIDA workflow
Fig. 1 summarizes the pseudotargeted SpecLipIDA workflow. Data processing includes these key steps: optimization of untargeted analysis conditions, primary and secondary spectral information acquisition using MS-DIAL 4.90 software, establishment of an information library of PUFA lipids based on analysis of product ion m/z values, and application of analysis based targeted analysis.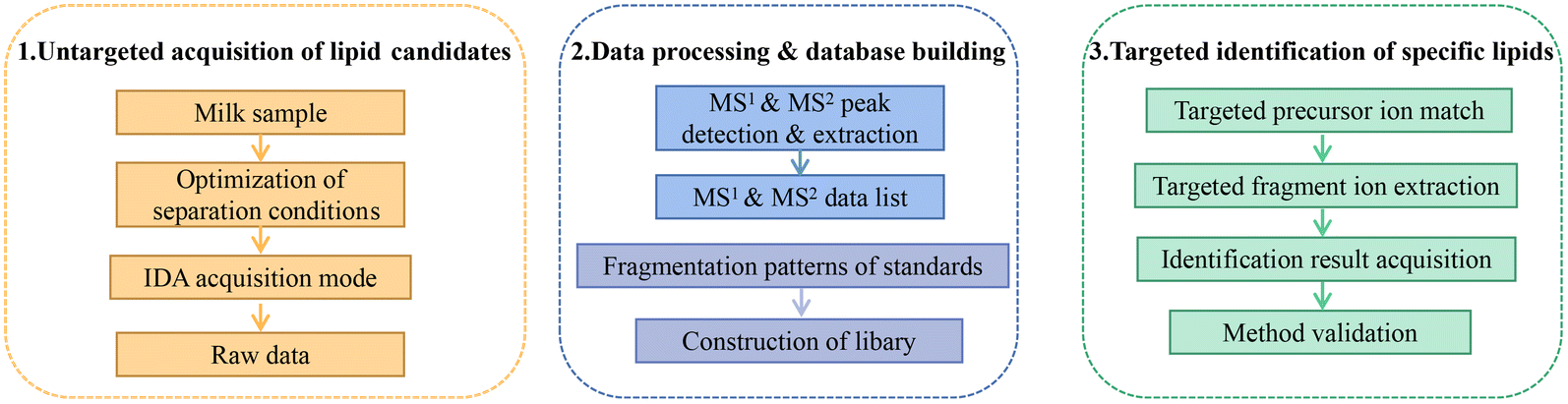 | ||
| Fig. 1 Schematic workflow for the pseudotargeted identification of specific polyunsaturated fatty acid lipids. | ||
2.6. Method validation
The method was evaluated for linearity, sensitivity, precision, and recovery. Standards were dissolved in chromatographic grade reagents to create 1 mg mL−1 solutions. Then standards were mixed, serially diluted for gradient concentrations. The limit of detection (LOD) represented the lowest detectable analyte concentration distinguishable from background noise, reported as a signal-to-noise ratio (S/N) of 3:1. The limit of quantification (LOQ) is the concentration yielding an S/N ratio of 10:1. Intraday and interday precision were verified by calculating relative standard deviation (RSD). Intraday precision involved six repetitions of the assay for each sample in one day, while interday precision repeated intraday precision once daily for six days. Recovery experiments introduced standards into the sample before lipid extraction, followed by complete experimental analysis in the same batch as the sample without standards.
2.7. Data statistics and analysis
Mass spectrometry raw data were processed using MS-DIAL 4.90 and SCIEX OS 2.0.1. Statistical analysis employed IBM SPSS Statistics 26. Multivariate analyses, specifically variable importance in projection (VIP) scores, were analyzed using SIMCA (version 14.1) and MetaboAnalyst 4.0. Figures were generated using Microsoft PowerPoint (version 2019) and GraphPad Prism (version 8.0).3. Results and discussion
3.1. Optimization of sample preparation
Milk lipids possess complex compositions, posing challenges in comprehensive compositional analysis. TGs constitute the main lipids, accounting for about 98% of the total, while PLs are minor, making up less than 2%. Consequently, the concentrations of different lipid types in milk lipids vary significantly.29 The presence of trace analytes in intricate matrices, along with numerous compounds for analysis, can lead to ion suppression.30 In this study, both TGs and PLs were studied. Matrix components primarily cause ion suppression, potentially affecting the analysis of low-abundance lipids.31 When milk lipids were directly analyzed without treatment, as depicted in Fig. 2E, no PUFA PLs were detected. One of the efficient strategies to lessen ion suppression is to remove interfering substances from the analytical process. Purification of PLs is an effective strategy to mitigate ion suppression. The positive ion mode was chosen for TG detection and the negative ion mode was chosen for PL detection. Fig. 2A–D depict the mapping diagram before and after milk lipid separation, highlighting a substantial increase in the number of PLs after separation. Fig. 2E also indicates the number of identified PUFA lipids after separation. Notably, efficient separation of milk lipids increased the detection of PUFA lipids. | ||
| Fig. 2 Optimization results of sample pretreatment methods. (A) Mapping in the positive ion mode before separation. (B) Mapping in the negative ion mode before separation. (C) Mapping in the positive ion mode after separation. (D) Mapping in the negative ion mode after separation. (E) Number of PUFA lipids identified before and after separation. (F) Recoveries of standards in different separation methods. (G) Number of PUFA lipids identified by different separation methods. | ||
Common lipid separation methods encompass solid-phase extraction and liquid–liquid extraction. Si cartridges and NH2 cartridges offer effective separation based on the lipid polarity difference,26,32,33 while liquid–liquid extraction exploits the characteristic of polar lipids’ insolubility in acetone to achieve swift separation of TGs and PLs in substantial quantities. Recovery of the standard mixture was utilized to evaluate these separation methods. Results in Fig. 2F demonstrate that NH2 cartridges exhibited recoveries of 69.31–92.42% for PUFA TGs, 77.16–85.35% for PCs, and 58.54–61.33% for PEs. Using Si cartridges, PUFA TG recoveries were 59.54–82.16%, PCs were 69.87–80.90%, and PEs were 29.01–40.48%. Acetone separation yielded PUFA TG recoveries of 64.36–83.65%, PCs of 10.89–27.42%, and PEs of 12.95–32.92%. NH2 cartridges outperformed Si cartridges and acetone separation in PUFA TGs, PCs, and PE recoveries. While PUFA TG recoveries were similar across the three methods, the recoveries of PUFA PEs from Si cartridges and acetone separation were extremely low. Notably, PUFA PCs recovered slightly less with Si cartridges compared to NH2 cartridges.
Solid phase extraction's elution mechanism depends on the interaction force between the target analyte and the adsorbent. Previous research indicated that NH2 cartridges excel in separating polar compounds in edible oils, partly due to their slightly lower polarity compared to Si cartridges.32 Si cartridges exhibited high affinity for polar lipids, possibly explaining the lower recovery of polar lipids. Acetone precipitation might not suit milk lipid separation due to the low PL content in milk and the temperature sensitivity of acetone precipitation for PLs.29 Gladkowski et al. found that the extraction yield of egg yolk PLs using acetone at 4 °C was significantly lower than that at 20 °C. They also noted that acetone temperature had an impact on PL solubility.34 In the process of repeated acetone extractions, certain PLs could be redistributed to the acetone phase, leading to suboptimal recovery of PLs and identification of species.
Furthermore, the number of identified PUFA lipids via the three separation methods was compared. NH2 cartridges outperformed others, independently identifying 49 PUFA TGs, 11 PUFA PCs, and 7 PUFA PEs (Fig. 2G). Considering both recovery rates and identification outcomes, NH2 cartridges proved optimal for TG and PL separation in milk lipids.
3.2. Optimization of data acquisition and processing
This study aimed to establish a high-coverage and high-sensitivity method for PUFA lipid analysis, comprehensively identifying PUFA TGs and PLs in milk lipids while mining MS-DIAL data deeply. The data acquisition mode significantly impacts lipid analysis. Mass spectrometry data rely on MS-DIAL preprocessing, and different acquisition modes greatly affect identification results. Data-independent acquisition (DIA) modes, such as SWATH, can obtain fragmentation data for all parent ions simultaneously, offering broad coverage and swift acquisition.35 While DIA modes have been commonly used in proteomics and metabolomics, their application in lipid analysis remains limited.14,36 The complex secondary spectra of SWATH and challenges in data processing with linkages between precursor and fragment ions make it difficult for use in lipid analysis.35 The IDA mode scans MS1 and MS2, with MS2 analysis dependent on chosen precursor ions, enhancing structural analysis. MS-DIAL's mathematical deconvolution can extract primary–secondary spectral data, matching with the silico library (LipidBlast) to annotate lipids.28 The SWATH mode, with its complex secondary spectra, makes MS-DIAL deconvolution less effective for accurate precursor-fragment ion matching. The IDA mode, on the other hand, yields higher-quality secondary spectra with unambiguous characteristic fragment information, as shown in Fig. S1.† Consequently, for data processing reliant on MS-DIAL for primary–secondary spectral data extraction, the IDA mode is recommended.To establish a PUFA lipid database, the PUFA lipid cleavage pattern was analyzed. The negative mode was preferred for efficient acquisition of characteristic fragments required for polar lipid structural analysis.13 An adduct ion was chosen to prevent the same lipid from being extracted in multiple forms: [M + NH4]+ for PUFA TGs, [M + OAc]− for PUFA PCs, and [M − H]− for PUFA PEs. Fig. S2† demonstrates that TG 20:4/20:4/20:4 produced a precursor ion at m/z 968.7703, with a fragment ion at m/z 647.5035 indicating ARA's neutral loss due to three identical fatty acid acyl chains. Fragment ions at m/z 269.2270, m/z 287.2377, and m/z 361.2794 reflected ARA loss as [RC = O–H2O]+, [RC = O]+, and [RC = O + 74]+. Similar cleavage rules applied to TG 20:5/20:5/20:5 and TG 22:6/22:6/22:6. Thus, the identification of PUFA TGs relied on characteristic fragments produced by their precursor ions and the neutral loss of fatty acyl chains. In negative ion mode, PC 20:4/20:4 formed [M + OAc]− at m/z 888.5759, and its precursor ion underwent a neutral loss of a methyl group at m/z 814.5388, which could be attributed to the fragmentation of the PC choline head. The loss of ARA was evident at m/z 303.2328, serving as a characteristic fragment for deducing the molecular structure of PC. Additionally, fragment ions at m/z 259.2431, m/z 510.2983, and m/z 528.3081 indicated the loss of ARA as well. Similar cleavage patterns were exhibited by PC 18:0/20:5 and PC 16:0/22:6. For PE 20:4/20:4, its precursor ion [M − H]− was observed at m/z 786.5071. The fragment ion at m/z 303.2329 corresponded to ARA, while m/z 259.2432 and m/z 500.2772 indicated the loss of [RCH2]− and [M − H–RCH = C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O]−, respectively. PE 18:0/20:5 and PE 16:0/22:6 showed identical cleavage rules. By profiling the cleavage rules, the database of PUFA lipids was established, and the identification of PUFA lipids was achieved through three key steps. Firstly, MS1 mass was accurately matched. Secondly, MS2 mass was further matched by screening of precursor ions. Finally, the molecular structure was ascertained based on the characteristic fragments reflecting fatty acyl chains. SpecLipIDA is founded on the preprocessing by MS-DIAL and the identification of characteristic fragments, which complements the MS-DIAL program, as specified in the Materials and Methods section.
O]−, respectively. PE 18:0/20:5 and PE 16:0/22:6 showed identical cleavage rules. By profiling the cleavage rules, the database of PUFA lipids was established, and the identification of PUFA lipids was achieved through three key steps. Firstly, MS1 mass was accurately matched. Secondly, MS2 mass was further matched by screening of precursor ions. Finally, the molecular structure was ascertained based on the characteristic fragments reflecting fatty acyl chains. SpecLipIDA is founded on the preprocessing by MS-DIAL and the identification of characteristic fragments, which complements the MS-DIAL program, as specified in the Materials and Methods section.
In this study, we compared the identification results of three data processing methods: SpecLipIDA, MS-DIAL, and GNPS (Fig. 3A). The raw data were processed using MS-DIAL, leading to the extraction of 219 TG features and 251 PL features in positive and negative ion modes, respectively. The lipid characterization by SpecLipIDA was congruent with the MS-DIAL results due to its dependence on MS-DIAL preprocessing. On the other hand, GNPS extracted 103 TG features and 143 PL features in positive and negative ion modes, respectively. Further targeting PUFA lipids, MS-DIAL, GNPS, and SpecLipIDA extracted 19, 7, and 38 PUFA TG features, as well as 4, 5, and 23 PUFA PL features (including isomers). While MS-DIAL managed to extract more lipid features compared to GNPS, SpecLipIDA demonstrated the ability to mine a greater number of PUFA lipid features than MS-DIAL.
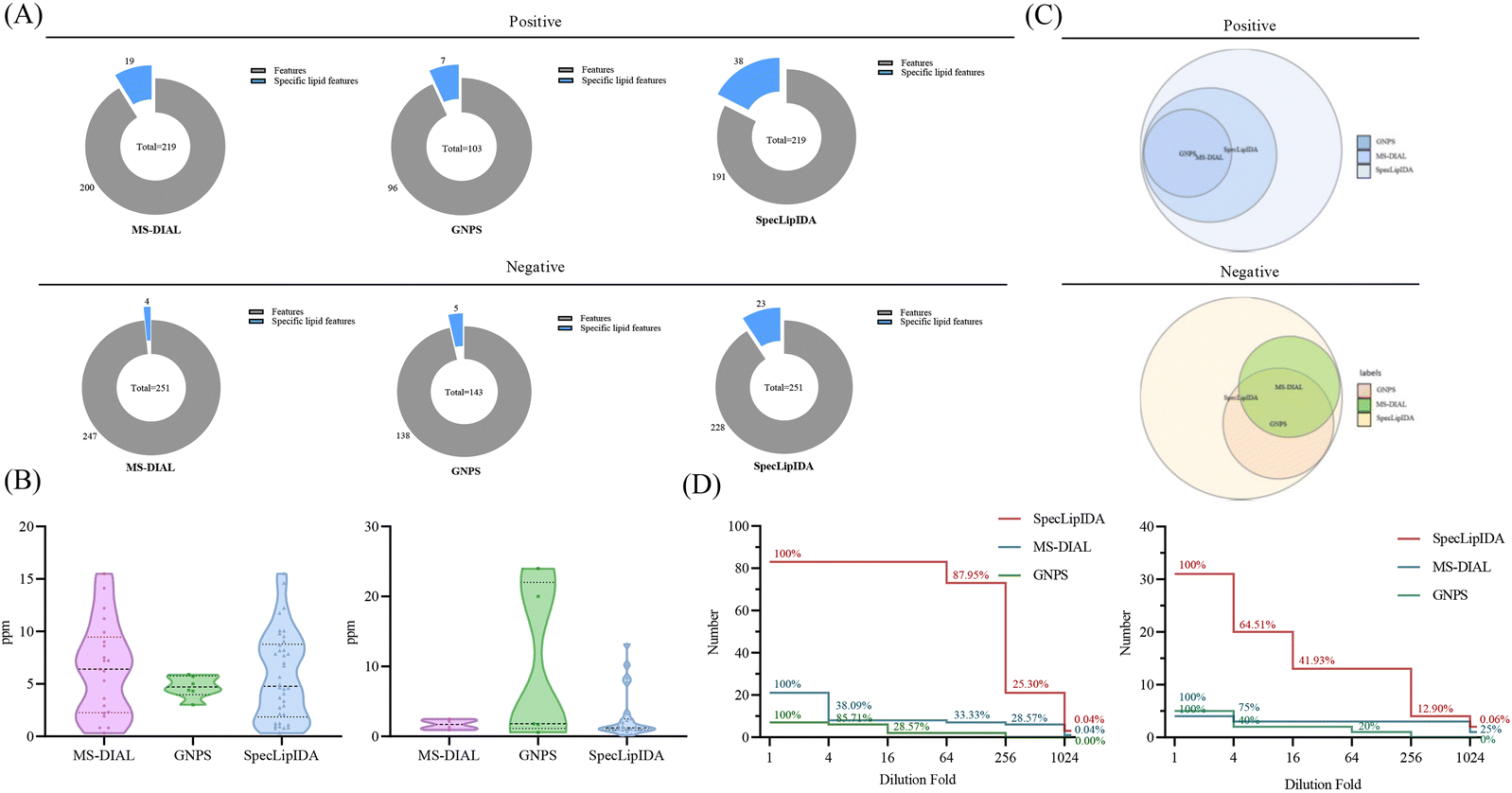 | ||
| Fig. 3 Comparison between different data processing methods, including (A) coverage, (B) mass deviation, (C) identification results, and (D) sensitivity. | ||
We compared the mass deviations in the identification results of the three methods, as depicted in Fig. 3B. The mass deviation of PUFA TGs identified by the three methods remained within 20 ppm. The majority of MS-DIAL and SpecLipIDA identifications were distributed within the range of 0–10 ppm, while most of the lipids identified by GNPS were distributed in the 0–5 ppm range. For PUFA PLs, MS-DIAL exhibited the lowest number of identifications with the least mass deviation, SpecLipIDA predominantly fell within the lower ppm range, and GNPS identified fewer numbers with more significant ppm fluctuations. In sum, despite their differences, the three methods exhibited acceptable mass deviations, affirming the reliability of the identification results.
When comparing the PUFA lipids identified by the three methods, SpecLipIDA identified 83 PUFA TGs and 32 PUFA PLs, encompassing identification from both MS-DIAL and GNPS, as shown in Fig. 3C. While GNPS relies on molecular network technology for lipid identification, its lipid database is relatively limited. Conversely, the MS-DIAL database is more extensive, including LipidBlast, MassBank, MetaboBASE, and other databases, which provides MS-DIAL an advantageous position in lipid identification.14 Therefore, utilizing MS-DIAL for lipid data preprocessing is advantageous. The C18 column and mass spectrometry posed challenges in separating co-effluents and lipids, leading to the accumulation of mass spectral information from multiple lipid molecules in the same secondary spectra. MS-DIAL effectively identified lipids at the sum composition level; however, it only annotated lipids with the highest matches at the molecular lipid level. This limitation resulted in fewer identifications for analyzing PUFA lipids. SpecLipIDA effectively addressing this challenge during PUFA lipid analysis by accurately matching PUFA lipid feature fragments. This approach facilitates precise identifications, enables profound data mining, and introduces a novel dimension to data analysis.
A series of diluted samples, with dilutions up to 1024-fold, were analyzed to evaluate the sensitivity of the three analytical methods. Each diluted sample underwent three replicates, and if an analyte was not detected in more than two runs, it was considered below the detection limit. The results, depicted in Fig. 3D, revealed that for PUFA TGs, SpecLipIDA continued to detect 87.95% of lipids at dilutions up to 64-fold, with a sharp decline beyond this point. In contrast, MS-DIAL's number of identifications sharply dropped at dilutions up to 4-fold, followed by a steady decrease, and GNPS exhibited a similar pattern. For PUFA lipids, both MS-DIAL and GNPS experienced significant declines in identification as the sample was progressively diluted, whereas SpecLipIDA, preprocessed by MS-DIAL, maintained robust sensitivity owing to its targeted feature fragment matching. It is important to note that if the sample concentration is sufficiently low, MS-DIAL data preprocessing may omit low-abundance fragments, thus affecting SpecLipIDA's identification results. For PUFA PLs, SpecLipIDA consistently sustained higher identification level compared to MS-DIAL and GNPS, despite a sharp drop in percentage at dilutions of up to 16-fold. The declining trend was smoother for MS-DIAL, while GNPS exhibited the least sensitivity. However, considering the relatively small base of MS-DIAL identifications, the decrease was gradual. Additionally, the injection concentration was found to have a more pronounced effect on the identification results for PUFA lipids with a low content.
To conclude, the pseudotargeted method employed in this study, labeled as “SpecLipIDA”, exhibited superior identification of specific lipids in IDA mode. It effectively harnessed deep data mining from MS-DIAL processed data, combining identification results from both MS-DIAL and GNPS methods. The method demonstrated commendable reliability and sensitivity, making it a suitable strategy for PUFA lipid analysis.
3.3. Method validation
The reliability of the SpecLipIDA method was further verified in terms of linearity, sensitivity, precision, and recovery. The results are presented in Table 1. The linear regression equations exhibited R2 values ranging between 0.9902 and 0.9997 for TGs within the 1–200 μg mL−1 range and PLs within the 0.5–100 μg mL−1 range, demonstrating a strong linear relationship between the peak area and standard concentration. Regarding sensitivity, the LOD and LOQ fell between 0.025–0.1 μg mL−1 and 0.05–0.25 μg mL−1, respectively, indicating the high sensitivity of the method. The RSD between ion chromatographic peak areas of standards and analytes in samples was below 12%, indicating the method's high stability and accuracy. For recovery experiments, TG and PL standards with concentrations of 50 μg mL−1 and 100 μg mL−1 were added before milk lipid extraction. Recoveries ranged from 58.54% to 92.42% for each standard. The lower recovery of PE could be due to the poor solubility of methanol as the solvent for PE. However, the reproducibility of its recoveries was better, so no change in solvent was needed.| Standard | Adduct ion | Calibration equation | R 2 | LOD (μg mL−1) | LOQ (μg mL−1) | Linear range (μg mL−1) | Intraday precision RSD% | Interday precision RSD% | Reproducibility RSD% |
|---|---|---|---|---|---|---|---|---|---|
| TG 20:4/20:4/20:4 | [M + NH4]+ |
y = 44250x + 17467 |
0.9991 | 0.050 | 0.150 | 1–200 | 2.29 | 6.68 | 1.91 |
| TG 20:5/20:5/20:5 | [M + NH4]+ |
y = 16482x + 776.23 |
0.9997 | 0.100 | 0.500 | 1–200 | 2.45 | 7.69 | 5.08 |
| TG 22:6/22:6/22:6 | [M + NH4]+ |
y = 31213x − 2643.3 |
0.9995 | 0.075 | 0.250 | 1–200 | 0.71 | 7.63 | 4.24 |
| PC 20:4/20:4 | [M + OAc]− |
y = 101377x + 40970 |
0.9927 | 0.025 | 0.060 | 0.5–100 | 1.51 | 8.10 | 5.46 |
| PC 18:0/20:5 | [M + OAc]− |
y = 124338x + 68686 |
0.9944 | 0.030 | 0.125 | 0.5–100 | 1.74 | 6.16 | 5.39 |
| PC 16:0/22:6 | [M + OAc]− |
y = 94988x + 74903 |
0.9902 | 0.050 | 0.125 | 0.5–100 | 4.34 | 7.46 | 3.39 |
| PE 20:4/20:4 | [M − H]− |
y = 190764x + 79489 |
0.9920 | 0.020 | 0.050 | 0.5–100 | 2.85 | 9.08 | 4.11 |
| PE 18:0/20:5 | [M − H]− |
y = 132944x + 100408 |
0.9945 | 0.040 | 0.200 | 0.5–100 | 3.86 | 9.38 | 6.38 |
| PE 16:0/22:6 | [M − H]− |
y = 189595x + 84297 |
0.9918 | 0.025 | 0.055 | 0.5–100 | 4.10 | 10.43 | 4.62 |
3.4. Method application
The established method was used to analyze PUFA lipids in different milk samples, which are widely used for lipid composition analysis. The distribution of PUFA lipids in the six types of milk samples is shown in Fig. 4. The diversity of PUFA TGs across different milk samples was due to the variation in fatty acyl chains present in TGs. HM had the highest number of identified PUFA TGs (47), followed by DR. HM and IFB exhibited similar identification results. As for PUFA PLs, there was substantial similarity in lipid species among milk samples, with the highest number (28) identified in HM and SN. Among PUFA lipids, ARA-containing lipids were the most prevalent, followed by DHA, and EPA was the least abundant, consistent with the findings of Jiang et al.13 We compared the analytical results of PUFA lipids with other milk studies. Jiang et al. detected 11 PUFA PEs and 5 PUFA PCs in donkey milk and camel milk, along with 4 PUFA PGs, 7 PUFA PIs, and 5 PUFA PSs.13 Aside from the variation in biological samples, opted for multiple adduct ions instead of one to identify the same lipid class. Tang et al. examined lipid distribution in human milk, animal milk, and formula, identifying 4 PUFA TGs, 2 PUFA PCs, and 2 PUFA PEs.37 Their findings were fewer than ours, as they conducted identification through manual proofreading, which was a laborious and incomplete approach. Given that the instrument's response intensity differs for various PUFA lipid types and finding corresponding standards is challenging, we chose a relatively quantitative method for subsequent analysis. This method involved dividing the peak area of each PUFA lipid by the total peak areas of all PUFA lipids within the category. Because isomers could not be eluted using the separation method, we employed MS2 as a mean of quantification. MS2 offered higher specificity and a lower isomer contribution compared to MS1.38 In SWATH mode, the OS-Q software provided the MS2 peak area by entering the MS1 mass and MS2 mass. The chemical structures and relative contents of the detected PUFA lipid molecular species in the six sample types are presented in Tables S1 and S2.† In addition, Fig. S4† illustrates the total relative content of PUFA lipids in the six milk types. For PUFA TGs, the highest content was TG 20:4/18:1/18:2 (14.24 ± 1.63%) in HM, TG 20:4/18:0/18:1 (43.08 ± 5.6%) in DR, TG 22:6/16:0/18:1 (22.61 ± 4.35%) in SN, TG 20:5/16:0/18:1 (28.14 ± 4.03%) in SH, and the highest in IFA (75.2 ± 2.47%) and IFB (35.86 ± 2.64%) was TG 20:4/16:0/16:1. The high TG 20:4/16:0/16:1 content in IFA and IFB might be due to the addition of microbial oils to commercial formulas to increase the PUFA content. Among PUFA PCs, PC 20:4/16:0 had the highest content in all six samples, ranging from 38.44 ± 6.06% to 55.9 ± 7.18%. Regarding PUFA PEs, PE 20:4/18:0 (26.35 ± 1.5%) was most abundant in HM, while the highest content of PE 20:4/18:1 was found in DR, SN, IFA, and IFB, ranging from 20.42 ± 0.59% to 23.09 ± 3.03%. In the SH, PE 20:4/16:0 (27.08 ± 0.48%) was the predominant variant. These results highlighted differences in PUFA lipid types and distribution between HM and other samples. To formulate infant formulas with a lipid composition and content similar to human milk, in-depth qualitative and quantitative analyses of milk lipids are essential.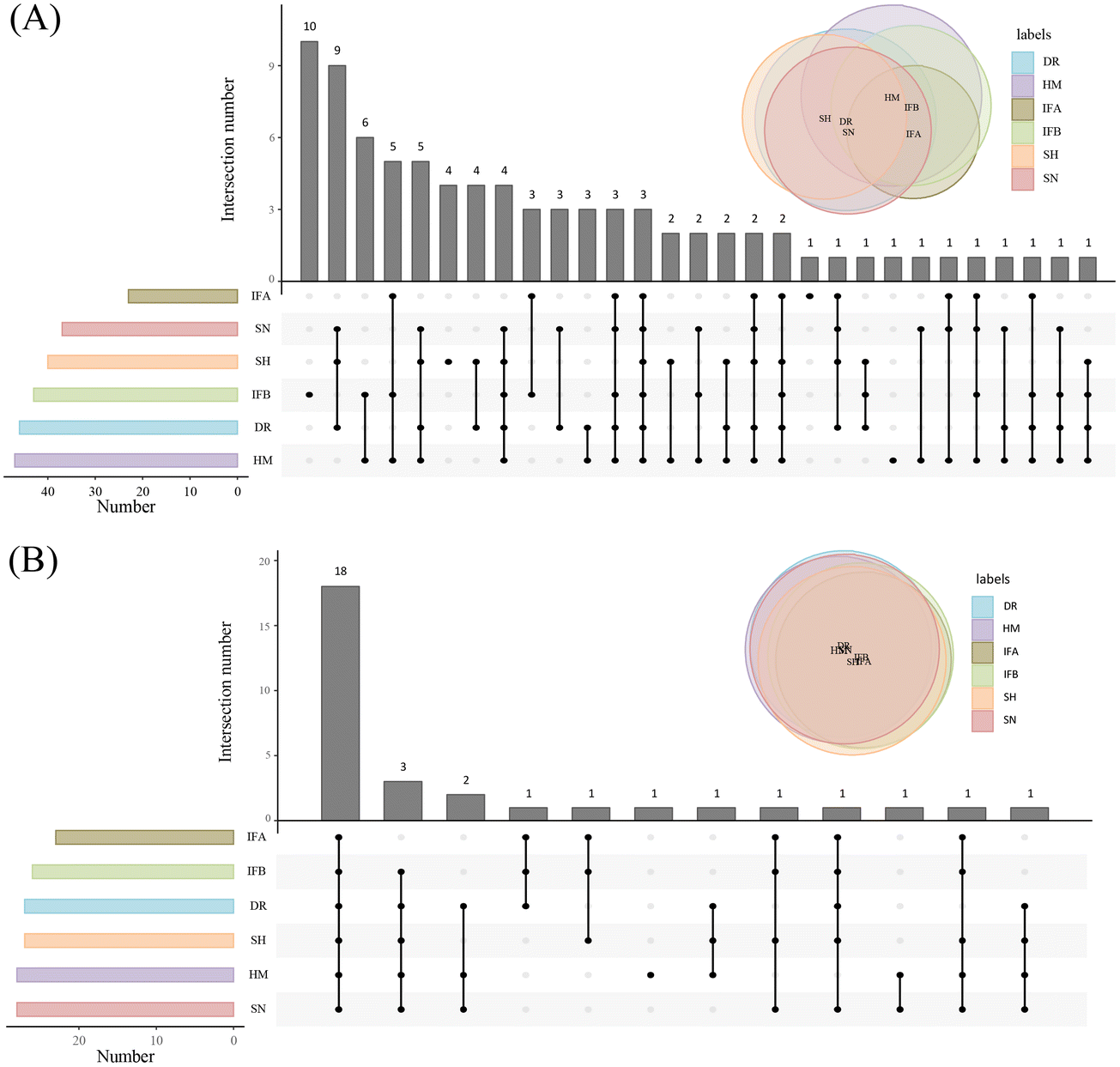 | ||
| Fig. 4 Distribution of PUFA lipids in six milk samples. (A) Distribution of PUFA TGs. (B) Distribution of PUFA PLs. | ||
Furthermore, orthogonal partial least squares discriminant analysis (OPLS-DA) and HeatMap models were used to illustrate the similarities and differences of PUFA lipids among different milk samples. The OPLS-DA score plots highlighted effective separation among milk species for both PUFA TGs and PUFA PLs, with the model validity confirmed through testing (Fig. S3†). Fig. 5A indicates that DR, SN, and SH were relatively similar in composition, while HM was well separated from IFA and IFB. For PUFA PLs, IFA and IFB exhibited similar compositions distinct from other samples (Fig. 5D). The heatmap supported these findings, revealing differences between human milk and other species’ samples and some similarities among three animal milk samples and two infant formulas. This result suggests that the species of organisms and their origins significantly influence the PUFA lipid composition. To assess the impact of these differences, we used the VIP value. PUFA lipids with VIP value >1.0 were highlighted. As shown in Fig. 5C and F, 28 PUFA TGs and 16 PUFA PLs emerged as potential markers for distinguishing diverse milk sources. Notably, ARA lipids stood out among the differential lipids, indicating their prominent role in shaping distinct sample variations.
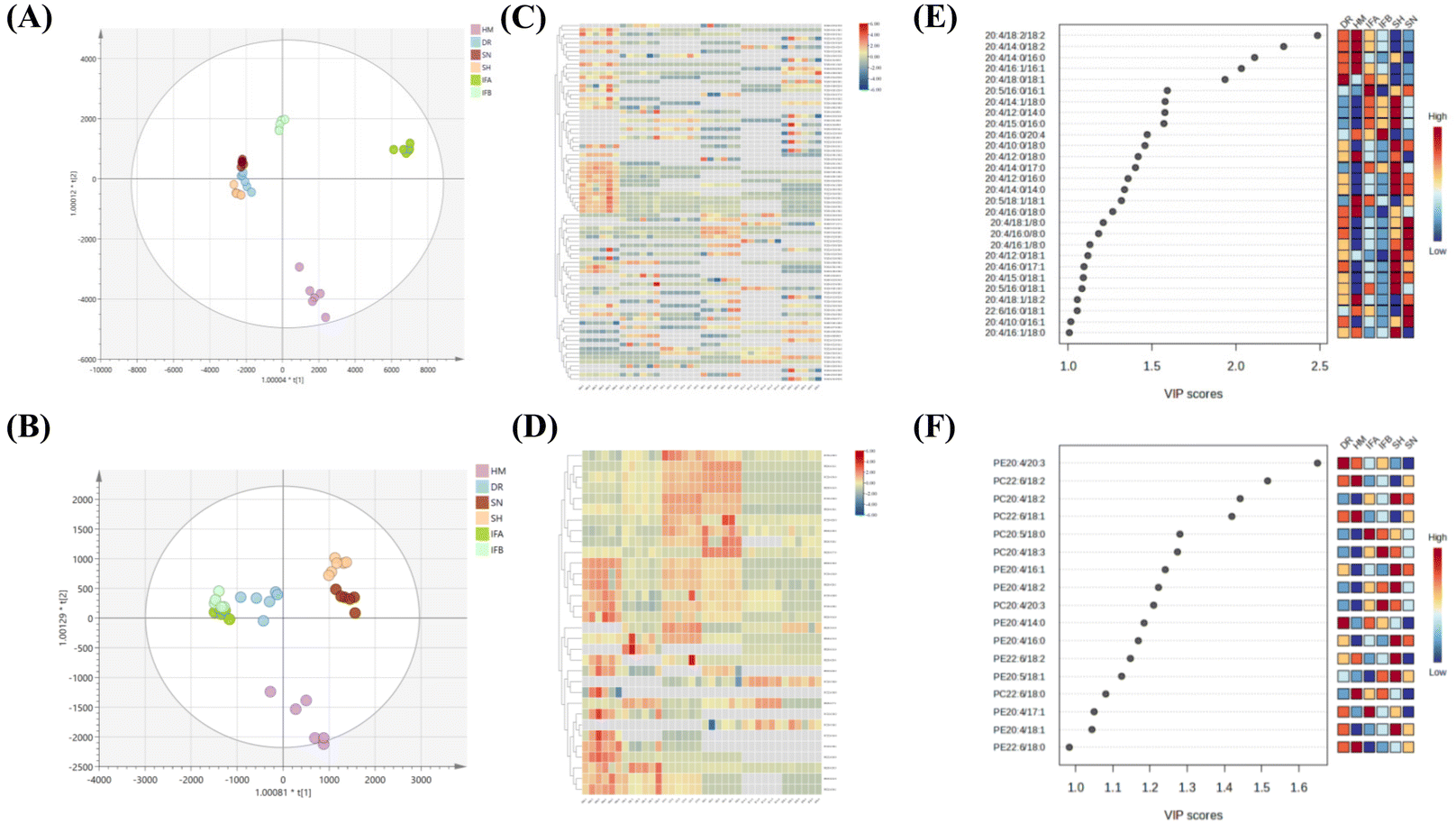 | ||
| Fig. 5 Statistical analysis of PUFA lipids from six milk samples. (A) OPLS–DA score plot of PUFA TGs. (B) OPLS–DA score plot of PUFA PLs. (C) Heat map of PUFA TGs. (D) Heat map of PUFA PLs. (E) VIP scores of PUFA TGs. (F) VIP scores of PUFA PLs. | ||
4. Conclusion
In contrast to the traditional MS-DIAL identification method, our approach enables comprehensive data mining and identification of specific lipids at the molecular lipid level. While the method offers speed and precision, it is essential to select suitable separation and data acquisition modes to achieve optimal spectra. Given the intricate and diverse nature of milk lipid composition, our future research will delve into in-depth milk lipid analysis, providing an analytical basis for infant formula development.Abbreviations
| PUFAs | Polyunsaturated fatty acids |
| ARA | Arachidonic acid |
| EPA | Eicosapentaenoic acid |
| DHA | Docosahexaenoic acid |
| TGs | Triglycerides |
| PLs | Phospholipids |
| IDA | Information-dependent acquisition |
| TLC | Thin-layer chromatography |
| NMR | Nuclear magnetic resonance |
| LC-MS | Liquid chromatography-mass spectrometry |
| LMSD | Lipid maps structure database |
| SH | Sanhe cattle |
| SN | Saanen goat |
| DR | DairyMeade sheep |
| HM | Human milk |
| ACN | Acetonitrile |
| IPA | Isopropanol |
| SWATH | Sequential window acquisition of all theoretical mass spectra |
| PCs | Phosphatidylcholine |
| PEs | Phosphatidylethanolamine |
| LOD | Limit of detection |
| S/N | Signal-to-noise ratio |
| LOQ | Limit of quantification |
| RSD | Relative standard deviation |
| VIP | Variable importance in projection |
| OPLS-DA | Orthogonal partial least squares discriminant analysis |
Author contributions
Jingnan Lei: methodology, software, formal analysis, data curation, and writing – original draft; Yuan He: software and data curation; Shuang Zhu: data curation; Jiachen Shi: methodology, Chin-Ping Tan: writing – review & editing; Yuanfa Liu and Yong-Jiang Xu: conceptualization, project administration, funding acquisition, validation, and writing – review & editing. All authors contributed to the final draft of the manuscript.Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (32172136), the Project of Jiangsu Provincial Center of Technology Innovation for Future Food (BM2020023), and the Collaborative Innovation Center of Food Safety and Quality Control in Jiangsu Province, Jiangnan University.References
- L. Wang, X. Li, L. Liu, H. da Zhang, Y. Zhang, Y. Hao Chang and Q. P. Zhu, Food Chem., 2020, 310, 125865 CrossRef CAS PubMed.
- Y. Sun, S. Tian, M. Hussain, S. Lin, Y. Pan, X. Li, L. Liu, X. Lu, C. Li, Y. Leng and S. Jiang, Food Res. Int., 2022, 161, 111872 CrossRef CAS PubMed.
- Q. Liu, J. Zhao, Y. Liu, W. Qiao, T. Jiang, Y. Wang, Y. Liu, Z. Luo, T. Yudron, J. Hou, Y. Liu and L. Chen, Food Res. Int., 2022, 157, 111025 CrossRef CAS PubMed.
- N. J. Andreas, B. Kampmann and K. Mehring Le-Doare, Early Hum. Dev., 2015, 91, 629–635 CrossRef CAS PubMed.
- N. Salem Jr. and P. Van Dael, Nutrients, 2020, 12, 626 CrossRef PubMed.
- J. T. Brenna, Nutr. Rev., 2016, 74, 329–336 CrossRef PubMed.
- C. I. Janssen and A. J. Kiliaan, Prog. Lipid Res., 2014, 53, 1–17 CrossRef CAS PubMed.
- X. Y. Cui, S. Jiang, C. C. Wang, J. Y. Yang, Y. C. Zhao, C. H. Xue, Y. M. Wang and T. T. Zhang, J. Agric. Food Chem., 2022, 70, 13327–13339 CrossRef CAS PubMed.
- M. Plourde and S. C. Cunnane, Appl. Physiol., Nutr., Metab., 2007, 32, 619–634 CrossRef CAS PubMed.
- X. Zhang, L. Liu, L. Wang, Y. Pan, X. Hao, G. Zhang, X. Li and M. Hussain, J. Agric. Food Chem., 2021, 69, 1146–1155 CrossRef CAS PubMed.
- H. Kikukawa, E. Sakuradani, A. Ando, S. Shimizu and J. Ogawa, J. Adv. Res., 2018, 11, 15–22 CrossRef CAS PubMed.
- S. Song, T. T. Liu, X. Liang, Z. Y. Liu, D. Yishake, X. T. Lu, M. T. Yang, Q. Q. Man, J. Zhang and H. L. Zhu, Food Chem., 2021, 348, 129091 CrossRef CAS PubMed.
- C. Jiang, X. Zhang, J. Yu, T. Yuan, P. Zhao, G. Tao, W. Wei and X. Wang, Food Chem., 2022, 393, 133336 CrossRef CAS PubMed.
- X. Chen, X. Peng, X. Sun, L. Pan, J. Shi, Y. Gao, Y. Lei, F. Jiang, R. Li, Y. Liu and Y. J. Xu, J. Agric. Food Chem., 2022, 70, 7815–7825 CrossRef CAS PubMed.
- S. Imperiale, K. Morozova, G. Ferrentino and M. Scampicchio, Eur. Food Res. Technol., 2023, 249, 861–902 CrossRef CAS.
- X. Zhang, H. Li, L. Yang, G. Jiang, C. Ji, Q. Zhang and F. Zhao, J. Food Compos. Anal., 2021, 101, 103988 CrossRef CAS.
- X. Yu, Q. Wang, W. Lu, M. Zhang, K. Chen, J. Xue, Q. Zhao, P. Wang, P. Luo and Q. Shen, J. Agric. Food Chem., 2021, 69, 7997–8007 CrossRef CAS PubMed.
- J. A. Bowden, C. Z. Ulmer, C. M. Jones, J. P. Koelmel and R. A. Yost, Metabolomics, 2018, 14, 53 CrossRef PubMed.
- E. Rampler, Y. E. Abiead, H. Schoeny, M. Rusz, F. Hildebrand, V. Fitz and G. Koellensperger, Anal. Chem., 2021, 93, 519–545 CrossRef CAS PubMed.
- J. P. Koelmel, N. M. Kroeger, C. Z. Ulmer, J. A. Bowden, R. E. Patterson, J. A. Cochran, C. W. W. Beecher, T. J. Garrett and R. A. Yost, BMC Bioinf., 2017, 18, 331 CrossRef PubMed.
- Z. Ni, G. Angelidou, M. Lange, R. Hoffmann and M. Fedorova, Anal. Chem., 2017, 89, 8800–8807 CrossRef CAS PubMed.
- H. Tsugawa, T. Cajka, T. Kind, Y. Ma, B. Higgins, K. Ikeda, M. Kanazawa, J. VanderGheynst, O. Fiehn and M. Arita, Nat. Methods, 2015, 12, 523–526 CrossRef CAS PubMed.
- V. B. O’Donnell, E. A. Dennis, M. J. O. Wakelam and S. Subramaniam, Sci. Signaling, 2019, 12, eaaw2964 CrossRef PubMed.
- J. Folch, M. Lees and G. H. S. Stanley, J. Biol. Chem., 1957, 226, 497–509 CrossRef CAS PubMed.
- M. A. Kaluzny, L. A. Duncan, M. V. Merritt and D. E. Epps, J. Lipid Res., 1985, 26, 135–140 CrossRef CAS.
- F. Teng, M. G. Reis, L. Yang, Y. Ma and L. Day, Food Chem., 2019, 297, 124976 CrossRef CAS PubMed.
- N. Price, Z. Wan, T. Fei, S. Clark and T. Wang, J. Am. Oil Chem. Soc., 2020, 97, 1043–1053 CrossRef CAS.
- X. Sun, J. Shi, R. Li, X. Chen, S. Zhang, Y. J. Xu and Y. Liu, J. Agric. Food Chem., 2022, 70, 3331–3343 CrossRef CAS PubMed.
- A. H. Ali, X. Zou, S. M. Abed, S. A. Korma, Q. Jin and X. Wang, Crit. Rev. Food Sci. Nutr., 2019, 59, 253–275 CrossRef CAS PubMed.
- A. Furey, M. Moriarty, V. Bane, B. Kinsella and M. Lehane, Talanta, 2013, 115, 104–122 CrossRef CAS PubMed.
- Y. Zhang, M. Zhang, L. Dong, J. Chang, H. Wang and Q. Shen, Eur. J. Lipid Sci. Technol., 2021, 124, 2100097 CrossRef.
- V. Ruiz-Gutierrez and M. C. Perez-Camino, J. Chromatogr. A, 2000, 885, 321–341 CrossRef CAS PubMed.
- F. Pernet, C. J. Pelletier and J. Milley, J. Chromatogr. A, 2006, 1137, 127–137 CrossRef CAS PubMed.
- W. Gładkowski, A. Chojnacka, G. Kiełbowicz, T. Trziszka and C. Wawrzeńczyk, J. Am. Oil Chem. Soc., 2011, 89, 179–182 CrossRef.
- J. Guo and T. Huan, Anal. Chem., 2020, 92, 8072–8080 CrossRef CAS PubMed.
- H. Zha, Y. Cai, Y. Yin, Z. Wang, K. Li and Z. J. Zhu, Anal. Chem., 2018, 90, 4062–4070 CrossRef CAS PubMed.
- Y. Tang, M. M. Ali, X. Sun, A. A. Debrah, M. Wang, H. Hou, Q. Guo and Z. Du, J. Chromatogr. A, 2021, 1658, 462606 CrossRef CAS PubMed.
- M. Raetz, E. Duchoslav, R. Bonner and G. Hopfgartner, Anal. Bioanal. Chem., 2019, 411, 5681–5690 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Secondary mass spectra in two acquisition modes; the fragmentation principle of PUFA lipids; OPLS-DA analysis of milk samples; the total relative content of PUFA lipids in the six milk samples (Fig. S1–S4); identification of PUFA lipids and the distribution of PUFA lipids in six milk samples (Tables S1 and S2). See DOI: https://doi.org/10.1039/d3an01536j |
| This journal is © The Royal Society of Chemistry 2024 |