
Ag nanoisland functionalized hollow carbon nanofibers as a non-invasive, label-free SERS salivary biosensor platform for salivary nitrite detection for pre-diagnosis of oral cancer†
Navami
Sunil
,
Rajesh
Unnathpadi
and
Biji
Pullithadathil
 *
*
Nanosensors and Clean Energy Laboratory, PSG Institute of Advanced Studies, Coimbatore-641004, India. E-mail: bijuja123@yahoo.co.in; pbm@psgias.ac.in
First published on 14th May 2024
Abstract
A highly selective, label-free, surface-enhanced Raman spectroscopy (SERS) based sensor platform employing hollow carbon nanofibers functionalized with silver nanoparticles (Ag@HCNFs) has been developed to monitor anomalous concentrations of potential biomarkers, such as salivary nitrite facilitating pre-diagnosis of oral cancer. Co-axial electrospinning was used for the fabrication of the nanofibrous Ag@HCNFs followed by thermal treatment of PAN/PVP core–shell nanofibers and chemical reduction of silver nanoislands. The developed plasmonic Ag@HCNFs was structurally and morphologically characterized using X-Ray diffraction, Raman spectroscopy, scanning electron microscopy, and transmission electron microscopy, which clearly demonstrated the successful anchoring of silver nanoparticles on hollow carbon nanofibers. The properties of Ag@HCNFs showed significant SERS enhancement of the order of 107 with a detection limit of 10−11 M with R6G, demonstrating its efficacy to investigate real-time salivary samples, particularly towards the detection of salivary nitrite within the clinically relevant range (50 μM–300 μM) towards the pre-diagnosis of oral cancer. The proposed SERS-based salivary platform has the potential to be used as a low-cost, non-invasive pre-diagnostic tool for early diagnosis and mass screening of oral cancer.
Introduction
Oral cancer is projected as one of the top ten most prevalent malignancies, which often becomes more risky with delayed clinical diagnosis, and has poor prognosis due to a lack of specific biomarkers, and expensive treatment options. It covers a wide range of tumours that are localized in the tongue base, tonsils, posterior pharyngeal wall, hard and soft palates, and buccal mucosa, and other oropharyngeal sub-sites.1,2 More than 50% of cases that are identified in oral cancer is still fatal, with one of the lowest five-year survival rates (about 50%).3 There is a considerable risk of malignant tumours being undiagnosed in hidden locations while using the current methods of diagnosis focusing on clinical investigations and histological examinations. Clinical examination, white light endoscopy, and histopathological analysis of needle biopsies have been the gold-standard diagnostic techniques for oral cancer. However, these methods have a major drawback of limited diagnosis accuracy and such investigations are usually conducted only in later stages of the disease when lesions appear abnormal. In this scenario, there is a significant demand for non-invasive, highly specific, and sensitive detection techniques.4,5Saliva has been actively researched as a potential diagnostic tool over the past ten years due to its non-invasive accessibility, ease of use, and availability of biomarkers such as genetic materials and proteins.6 Saliva contains a variety of biomolecules that enter the mouth through the blood by passive diffusion, active transport, or extracellular ultra-filtration. These biomolecules include hormones, antibodies, growth factors, enzymes, and microorganisms making it as a desirable diagnostic candidate for the detection and monitoring of various biomarkers in new-borns, children, adults, and uncooperative patients because it can be seen in many instances as a reflection of the physiological function of the body.7,8 Among them, salivary nitrite is being extensively investigated as a potential biomarker for the diagnosis of oral cancer. The presence of nitrite ions could be used to measure the concentration of nitric oxide (NO) molecules in cell cultures, which is an indication of oxidative stress in cells and could be potentially associated with oral and nasopharyngeal cancer.9,10 Foods containing nitrite ions, such as drinking water, fruits, vegetables, and salty sausages, enter the digestive tract through the oral cavity, where they further travel to the salivary glands and the bloodstream. According to numerous reports, patients with oral cancer have salivary nitrite concentraton levels much greater (>200 μg L−1) than those in control groups, which range from 30 to 100 μg L−1.11,12 Numerous analytical techniques, including chromatography,13 fluorometry,14 electrochemistry,15 colorimetry,16 and electrophoresis,17 have been deployed for the detection of nitrite ions. However, all these techniques are more suited for extensive laboratory testing since they demand time-consuming, difficult sample pre-treatment procedures and high levels of operating and experimental expertises. Such challenges have been resolved by the development of surface-enhanced Raman scattering (SERS) based sensors, which are highly efficient tools for detecting trace-level biomolecules/substances. Raman spectroscopy provides the fingerprint data that allow us to recognise multiple components in a mixture without extensive separation.18,19 Significant Raman scattering enhancement occurs when the targeted molecules approach the hot spots formed at the interstitial junctions between plasmonic metallic nanoparticles revealing its potential for extremely sensitive, non-invasive, and label-free detection of even single molecules. Many reports have proven SERS as a promising detection method to distinguish between healthy controls and oral cancer patients by enhancement in the characteristic salivary Raman bands extending its application towards cancer diagnosis.20–22 SERS is a suitable detection method with high selectivity and specificity for salivary biomarkers because trace-level concentrations of analyte present in saliva usually require assays with high sensitivity and low limit of detection.
The presence and spatial distribution of multiple hot junctions between the densely packed metallic nanocrystals are classic characteristics of the highly active SERS substrates. Typical SERS substrates have been made of noble metal nanoparticles like silver and gold nanoparticles.23 Although the aggregates of these nanoparticles are desirable in SERS, the poor dispersity of nanoparticles in complex samples leads to poor reproducibility and limit their wider applicability. In order to overcome these challenges, solid-state SERS active substrates with high sensitivity and consistent Raman boosting capability have been created by growing these plasmonic nanoparticles on some solid supports which are sensitive to the target molecules leading to repeatable and stable SERS signals.24,25 In this context, electrospun carbon nanofibers can be used as a promising solid support. The new generation CNF structures, such as hollow carbon nanofibers (HCNFs), possess twice as much surface area owing to the availability of both inner and outer surfaces, which can be used as the active sites for anchoring of the nanoparticles/functional molecules and thereby contributing towards SERS enhancement. Since HCNFs have a large surface area and can accommodate a high density of plasmonic nanoclusters, they are therefore recommended as an excellent candidate for SERS substrates. One of the most widely used methods for creating core–shell nanofibers is co-axial electrospinning because of its ease of use, low cost, and high throughput. Incorporating plasmonic nanoparticles such as Ag onto electrospun hollow carbon nanofibers opens a broad avenue of applications in SERS sensing.
Herein, we report a highly sensitive, label-free salivary sensor platform based on hollow carbon nanofibers anchored with Ag nanoparticles (Ag@HCNFs) for pre-diagnosis of oral cancer by monitoring the trace-level concentration of salivary nitrites. To the best of our knowledge, no studies on the development of SERS substrates utilising silver nanoparticles anchored on hollow carbon nanofibers have been reported. Co-axial electrospinning was utilised to create the fibrous mat, which was then transformed into hollow carbon nanofibers utilising a two-step procedure that included stabilisation and carbonization and a chemical reduction method was adopted for the synthesis of Ag nanoparticle anchored HCNFs. The SERS-based salivary biosensor was utilised to investigate the SERS enhancement properties of salivary nitrite within the clinically relevant range. This study demonstrates the potential of the developed plasmonic SERS material for simple, low-cost, non-invasive, and label-free salivary biomarker detection for the early stage screening and diagnosis of oral cancer by monitoring the salivary nitrite level.
Experimental section
Materials and reagents
Polyacrylonitrile (PAN, average molecular weight = 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Sigma Aldrich), polyvinylpyrrolidone (PVP, average molecular weight = 360000), dimethyl formamide (DMF, 99%, Merck), sulphuric acid (H2SO4, 98.08%, Merck), nitric acid (HNO3, 70%, Merck), silver nitrate (AgNO3, 99.5%, Merck), and sodium borohydride (NaBH4, 97%, Merck) were used for the synthesis of Ag@HCNFs. All reagents were of analytical grade and directly used without any further purification.
000, Sigma Aldrich), polyvinylpyrrolidone (PVP, average molecular weight = 360000), dimethyl formamide (DMF, 99%, Merck), sulphuric acid (H2SO4, 98.08%, Merck), nitric acid (HNO3, 70%, Merck), silver nitrate (AgNO3, 99.5%, Merck), and sodium borohydride (NaBH4, 97%, Merck) were used for the synthesis of Ag@HCNFs. All reagents were of analytical grade and directly used without any further purification.
Fabrication of hollow carbon nanofibers using co-axial electrospinning
The hollow carbon nanofibers were fabricated using a co-axial electrospinning technique by modifications of the previously reported processes in the literature.26–28 During coaxial electrospinning, hollow carbon nanofibers were developed from a stable suspension of PAN, which served as the shell material and PVP acted as a sacrificial core polymer feed solution. PAN was dissolved in a DMF (8% wt/v) solution and heated up to 60 °C for 2 h to create a homogeneous feed solution for the outer shell fluid. The inner core solution was prepared by dissolving PVP in DMF (12 wt/v%) followed by stirring for two hours at 60 °C. The co-axial electrospinning set-up consists of two stainless-steel capillaries with blunt tip needles with diameters of 18-gauge (outer) and 21-gauge (inner) installed coaxially as the core and shell, respectively, equipped with two plastic syringe connectors. A syringe pump (World Precision Instruments Inc, USA, SP200I) was used to dispense the two feed solutions at a constant flow rate of 0.5 ml h−1 and a constant voltage of 18–20 kV was applied between the needle and collector using a high voltage power source (Glassman High Voltage Inc., USA, EH30P3). A fixed distance of 30 cm was used between the needle tip and the collecting plate. The electrospun PAN/PVP fibers were deposited on a clean copper sheet to obtain a thick fibrous mat. All experiments were carried out in natural environments. The as-spun nanofibers were converted into hollow carbon nanofibers using a two-step thermal process comprising stabilization followed by carbonization. After being stabilised for 2 h at 300 °C in a horizontal tubular furnace under an air atmosphere, the electrospun co-axial nanofibers were further carbonized for 1 h at 800 °C under a nitrogen atmosphere (5 °C min−1) and nitrogen gas was continuously supplied throughout the process to expel the volatile by-products throughout the carbonization process without generating any structural defects.Functionalization of hollow-carbon nanofibers
The developed nanofiber mats were immersed in 1 M HCl and extensively rinsed with deionized water and ethanol in order to remove the carbon nanofibers from impurities. The functionalization of the electrospun carbon nanofibers was accomplished using acid treatment by refluxing with a 3:1 (v/v) solution of H2SO4 (3 M) and HNO3 (3 M) for 8 h while maintaining a temperature of 120 °C. To achieve a neutral pH, the resultant solution was continuously rinsed with distilled water, filtered, and dried overnight at 100 °C in a hot air oven to obtain functionalized carbon nanofibers.
Synthesis of Ag@HCNFs
After the functionalization of the hollow carbon nanofibers, silver anchored carbon nanofibers were synthesised through a chemical reduction method using sodium borohydride as the reducing agent. Briefly, 60 mg of HCNFs was dispersed in de-ionized water and sonicated for 30 min. It is followed by the addition of 30 mM AgNO3 and the reaction mixture was agitated for 1 h at room temperature. After that, silver nitrate was reduced to silver nanoparticles (Ag0) by adding drops of an ice-cold solution containing 50 mM sodium borohydride to the above solution. The resulting slurry was filtered, thoroughly washed with distilled water, and then dried overnight at 80 °C in a hot air oven to obtain the Ag@H-CNFs. A similar synthetic process was adopted to produce hollow carbon nanofibers having varied silver loadings.Fabrication of the SERS substrate
5 mg of Ag@HCNFs was dispersed in ethanol by sonication for 30 min for the fabrication of the SERS substrates. The glass slides were thoroughly cleaned with ethanol and ultrapure water several times and dried at 40 °C for 30 min. Then, 20 μL of the above nano-composite dispersion was dropcast onto the surface of the glass slide. Rhodamine 6G was used as a common probe molecule in order to evaluate the efficacy of the developed Ag@HCNF based SERS substrates. The R6G solutions with concentrations ranging from 10−6 M to 10−12 M in DI water were prepared using the standard R6G stock solution (0.1 M). 20 μL solution of R6G with a specific concentration (1 × 10−6 to 1 × 10−12 M) was dropcast onto the Ag@HCNF based SERS substrate, dried at 50 °C and used for the SERS analysis.Preparation of healthy saliva samples
After rinsing the mouth with clean water at least three times, saliva samples from healthy volunteers were collected 1 h after the food intake. Saliva was directly collected into a disposable cup using a non-stimulated collection technique. The salivary samples were then transferred into 2 mL centrifuge tubes and centrifuged at 4 °C for 15 min at 12000 rpm to eliminate the food debris and oral mucous epithelial cells. After centrifuging, the supernatant was collected and kept at 4 °C until analysis to obtain pure saliva.
Spiked sample preparation
Different nitrite concentrations ranging from 50 μM to 300 μM were prepared in distilled water. It was further spiked in the healthy saliva samples for analysis. In other words, 0.5 mL of saliva was mixed with 0.5 mL of various nitrite concentrations, and 5 μL of the spiked sample was dropped onto the SERS substrate followed by overnight atmospheric drying prior to analysis.Characterization techniques
X-ray diffraction (XRD) measurements were performed using a PANalytical X-pert diffractometer (Malvern) with a Cu Kα radiation (λ = 1.54 Å) source. The surface morphology of Ag@HCNFs was analysed using scanning electron microscopy (SEM, Carl Zeiss). The morphological characterization of Ag@HCNFs was carried out using transmission electron microscopy (JEM-2010, 200 kV, JEOL, Japan) integrated with EDAX. FESEM analysis was carried out using a field emission scanning electron microscope (Carl Zeiss, USA; model: SIGMA with GEMINI column), resolution 1.5 nm, and SERS spectra were acquired using a confocal Raman microscope (WITec alpha300 RA, Ulm, Germany) equipped with a 532 nm laser. The laser power used at the sample position was 1 mW and a 50× objective was used for SERS analysis. The spectra were randomly acquired at three distinct locations, with 10 s accumulations, at 20 different points.Results and discussion
Co-axial electrospinning followed by a chemical reduction method was used to prepare silver nanoparticle anchored hollow carbon nanofibers. After the co-axial electrospinning process, the fabricated core–shell structured PAN/PVP nanofibers were initially stabilised in air for 2 h at 300 °C, followed by carbonization for 1 h under a nitrogen atmosphere at 800 °C. The stabilisation under air leads to oxidation, aromatization, cyclization, dehydrogenation, and crosslinking, which not only serve as prerequisites for the subsequent carbonization process, but also have a significant impact on the functional properties of the carbon nanofiber.29,30 Hence, the primary goal of thermal stabilisation is to stabilise the PAN precursor fibers for the upcoming heat treatment procedure, preventing the melting of PAN during the carbonization process.31The major mechanism underlying the retention of the nanofibrous structure during carbonization is the cleavage and transformation of the nitrile bond into non-meltable cyclic or ladder-like structures. During the carbonization process, the linear PAN molecules dissociate two hydrogen atoms in the monomeric unit, creating a double bond between the carbon atoms. Also, during this step, the sacrificial core material, PVP, was decomposed and burnt out, retaining the hollow nanofiber structure in the outer shell. Scheme 1 shows the schematic illustration adopted for the synthesis of silver nanoparticle functionalized hollow carbon nanofibers.
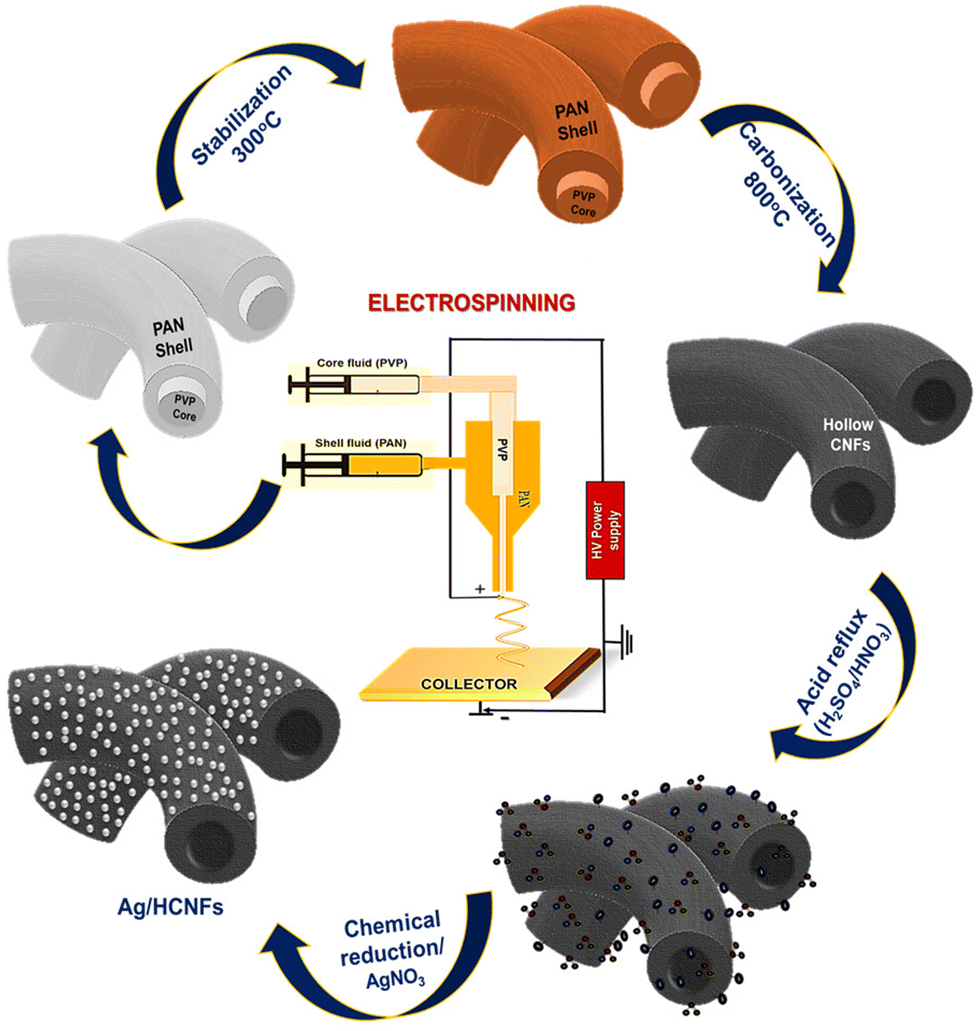 | ||
| Scheme 1 Schematic diagram illustrating the fabrication of silver nanoparticles anchored on hollow carbon nanofibers. | ||
The chemical reduction method was used to prepare stable silver nanoparticles on hollow carbon nanofibers using NaBH4 as the reducing agent.32 Silver nitrate is reduced by an ice-cold solution of sodium borohydride, resulting in the formation of stable silver nanoparticles as per the following equation:
| Ag+ + BH4− + 3H2O → Ag0 + B(OH)3 + 3.5 H2 | (1) |
The chemical reduction process with sodium borohydride as the reducing agent resulted in the formation of the silver nanoparticles uniformly anchored on the surface of HCNFs forming Ag@HCNFs.
Structural and morphological characterization
The structural characterization of HCNFs and Ag@HCNFs was carried out using XRD analysis and Raman analysis as shown in the Fig. 1. The XRD analysis of the HCNFs and Ag@HCNFs is shown in Fig. 1(a). The XRD pattern of the synthesised material revealed the presence of amorphous carbon nanofibers with crystalline Ag nanoparticles. According to JCPDS card 41-1487, the broad diffraction peak centred at around 25.6° can be indexed to the (002) plane of the amorphous phase of the hexagonal graphitic carbon found in the hollow carbon nanofibers. The (111), (200), (220), and (311) crystal planes of the face-centered cubic structure of Ag correspond to the crystal peaks centered at 2θ = 38.1°, 44.3°, 64.5°, 77.4°, and 82.5° according to JCPDS file No. 04-0783. The absence of any other crystalline phases observed in the XRD patterns revealed that no other detectable impurities were present in Ag@HCNFs.
 | ||
| Fig. 1 (a) XRD patterns and (b) Raman spectra of HCNFs and Ag@HCNFs. | ||
The crystallite size of the Ag nanoparticles present on HCNFs was calculated from the XRD pattern using the Scherrer equation:
| D = Kλ/βcosθ, | (2) |
The Raman spectra of HCNFs and Ag@HCNFs are displayed in Fig. 1(b). The D and G bands of HCNFs and Ag@HCNFs are observed at 1365 cm−1 and 1597 cm−1, respectively. According to Fig. 1(b), these bands are caused by the breathing mode with A1g symmetry of defects with the presence of sp3 carbon atoms and the E2g phonon mode of sp2 carbon.33Fig. 1(b) displays the Raman spectrum of Ag@HCNFs, which contains vibrational modes at 445 and 627 cm−1 as a result of the stretching vibrations of C–S–C and C–N–C. The other bands correspond to the carbon peaks present at 1365 cm−1 (D band) and 1597 cm−1 (G band) produced due to the symmetric and anti-symmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the carboxylic group respectively. The structural flaws in the HCNFs and Ag@HCNFs were responsible for the D band at 1365 cm−1, and the modification with Ag also caused the peak to broaden. Because of the significant charge transfer interaction between the Ag nanoparticles and HCNFs in the Ag@HCNFs, the G band was found to be shifted to a slightly higher wavenumber.34 The UV-vis absorption spectra of Ag nanoparticles are shown in ESI S2.†
O stretching vibrations of the carboxylic group respectively. The structural flaws in the HCNFs and Ag@HCNFs were responsible for the D band at 1365 cm−1, and the modification with Ag also caused the peak to broaden. Because of the significant charge transfer interaction between the Ag nanoparticles and HCNFs in the Ag@HCNFs, the G band was found to be shifted to a slightly higher wavenumber.34 The UV-vis absorption spectra of Ag nanoparticles are shown in ESI S2.†
The morphological characterisation was carried out utilising TEM analysis to infer about the microstructural and morphological characteristics of the hollow carbon nanofibers as well as to investigate the distribution of silver nanoparticles anchored on HCNFs. The continuous fibrous structure of hollow carbon nanofibers with a uniform diameter (∼200 nm) is depicted in Fig. 2(a and b). Due to the poor thermal stability properties, PVP completely disintegrated at high temperatures, leaving no residual material leading to the formation of hollow carbon nanofibers.
 | ||
| Fig. 2 TEM mage of (a and b) hollow CNFs, (c) SAED pattern of HCNFs (d and e), TEM image of Ag@HCNFs, (f) SAED pattern of Ag@HCNFs, and (g) HR-TEM image and IFFT profile (inset) and (h) EDS spectrum of Ag@HCNFs. | ||
Fig. 2(c) shows the SAED pattern of the HCNFs clearly illustrating the amorphous diffusion rings. The lower magnification images depict the homogeneous distribution of silver nanoparticles, which were found to be well-dispersed and firmly adhered to the surface of hollow carbon nanofibers as depicted in Fig. 2(d and e). The huge number of surface functional groups on HCNFs act as nucleation sites for the formation of Ag nanoparticles, and could be primarily attributed to the small range of particle sizes of silver nanoparticles.35,36 TEM images demonstrating the uniformity and high particle distribution of the silver nanoparticles anchored on HCNFs are depicted in ESI Fig. S3.† The presence of nitrogen species on the carbon lattice of the hollow carbon nanofibers resulted in more defects, and the high surface area to volume ratio of HCNFs can provide greater electron density sites, which helps to homogeneously anchor the silver nanoparticles due to the strong nanoparticle–carbon support interaction. TEM images corresponding to different concentrations of AgNO3 (10 mM, 20 mM and 30 mM) used for the preparation of Ag@HCNFs with varied Ag loading are given in ESI S1.†
Fig. 2(f) shows the selected area diffraction (SAED) pattern analysis of Ag@HCNFs. The spot patterns assigned to the (111), (200), (220), (311) and (222) planes of fcc Ag crystals demonstrate the polycrystalline nature of Ag, which is in good agreement with the XRD analysis results of Ag@HCNFs confirming the successful formation of silver nanoparticles tethered on hollow carbon nanofibers. The lattice d-spacing value of 0.23 nm obtained from the Inverse Fast Fourier Transform (IFFT) analysis of the HRTEM images of Ag@HCNFs, depicted in Fig. 2(g), was compatible with the (111) crystalline plane of fcc Ag. The reported lattice spacing of 0.23 nm was in good agreement with that in the JCPDS database, which corresponds to the (111) crystal planes of fcc Ag, indicating that the silver nanoparticles were successfully formed on the surface of hollow carbon nanofibers. Fig. 2(h) shows the EDS spectrum of Ag@HCNFs, which also confirms the formation of the silver nanoparticle anchored hollow carbon nanofibers.
The FESEM images of CNFs and hollow CNFs are depicted in Fig. 3. Both CNFs and HCNFs were found to have long, continuous cylindrical morphologies, with diameters uniformly distributed between 180 and 200 nm. From Fig. 3a, it is obvious that the CNFs have a smooth surface and the cross-sectional view indicates the absence of a hollow structure. The cross-sectional field-emission SEM image of HCNFs clearly shows their hollow nature, as depicted in Fig. 3b. The hollow core was found to be generated within nanofibers during the thermal decomposition process by the sacrificial removal of the inner PVP phase, whereas the continuous PAN outer phase was easily converted into carbon during the carbonization process.
 | ||
| Fig. 3 Cross-sectional FESEM images of (a) carbon nanofibers and (b) hollow carbon nanofibers. | ||
Ag@HCNFs were subjected to confocal Raman microscopic imaging in order to comprehend the chemical identity of the material as shown in Fig. 4. The breathing mode with A1g symmetry of defects with the presence of an sp3 carbon atom and E2g phonon mode of sp2 carbon are responsible for the D and G bands of Ag@HCNFs, which were observed at 1353 cm−1 and 1590 cm−1, respectively. Fig. 4a depicts the optical image of Ag@HCNFs. Fig. 4b and c show the Raman images of the D and G bands of Ag@HCNFs, which were observed at 1353 and 1590 cm−1, respectively. The fibrous structure of the hollow CNFs was clearly visible in the Raman mapping images. A uniform distribution of silver nanoparticles formed by the stretching vibration of C–N–C and C–S–C over the solid support material of the HCNFs was visible in the Raman image (Fig. 4d and e) corresponding to the peaks at 445 and 670 cm−1. Further evidence for the uniform distribution of silver nanoparticles over the hollow CNFs can be found in the combined Raman image (Fig. 4f) corresponding to the spectra of Ag@HCNFs.
 | ||
| Fig. 4 (a) Optical image of Ag@HCNFs, isolated confocal Raman images corresponding to (b) the D band at 1353 cm−1 (scale bar = 20 μm), (c) G band at 1590 cm−1 (scale bar = 20 μm), silver NPs at (d) 445 cm−1 (stretching vibration of C–N–C) and (e) 670 cm−1 (stretching vibration of C–S–C) and (f) combined Raman mapping (scale bar = 20 μm). | ||
A closer investigation of the FESEM, TEM and Raman mapping images reveals the successful formation of hollow carbon nanofibers having a high surface area to volume ratio that facilitated better binding to the analyte molecules. Additionally, Ag nanoparticles were uniformly coated on the smooth surfaces of the CNFs, enabling the successful anchoring of Ag nanoparticles which created numerous hotspots to produce high SERS enhancement. XRD results were corroborated with TEM analysis results, demonstrating the successful integration and formation of Ag@HCNFs.
Evaluation of the SERS enhancement properties of Ag@HCNFs
The common Raman reporter molecule, rhodamine 6G (R6G), was used to evaluate the performance of the Ag@HCNF based SERS substrate, as shown in Fig. 5. The SERS spectra were acquired using a confocal Raman microscope equipped with a 532 nm laser. In order to ensure reproducibility, the spectra were randomly acquired at three distinct locations, with 10 s accumulations, and 20 points were taken. The laser power used at the sample position was 1 mW. | ||
| Fig. 5 Comparison of the Raman spectrum and SERS spectrum of R6G using Ag@HCNF based SERS substrates. | ||
The normal Raman spectrum and SERS spectrum of R6G molecules were acquired using glass substrates coated with and without the SERS active material, Ag@HCNFs, with individual concentrations of 10−1 M and 10−6 M, respectively (Fig. 5). The distinctive bands of R6G that emerged at 615 cm−1, 775 cm−1, 1188 cm−1, 1315 cm−1, 1365 cm−1, 1510 cm−1, 1574 cm−1, and 1645 cm−1 are attributed to the C–C–C ring in-plane bending, the C–H out-of-plane bending, the C–C stretching mode, and the C–C stretching of xanthene, respectively. Additionally, the extra peaks observed at 1365 cm−1, 1510 cm−1, and 1645 cm−1 are caused by the aromatic C–C stretching vibrations which are in good agreement with the earlier reports.37,38 In Fig. 5, only low intensity peaks of rhodamine 6G are evident in normal Raman spectra in the absence of the SERS active material, Ag@HCNFs, whereas all the distinct bands corresponding to rhodamine 6G are found to be highly enhanced in the presence of Ag@HCNFs. This significant improvement could be a result of the strong localization of the near field intensity producing hot spots, where the Raman signal from the analyte molecules can be amplified by several orders of magnitude. Such hot spots are created between nearly in contact silver nanoparticles, which promotes the activation of abnormally strong electromagnetic fields that serve to significantly amplify the Raman signals leading to high enhancement.
The intensity of the appropriate rhodamine 6G peak (1365 cm−1) measured in the SERS experiments was compared to the equivalent peak measured from an aqueous solution of rhodamine 6G in normal Raman spectra to determine the SERS enhancement factor values. While the SERS measurements employ a micromolar concentration, the typical Raman spectra were obtained at a concentration of 10−1 M. Prior to the calculation of the enhancement factor, baseline correction was done to the Raman and SERS spectra of R6G to ensure accurate results. The SERS enhancement factor per molecule was calculated using the following equation:
| Enhancement factor = ISERS × NRaman/IRaman × NSERS | (3) |
| Number of molecules = πr2hcNA | (4) |
184, respectively, in the obtained Raman and SERS spectra. As a result, the intensity ratio ISERS/IRaman = 13176/313 was estimated as 163.81. Consequently, by substituting the values in the aforementioned equation, an enhancement factor of ∼16.3 × 107 was calculated. The enhancement factor was calculated at different spots to ensure the reproducibility of the obtained results. This EF value in the range of ∼107 to 108 using R6G molecules proves the efficiency of the Ag@HCNF based SERS substrate for the detection of the trace level concentration of the analyte extending its application towards the detection of salivary biomarkers.
The comparison of the performance of the SERS substrates using Ag NPs anchored on normal carbon nanofibers and Ag NPs anchored on hollow carbon nanofibers was carried out using R6G (ESI S4(a)†) which showed a higher enhancement in Ag@HCNFs as compared to that of Ag@CNFs. This is attributed to the large surface area to volume ratio of the hollow carbon nanofibers as compared to the normal CNFs so that the number of Ag NPs anchoring on the surface of HCNFs also increases, leading to the formation of a large number of hotspots contributing to enormous enhancement. Also, in the case of HCNFs, the inner and outer surfaces can act as the active sites since both the surfaces will have –OH groups imparted on their surface as a result of activation due to acid treatment. Later, after the chemical reduction of AgNO3 using sodium borohydride, the silver nanoparticles were anchored on the outer and inner surfaces of the HCNFs leading to a tremendous number of hotspot generation on the outer and inner surfaces, contributing towards the higher SERS enhancement, whereas, in the case of normal CNFs, only the outer surface is available for the silver nanoparticles which could be the reason for the comparatively lower SERS enhancement. Hence, it can be concluded that the SERS signal can be enhanced several times using HCNFs as the support material for the plasmonic sites, demonstrating them as a potential solid base substrate for SERS applications.
The SERS performance of the developed substrate was also assessed by varying the AgNP concentrations (10 mM, 20 mM, and 30 mM) on the HCNFs, which demonstrated a good enhancement up to 35000 counts using the 30 mM concentration of Ag NPs (ESI S4(b)†). The synergistic effect between the plasmonic Ag NPs produces hotspots and these numerous interparticle gaps which may behave as Raman hotspots with enhanced local electromagnetic fields may also be the reason for the enormous SERS enhancement.39,40 Lower enhancement was observed for the Ag@HCNFs with precursor molar ratios of 10 mM and 20 mM, suggesting that there were insufficient silver nanoparticles to be deposited on the surface of the HCNFs leading to a lower number of hotspots contributing to lower enhancement. Hence, it can be concluded that, the structure and amount of silver loading on the surface HCNFs are significantly influenced by the silver precursor molar ratio, which also contributes towards the SERS performance. The SERS spectra of various concentration ranges of R6G, ranging from 10−6 M to 10−11 M, were recorded to determine the limit of detection of the Ag@HCNF based SERS substrate (ESI S4(c)†). This showed all of the main characteristic peaks of the probe molecules even at concentrations down to as 10−11 M, indicating the efficiency of the SERS substrate in detecting a trace level concentration of the analyte. The relationship between concentration and peak intensity at 1365 cm−1 shown in ESI S4(d)† shows a linear trend with an R2 value of 0.9953 and a standard deviation of about 1.381. The above findings show that the proposed Ag@HCNF based SERS substrate exhibits the lowest LOD of 10−11 M and the highest EF on the order of 107, indicating that the method provides a sensitive protocol for the trace level R6G detection down to 10−11 M. This indicates the efficiency of the SERS substrate to detect the analytes at very low concentrations extending its application to real-time sample analysis even in a complex saliva mixture.
SERS detection of salivary nitrite
Oral cancer continues to be the leading cause of oral disease-related death worldwide and is a serious and expanding global public health issue. Nitrite in saliva is a biomarker used to diagnose oral diseases, including obstructive lipoproteinaemia and nasopharynx carcinoma. Nitrite ions from foods such as drinking water, fruits, vegetables, and salty sausages reach the digestive tract through the oral cavity and finally reach the saliva through the blood circulation via the salivary glands.41–43 Haemoglobin in the blood undergoes an irreversible conversion to achieve oxygen uptake as a result of nitrite reactions, which lowers the amount of oxygen that can be carried by the blood.44,45 Although nitrates themselves are not toxic, they may occasionally serve as a catalyst for a series of chemical reactions that turn them into toxic compounds. Nitrate is converted to nitrite as a result of the microbial environment.In comparison with nitrates, nitrites are more hazardous. Since the saliva of habitual tobacco chewers and smokers contains nitrosamines that are likely leached from the tobacco or formed in situ by the nitrosation of tobacco alkaloids and that of secondary amines, many studies have shown that oral carcinoma patients will have a significant increase in salivary nitrate and nitrite levels compared to the control group as shown in Scheme 2. It is also suggested that tobacco chewers might be continuously exposed to nitrosamines and other nitroso compounds through their oral mucosa.
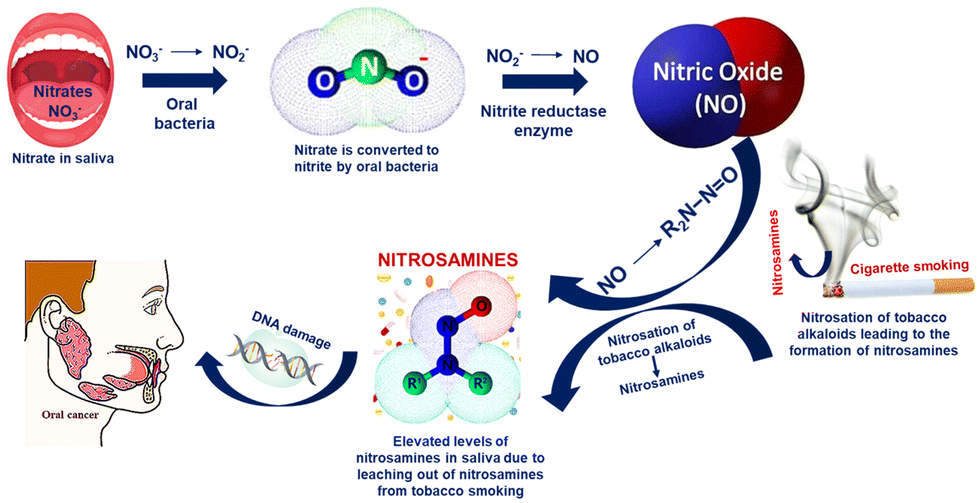 | ||
| Scheme 2 Mechanism of the conversion of nitrates to carcinogenic nitrosamines. | ||
Additionally, human whole saliva may contain substances that inhibit the nitrosation reactions. Individual differences in the concentrations of these inhibitory substances may have an indirect relationship with a person's susceptibility to oral cancer. By using nitrate reductase enzymes, the oral bacterial species that are located at the posterior part of the tongue convert nitrate to nitrite. These bacteria use nitrate and nitrite as final electron acceptors in their respiration and convert nitrate to NO. Additionally, in the case of habitual tobacco chewers and smokers, the nitrosation of the tobacco alkaloids leads to the formation of nitrosamines.46 Overproduction of NO in the saliva of tobacco users is thought to be a factor in the development of cancer-causing nitrosamines or other pathological processes in the oral cavity by inhibiting DNA repair mechanisms or developing other pathological processes. The salivary nitrite level in the control group varied from 30 to 100 μg L−1, whereas the nitrite level was significantly higher (>200 μg L−1) for oral cancer patients according to numerous reports.47 Hence, the detection of nitrite in saliva can be used as a detection method for the pre-diagnosis of oral carcinoma.
In order to comprehend the effectiveness of Ag@HCNFs for clinical applications, the viability of SERS for the detection of nitrite was initially investigated. In Fig. 6(a), the Raman (0.1 M) and SERS (100 μM) spectra for nitrite are compared using the Ag@HCNFs-based SERS substrate. As anticipated, SERS spectra showed a clearly distinguishable peak of nitrite centred at around 1330 cm−1, which is caused by the symmetric N–O stretching vibrations of the nitrite.48
 | ||
| Fig. 6 (a) Raman and SERS spectra of nitrite using Ag@HCNF based SERS substrates and (b) comparison of saliva and saliva spiked with nitrite. | ||
This strong intensity enhancement of the 1330 cm−1 peak proves the efficacy of SERS active Ag@HCNFs to detect very low concentrations of nitrite especially in complex mixtures such as saliva. The limit of detection of the SERS substrate was found to be 10−10 M using the analyte nitrite and is given in ESI S5.†
SERS analysis of spiked healthy saliva samples
The reproducibility of the SERS sensors is significantly impacted by the complexity of the real saliva sample matrix. In order to assess the viability of salivary biomarker detection, real saliva samples from healthy individuals were analysed without any tedious pre-treatment procedures like dilution, pH adjustment, and so on. Instead, a simple centrifugation technique was employed to remove food debris and mucus to acquire pure salivary samples. The SERS method was utilised as a practical application to identify nitrite in salivary samples as a pre-diagnostic tool for oral cancer.The SERS spectral analysis was utilised to compare control samples of saliva with healthy saliva that had been spiked with various concentrations of nitrite, between 1100 cm−1 and 1500 cm−1. Fluorescence subtraction and background trace elimination were used to further process the data. Fig. 6b compares the SERS spectra of unadulterated saliva with saliva that was spiked with 100 μM nitrite.
The spectra showed Raman signals at 1260 cm−1 and 1460 cm−1, which were attributed to amide III and amide II/CH2 scissoring/CH deformation present in the saliva. Low intensity peaks at 1286 cm−1 and 1380 cm−1 were also observed in the SERS spectra of saliva which correspond to the L-tryptophan, amide III and δ (CH3) of lipids.49
The Raman spectra of a healthy saliva sample spiked with nitrite showed strong SERS bands at 1260 cm−1 attributed to the amide III groups in saliva and the peak centred at 1330 cm−1, caused by the symmetric N–O stretching vibrations of the nitrite, was used for the quantitative detection of nitrite present in saliva.
Fig. 7(a) shows the SERS spectral comparison of healthy saliva spiked with various concentrations of nitrite within the clinically relevant range from 50 μM to 300 μM in order to discriminate between healthy people and oral cancer patients. Raman signals at 1268 cm−1 were observed in the SERS spectrum of pure saliva spiked with nitrite and are attributed to amide III groups of proteins present in the saliva. As expected, the intensity of the characteristic SERS peak centred at 1330 cm−1 which is caused by the symmetric N–O stretching vibrations of the nitrite, was found to increase as a function of the concentration of the target analyte between 50 μM and 300 μM. Hence, this peak can be used for the quantitative detection of nitrite in saliva. Moreover, from Fig. 7(b), it can be inferred that the peak intensity of nitrite as a function of concentration followed a good linear trend with the root mean square (R2) value equal to 0.9983, and standard deviation values of ±2.669, which demonstrates the efficiency of the Ag@HCNFs based SERS substrate to detect the nitrite in salivary samples within the clinically relevant range, thereby extending its application towards oral cancer diagnosis.
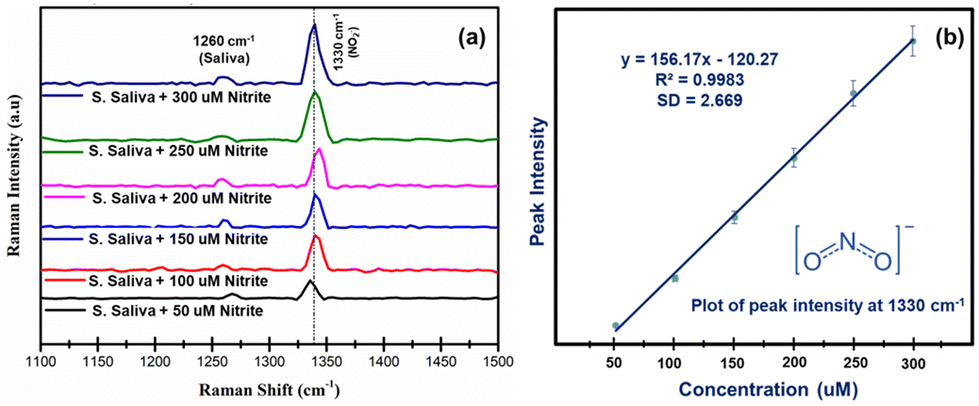 | ||
| Fig. 7 (a) SERS spectra of saliva spiked with different concentrations of nitrite ranging from 50 μM to 300 μM and (b) the corresponding plot of peak intensity as a function of concentration in the presence of Ag@HCNFs. | ||
In order to prove the selectivity of the developed sensor, the effects of other interferents present in the saliva were investigated. The common anions that interfered with salivary nitrite were acetate, carbonate, chloride, nitrate, thiocyanate, and sulphate. Ag@HCNFs were thus combined with solutions containing 100 μM of various anions, such as CH3COOH−, Cl−, CO32−, NO3−, SCN−, and SO42−, in order to evaluate the impact of interferents and to prove the selectivity of the Ag@HCNFs based SERS substrates as depicted in Fig. 8.
 | ||
| Fig. 8 Effect of common interfering anions with salivary nitrite (a) in the spectral range of 200 cm−1 to 4000 cm−1 and (b) in the spectral range of 900 cm−1 to 1600 cm−1. | ||
According to the findings in Fig. 8(a and b), the presence of these anions does not significantly affect the peak intensity at 1300 cm−1. The Raman bands of the majority of common chemical bonds are typically within the range far away from the peak responsible for the symmetric N–O stretching vibrations of the nitrite (1300 cm−1) and no overlapping was observed between these interference peaks and the NO2− signal, making the SERS sensor detect nitrite in saliva extremely selectively. As a result, this peak can be confidently used to indicate the concentration of NO2− with high specificity.
Plausible mechanism for SERS enhancement
The electromagnetic (EM) and chemical (CM) mechanisms contribute to the explanation of the overall SERS effect. The former requires the excitation of the localised surface plasmon resonance and entails the enhancement of optical fields. It does not require specific bonds between the adsorbate and the substrate and it is a long-range effect mostly contributing to the noble metal substrate producing SERS enhancement up to 105 to 1010. The latter is the chemical enhancement mechanism which is a short-range effect that requires a bond between the adsorbate and substrate and involves the charge transfer between the adsorbate and substrate to achieve SERS enhancement up to 102 to 103.50 Here, the SERS enhancement by Ag@HCNFs was due to the combined effect of both electromagnetic enhancement due to the numerous hotspots created between the neighbouring closely packed silver nanoparticles on the surface of Ag@HCNFs, and the chemical enhancement resulting from the charge transfer effect or dipole−dipole interaction created by the Ag@HCNFs and the analyte molecules.The major contribution of the SERS enhancement in Ag@HCNFs was from the EM mechanism. It has been demonstrated that the Ag nanoparticles in HCNFs cause hot spots that provide a powerful SERS effect based on electromagnetic enhancement, especially when the gaps get reduced to a few nanometers. When the laser strikes the surface of the HCNFs modified by Ag nanoparticles, the localised surface plasmon is excited, creating a potent local electromagnetic field near the Ag. Because of the close proximity of the Ag nanoparticles on the HCNF surface, there may be strong interparticle plasmon interaction in this instance.51 Additionally, the acid refluxing of the HCNFs following the carbonization process will activate both the inside and outside surfaces of hollow carbon nanofibers leading to the large number of active sites for the silver nanoparticles to attach, contributing towards SERS enhancement. Due to the close packing and uniform decoration of the Ag NPs in HCNFs, plasmonic field coupling can result in the formation of a high density of hot spots that can enhance the Raman signal to several orders of magnitude as illustrated in Scheme 3.
 | ||
| Scheme 3 Plausible SERS enhancement mechanism of the Ag@HCNF based SERS substrate towards the analyte molecules. | ||
The chemical adsorption that occurs between the molecule and the substrate contributes to the chemical enhancement of SERS in Ag@HCNFs. The molecule needs to be in close proximity to the substrate for the charge transfer to occur since chemical enhancement is a short-range effect. The higher polarizability of the molecule caused by this charge transfer results in the increased cross section of Raman scattering. A change in the adsorption orientation of the molecule would have an impact on the polarisation tensor change of some vibrations induced by charge transfer, which would change the SERS spectral behaviour. The π–π bond interaction makes the bonding between R6G molecules and HCNFs stable.52 Most significantly, CNFs exhibit exceptional affinity for adsorbing and concentrating a range of target molecules using the π–π stacking mechanism. The benefits of CNFs, such as minimising the analyte's background fluorescence, their ability as potential surface plasmon enhancers exhibiting a chemical charge transfer between the analyte molecules and carbon nanofibers, etc., greatly contribute to the SERS enhancement, revealing that the developed Ag@HCNF based SERS substrates exhibit an extensive combination of enhancement from the electromagnetic mechanism of closely packed Ag nanoparticles and chemical mechanism of CNFs, which will be very advantageous for SERS biosensors. The morphological characterization and SERS performance of Ag@HCNF-based sensors have expanded their use in clinical diagnosis by providing a better understanding of the mechanisms underlying enhancement and the detection of oral cancer biomarkers in saliva within the clinically relevant range. As a result, Ag@HCNFs represent a novel class of sensing materials that can be employed as a SERS substrate for the label-free detection of oral cancer biomarkers.
Conclusions
A highly selective, label-free, Surface-Enhanced Raman Spectroscopy (SERS) based salivary sensor platform using silver nanoparticles functionalized on hollow carbon nanofibers (Ag@HCNFs) was developed for pre-diagnosis of oral cancer by monitoring the concentraton of salivary nitrite. Co-axial electrospinning was used for the fabrication of the fibrous mat and was converted to hollow carbon nanofibers using a two-step process involving stabilization and carbonization. The hollow carbon nanofiber structure was formed by the decomposition of the sacrificial core component, PVP, during the carbonization process. The smooth surface and high surface area to volume ratio of the HCNFs enabled the uniform decoration of the silver nanoparticles, resulting in numerous homogeneous hot spots resulting in stable SERS signals. The developed Ag@HCNFs based SERS substrate was structurally and morphologically characterized using XRD, Raman, SEM and TEM analyses which evidently showed the successful formation of silver nanoparticles anchored on hollow carbon nanofibers. The SERS performance evaluation of Ag@HCNFs with R6G demonstrated a strong SERS enhancement of the order of 107 and a detection limit of 10−11 M, proving its effectiveness in analysing real-time salivary samples. Additionally, the SERS enhancement of nitrite was examined with Ag@HCNFs by spiking nitrite in samples of healthy saliva. The enhancement of the distinctive nitrite SERS spectra demonstrated the feasibility of its trace-level detection in the clinically significant range (50 μM–300 μM) even in the complex matrix of real saliva. Additionally, the impact of other common interfering anions in saliva was also studied which showed no overlapping peaks with the NO2− signal, making the SERS sensor highly selective to detect nitrite in saliva, demonstrating the promising potential of the developed SERS sensor for the pre-diagnosis and mass screening of oral cancer.Author contributions
The manuscript was written through contributions from all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge DST-INSPIRE, Government of India, Ministry of Science and Technology, Department of Science and Technology, for the financial support by funding through a DST-INSPIRE Fellowship (No. DST/INSPIRE Fellowship/2020/IF200094). The authors also acknowledge the facilities and support provided by the management, PSG Sons and Charities, Coimbatore.References
- C. Rivera, Int. J. Clin. Exp. Pathol., 2015, 8(9), 11884 CAS.
- P. Boyle and B. Levin, International Agency for Research on Cancer, IARC Press, 2008 Search PubMed.
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, Ca-Cancer J. Clin., 2011, 61(2), 69–90 CrossRef PubMed.
- E. M. O'Sullivan, Ir. Med. J., 2005, 98(4), 102–105 Search PubMed.
- S. Warnakulasuriya, Oral Oncol., 2000, 45(4–5), 309–316 Search PubMed.
- Y. H. Lee and D. T. Wong, Am. J. Dent., 2009,(4), 241 Search PubMed.
- D. P. Lima, D. G. Diniz and S. A. Moimaz, Int. J. Infect. Dis., 2010, 14(3), e184–e188 CrossRef PubMed.
- S. Chiappin, G. Antonelli, R. Gatti and F. Elio, Clin. Chim. Acta, 2007, 383(1–2), 30–40 CrossRef CAS PubMed.
- L. Retterstol, T. Lyberg, T. Aspelin and K. Berg, Twin Res Hum Genet, 2006, 9(2), 210–214 CrossRef PubMed.
- B. Bartholomew and M. J. Hill, Food Chem. Toxicol., 1984, 22(10), 789–795 CrossRef CAS PubMed.
- H. Yang, Y. Xiang, X. Guo, Y. Wu, Y. Wen and H. Yang, Sens. Actuators, B, 2018, 271, 118–121 CrossRef CAS.
- V. Shende, A. T. Biviji and N. Akarte, J. Oral Maxillofac. Pathol., 2013, 17(3), 381 CrossRef PubMed.
- J. M. Doyle, M. L. Miller, B. R. McCord, D. A. McCollam and G. W. Mushrush, Anal. Chem., 2000, 72(10), 2302–2307 CrossRef CAS PubMed.
- T. Pérez-Ruiz, C. Martinez-Lozano and V. Tomás, Anal. Chim. Acta, 1992, 265(1), 103–110 CrossRef.
- C. H. Kiang, S. S. Kuan and G. G. Guilbault, Anal. Chim. Acta, 1975, 80(2), 209–214 CrossRef CAS PubMed.
- J. Nam, I. B. Jung, B. Kim, S. M. Lee, S. E. Kim, K. N. Lee and D. S. Shin, Sens. Actuators, B, 2018, 270, 112–118 CrossRef CAS.
- F. Guan, H. Wu and Y. Luo, J. Chromatogr., A, 1996, 719(2), 427–433 CrossRef CAS.
- J. Chen, Y. Li, K. Huang, P. Wang, L. He, K. R. Carter and S. R. Nugen, ACS Appl. Mater. Interfaces, 2015, 7(39), 22106–22113 CrossRef CAS PubMed.
- P. Wang, S. Pang, J. Chen, L. McLandsborough, S. R. Nugen, M. Fan and L. He, Analyst, 2016, 141(4), 1356–1362 RSC.
- A. Fălămaş, H. Rotaru and M. Hedeşiu, Laser Med. Sci., 2020, 35(6), 1393–1401 CrossRef PubMed.
- E. Farnesi, S. Rinaldi, C. Liu, J. Ballmaier, O. Guntinas-Lichius, M. Schmitt and J. Popp, Sensors, 2023, 23(21), 8915 CrossRef CAS PubMed.
- G. Calado, I. Behl, A. Daniel, H. J. Byrne and F. M. Lyng, Transl. Biophotonics, 2019, 1(1–2), e201900001 CrossRef.
- V. K. Rao and T. P. Radhakrishnan, ACS Appl. Mater. Interfaces, 2015, 7(23), 12767–12773 CrossRef CAS PubMed.
- Z. Li, G. Meng, Q. Huang, X. Hu, X. He, H. Tang, Z. Wang and F. Li, Small, 2015, 11(40), 5452–5459 CrossRef CAS PubMed.
- Z. Wang, G. Meng, Z. Huang, Z. Li and Q. Zhou, Nanoscale, 2014, 6(24), 15280–15285 RSC.
- K. K. Karuppanan, A. V. Raghu, M. K. Panthalingal and B. Pullithadathil, ChemElectroChem, 2019, 6(7), 2029–2042 CrossRef CAS.
- A. G. El-Deen, N. A. Barakat, K. A. Khalil and H. Y. Kim, New J. Chem., 2014, 38(1), 198–205 RSC.
- P. Zhao, M. Yao, H. Ren, N. Wang and S. Komarneni, Appl. Surf. Sci., 2019, 463, 931–938 CrossRef CAS.
- Z. Bashir, Carbon, 1991, 29(8), 1081–1090 CrossRef CAS.
- E. Ismar and A. S. Sarac, Polym. Bull., 2018, 75, 485–499 CrossRef CAS.
- J. B. De Oliveira, L. M. Guerrini, S. S. Oishi, L. R. de Oliveira Hein, L. dos Santos Conejo, M. C. Rezende and E. C. Botelho, Mater. Res. Express, 2018, 5(2), 025602 CrossRef.
- K. C. Song, S. M. Lee, T. S. Park and B. S. Lee, Korean J. Chem. Eng., 2009, 26, 153–155 CrossRef CAS.
- K. G. Nair, V. Ramakrishnan, R. Unnathpadi, K. K. Karuppanan and B. Pullithadathil, J. Phys. Chem. C, 2020, 124(13), 7144–7155 CrossRef CAS.
- G. J. Melvin, Q. Q. Ni, Y. Suzuki and T. Natsuki, J. Mater. Sci., 2014, 49, 5199–5207 CrossRef CAS.
- J. J. Shao, Z. J. Li, C. Zhang, L. F. Zhang and Q. H. Yang, J. Mater. Chem. A, 2014, 2(6), 1940–1946 RSC.
- A. J. Plomp, D. S. Su, K. D. Jong and J. H. Bitter, J. Phys. Chem. C, 2009, 113(22), 9865–9869 CrossRef CAS.
- N. R. Barveen, T. J. Wang and Y. H. Chang, Microchem. J., 2020, 159, 105520 CrossRef.
- E. A. Kumar, N. R. Barveen, T. J. Wang, T. Kokulnathan and Y. H. Chang, Microchem. J., 2021, 170, 106660 CrossRef.
- Y. Yang, Z. Zhang, Y. He, Z. Wang, Y. Zhao and L. Sun, Sens. Actuators, B, 2018, 10(273), 600–609 CrossRef.
- J. Wei, A. Qayum, X. Jiao, T. Wang and D. Chen, Appl. Surf. Sci., 2021, 568, 150936 CrossRef CAS.
- L. Retterstol, T. Lyberg, T. Aspelin and K. Berg, Twin Res Hum Genet, 2006, 9(2), 210–214 CrossRef PubMed.
- R. Weller, Br. J. Dermatol., 1997, 137(5), 665–672 CAS.
- H. Yang, Y. Xiang, X. Guo, Y. Wu, Y. Wen and H. Yang, Sens. Actuators, B, 2018, 271, 118–121 CrossRef CAS.
- M. Gao, W. Fang, J. Ren, A. Shen and J. Hu, Analyst, 2016, 141(17), 5195–5201 RSC.
- C. T. Tran, H. T. Tran, H. T. Bui, T. Q. Dang and L. Q. Nguyen, J. Sci.: Adv. Mater. Devices, 2017, 2(2), 172–177 Search PubMed.
- X. M. Qu, Z. F. Wu, B. X. Pang, L. Y. Jin, L. Z. Qin and S. L. Wang, J. Dent. Res., 2016, 95(13), 1452–1456 CrossRef CAS PubMed.
- V. Shende, A. T. Biviji and N. Akarte, J. Oral Maxillofac. Pathol., 2013, 17(3), 381 CrossRef PubMed.
- A. Ianoul, T. Coleman and S. A. Asher, Anal. Chem., 2002, 74(6), 1458–1461 CrossRef CAS PubMed.
- M. Czaplicka, A. A. Kowalska, A. B. Nowicka, D. Kurzydłowski, Z. Gronkiewicz, A. Machulak and A. Kamińska, Anal. Chim. Acta, 2021, 1177, 338784 CrossRef CAS PubMed.
- X. Jiang, Y. Chen, J. Du, X. Li, Y. Shen, M. Yang, X. Han, L. Yang and B. Zhao, J. Raman Spectrosc., 2018, 49(8), 1257–1264 CrossRef CAS.
- Y. Qian, G. Meng, Q. Huang, C. Zhu, Z. Huang, K. Sun and B. Chen, Nanoscale, 2014, 6(9), 4781–4788 RSC.
- X. Liang, B. Liang, Z. Pan, X. Lang, Y. Zhang, G. Wang, P. Yin and L. Guo, Nanoscale, 2015, 7(47), 20188–20196 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images of Ag@HCNFs at different concentrations of AgNO3, UV-visible absorption spectra of Ag NPs, TEM images showing the uniformity and particle distribution of silver nanoparticles anchored on HCNFs, comparison of the SERS spectra of R6G using Ag@CNFs and Ag@HCNFs, evaluation of the SERS performance at various silver loadings and at different concentrations of R6G ranging from 10−6 to 10−11 M and the limit of detection of nitrite using Ag@HCNFs based SERS detection. See DOI: https://doi.org/10.1039/d4an00641k |
| This journal is © The Royal Society of Chemistry 2024 |