
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Local electrochemical sample acidification for the detection of Pb2+ traces†
Amira
Mahmoud
 ,
Július
Gajdár
,
Mariela
Brites Helú
,
Mathieu
Etienne
and
Grégoire
Herzog
*
,
Július
Gajdár
,
Mariela
Brites Helú
,
Mathieu
Etienne
and
Grégoire
Herzog
*
Université de Lorraine, CNRS, LCPME, F-54000 Nancy, France. E-mail: gregoire.herzog@cnrs.fr
First published on 4th September 2024
Abstract
Electrochemical detection of pollutants (e.g. heavy metals) in real samples often requires the adjustment of pH to allow optimal sensitivity. Such sample pretreatment can be challenging for on-site applications as it implies the use of valves, pumps and storage of base or acid solutions. We report here the use of an electrochemical approach for the control of water sample pH. It offers the possibility for local pH adjustment while simultaneously detecting Pb2+, whose detection sensitivity is pH dependent. An effective electrochemical method through local electrochemical acidification is performed to detect Pb2+ within a desired pH range without the need to add chemical reagents. Local acidification is based on water electrolysis. An anodic potential is applied to an acidifier to rapidly electrogenerate protons. This allows the sample pH to be tailored to the optimal detection condition. Reduction of the Pt oxide layer formed on the acidifier is key to obtain repeatable results in Pb2+ detection. On-site sample acidification is combined with anodic stripping voltammetry to reach a detection limit of 6 ppb (30 nM), which is lower than the World Health Organization guideline value for Pb2+ level in drinking water.
1. Introduction
Anodic Stripping Voltammetry (ASV) is a powerful technique for the electrochemical determination of metallic cations in aqueous solutions.1–3 In electroanalysis, the pH of reaction medium is a fundamental parameter that needs to be adjusted for the optimal detection of the target analyte. Variation in pH can trigger dramatic changes in chemical reaction rates and thus limit metal deposition.4 The classic method for adjusting the sample pH is the rudimentary addition of buffers or strong acids and bases. Recently, several approaches were developed to control pH, including electrochemical redox reaction without the need of adding chemical reagents. Strakosas et al. developed a non-enzymatic glucose sensor based on cobalt oxide able to monitor glucose levels in neutral bodily fluids such as sweat and tears.5 As metal oxides require alkaline medium for glucose detection, an electrochemical strategy to control locally the pH was developed to sense glucose in fluids at neutral pH. The developed strategy was based on the integration of palladium contact that absorbs H+ from the neutral medium and generates a constant OH− concentration to increase the pH up to 10 and perform an effective glucose detection. Wiorek et al. recently showed that using polyaniline PANI film acted, through an electrochemical pulse, as a proton pump for rapid sample acidification and alkalinity detection (5 min).6–8 Despite excellent results, it is necessary to improve the chemical composition of the proton pump to avoid the need for regeneration after acidification process. Fomina and coworker employed a combination of solution-borne quinones and galvanostatic excitation to demonstrate highly reproducible acidification or alkalinisation of pH environments localized to an electrode surface.9 In this case, pH monitoring is realized through quinones as electroactive agents, known by their chemically reversible proton production/consumption upon electrochemical stimulation. Most frequently, controlling pH by in situ electrochemical approach can also involve water electrolysis.10 During this process, water is split to generate flows of protons or hydroxyl ions to radically alter medium pH. Protons generated by the oxidative water splitting reaction lead to local acidification (eqn (1)). However, hydroxyl ions produced by the reduction of water reaction serve to increase the solution pH to alkaline condition (eqn (2)).Oxidation of water reaction:
| 2H2O → O2 + 4H+ + 4e− | (1) |
Reduction of water reaction:
| 2H2O + 2e− → 2OH− + H2 | (2) |
Coupling the process of water electrolysis with sensing application allows the rapid and efficient recognition of analytes by generating protons or hydroxyl ions optimal for the detection mechanism targeted. Read et al. control electrochemically the pH, using a boron-doped ring disc electrode system, to detect mercury in neutral solutions.11 This approach is based on water electrolysis by applying an appropriate oxidative potential to the ring electrode to generate protons. Acidification of the electrolyte solution occurs in the vicinity of the disc electrode at which mercury is detected. Similarly, Seymour and coworkers recently used gold interdigitated microband electrodes.12 Water was electrolysed at acidifier electrodes, changing the local pH near the sensing electrodes. An effective chlorine detection was demonstrated in water samples with electrochemical pH control.12 Similarly, Ag+ ions were detected in sodium acetate and in tap water with simultaneous electro-production of protons at an acidifier interdigitated electrode array to adjust the sample pH to the optimal condition.13 Huseinov et al. recently reported the use of membrane electrolysis for a reagent-free acidification.14 This approach is based on an in situ generation of nitric acid to produce an acidic environment in water sample enabling an effective detection of lead. On the other hand, Zhao et al.15 achieved optimal Pb2+ detection at pH 4.5 with a carbon nanotube-based electrode using a 0.1 M acetate buffer as supporting electrolyte. Similarly, Kang et al.16 reported pH optimization in acetate buffers with pHs in the 4.6–6.5 range. The LODs achieved in these conditions were in the nanomolar or subnanomolar range. However, this was possible in slightly acidic conditions thanks to the removal of dissolved oxygen through nitrogen bubbling.
Lead (Pb) is one of the heavy metals that poses damaging impacts on the environment and human health due to its extreme poisoning and bioavailability.17 Lead has been intensively used in industrial and mining activities, coal combustion, leaded paints, fuels,18 and as a material for household plumbing such as fittings, pipes, and solder.19 Lead contamination could be present on tap water that has been in contact with a lead pipe for a long time. Subsequently, lead can readily access the human body, yielding to serious health complications, such as kidney cancer, teeth disease and trouble in brain functions.20 The World Health Organization (WHO) recommended a concentration below 10 μg L−1 (48 nM) for Pb2+ in drinking water.21 Electroanalytical techniques are perfectly suitable to reach these concentration levels in aqueous samples to monitor drinking water.1,2 However, the pH level of drinking water is normally varying between 6.5 and 8.522 with an average of 7.5.23 For this reason, the challenge is to develop an effective lead sensor that allows the detection of lead in drinking water within this pH range without addition of reagents.
We report here on the development of a pH control approach based on an in situ electrochemical sample acidification. An electrochemical cell involving two working electrodes was used. The first working electrode is used for Pb sensing while the second one serves to acidify the electrolyte solution. Applying a positive potential to the acidifier, allows water oxidation and the electro-generation of a flux of protons, which diffuse to the vicinity of the sensor surface. This flux of protons is sufficient to lower the sample pH to the optimal acidic condition. Lead is used as a model analyte to demonstrate the applicability of the electrochemical pH control method. Thus, an effective electrochemical detection with in situ acidification was demonstrated to detect concentration of lead in agreement with WHO recommendations.
2. Experimental section
2.1. Chemicals and reagents
Nitric acid (HNO3, ≥99.999% trace metals basis), Sodium nitrate (NaNO3, 99.0%) and lead standard for ICP (1000 ± 2 mg L−1) were purchased from Sigma-Aldrich. All solutions were prepared using an ultrapure Millipore Milli-Q (18.2 MΩ) water. A stock solution of Pb2+ (10 ppm) was prepared in 10 mL of ultrapure water. Supporting electrolyte solutions of different pH values with constant ionic strength were tested for Pb2+ detection: 0.1 M HNO3 (pH 1), 0.01M HNO3 + 0.1 M NaNO3 (pH 2), 0.001 M HNO3 + 0.1 M NaNO3 (pH 3), 0.1 M NaNO3 (pH 5.6).2.2. Electrochemical experiments
All the electrochemical measurements were carried out in a cell involving a four-electrode system: two working electrodes (i) a sensing electrode (gold screen printed electrode, Au-SPE, for lead detection), (ii) an acidifier electrode for the pH control, (iii) a reference electrode and (iv) a counter electrode. Au-SPE 220BT, used as the sensing electrode (Ø = 4 mm), and a silver/silver chloride pseudo-reference electrode, were purchased from Dropsens/Metrohm. An external platinum electrode positioned above the acidifier was used as auxiliary electrode. A platinum mesh (aperture: 0.012 cm, wire diameter: 0.004 cm) was obtained from Goodfellow France and used as acidifier. A 3D printed cell was specifically designed to introduce the SPE with the inserted Pt grid as acidifier. The Pt grid was kept at a distance of approx. 250 μm by a rubber O-ring purchased from Coruba, UK. The fabrication of the electrochemical cell by 3D printing was explained in detail in our previous publications.24,25 A simplified schematic representation of the designed cell and the principle of lead detection is shown in Fig. 1. | ||
| Fig. 1 (A) Schematic representation, (B) photograph, and (C) operating principle of the 3D printed electrochemical cell with integrated pieces: (1) external auxiliary Pt electrode, (2) acidifier (Pt grid), (3) spacer, (4) screen printed electrode 220BT (gold sensing electrode and silver reference electrode). | ||
The electrochemical cell was connected to a Metrohm/Eco Chemie Autolab PGSTAT100 Bipotentiostat/Galvanostat operated by Nova 2.1.4 software. All the experiments were performed using 500 μL as sample volume. Cyclic voltammetry (CV) at scan rate of 50 mV s−1 was initially performed in the presence of 100 ppm Pb2+ to determine the potential window and the position of the Pb deposition and stripping peak. CV analyses were also carried out to optimize the pH control conditions. Anodic stripping voltammetry using square wave voltammetry (SVW) was performed to analyse samples containing different concentrations of Pb2+ in NaNO3 (0.1 M, pH 5.6) with in situ acidification. SWV analyses were recorded from −0.7 V to 0 V using the following parameters: −0.7 V as deposition potential, 120 s as time of deposition, 5 mV as potential step, 25 mV as amplitude and 20 Hz as frequency. During analysis, the sample was stirred to avoid the accumulation of oxygen bubbles. Calibration curves were obtained by detecting different Pb2+ concentration going from 10 to 100 ppb. The limit of detection (LOD) was calculated using the 3.3σ/S equation, with S is the slope of the calibration curve equation and σ is the standard error of the intercept.
To assess the influence of pH changes on the reference potential of the pseudo reference electrode, open circuit potential (OCP) measurements across a range of pH values were performed (Fig. S1†). The results indicate that the pseudo reference electrode presents relative stability with minimal potential variations across the tested pH (1; 2; 3 and 5.6), ensuring reliable sensor response with varying pH.
3. Results and discussion
3.1. Optimization of the electrochemical pH control
We have investigated the impact of the potential applied at the acidifier on the pH in the vicinity of the sensor. We used the peak potential of the gold oxide reduction to monitor and optimize the local pH changes.26 We are aiming to reach a local pH of 1 as it is reported as an optimal pH for the Pb2+ detection with minimised interferences from Cu2+ ions.19 Gold oxide was produced and then reduced by cyclic voltammetry recorded from −0.1 V to +1 V on Au-SPE at a scan rate of 50 mV s−1. Fig. S2A† shows the cyclic voltammograms obtained at different solutions over the pH range: 1 < pH < 5.6 as described on the Experimental section. The voltammetric response of a gold electrode immersed at pH 1 shows typical characteristic peaks in both anodic and cathodic scans (red curve of Fig. 2A).27 The peaks in the anodic scan are attributed to the formation of different forms of gold oxides on the electrode surface. When reversing the scan direction, a single sharp reduction peak in the cathodic scan is shown and correspond to the reduction of the oxide layer. The position of the reduction peak is strongly pH-dependent and is shifted to lower potentials with the increase of pH. The corresponding plot illustrating this dependence is shown in Fig. S2B.† The peak potential value shifts from +0.47 V at pH 1 to +0.25 V at pH 5.6. An intense decrease in peak current is also noticed when comparing the peak reduction current obtained at pH 1, with the one attained at pH 5.6. These results reveal that the peak potential for gold oxide reduction can be used to monitor the pH in acidic range.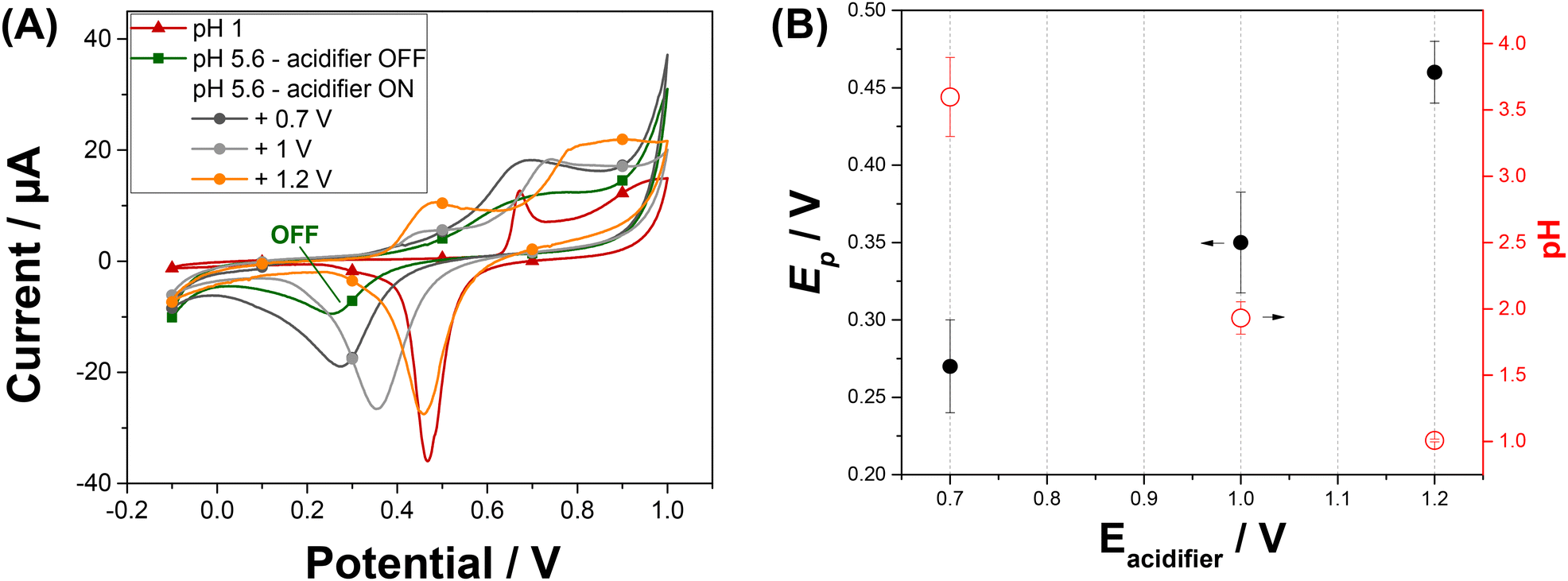 | ||
| Fig. 2 (A) Cyclic voltammogram of the gold electrode obtained in 0.1 M HNO3 (pH 1) solution and in 0.1 M NaNO3 (pH 5.6) solution, before and after applying different potentials (+0.7; +1.0, and +1.2 V) to the acidifier for 5 s before the scan. Scan rate 50 mV s−1. (B) Variation of the Au cathodic peak potential, Ep, (filled discs) and of the calculated pH (circles) as a function of the potential applied to the acidifier, Eacidifier. | ||
Cyclic voltammetry at Au-SPE was recorded at pH 5.6 simultaneously with the application of an oxidative potential at the acidifier electrode. Fig. 2A shows the cyclic voltammogram of Au-SPE sensing electrode obtained before and after applying different potentials ranging from +0.4 to +1.2 V to the acidifier. The reduction peak current increases and its position shift to higher potentials with the potential to the acidifier, confirming that the pH of the solution varies with the applied potential. The variation in the reduction gold oxide potential and the corresponding calculated pH are plotted versus the potential applied at the acidifier in Fig. 2B. The reduction peak potential value shifts from +0.25 V when the acidifier is switched off, to +0.46 V when it is biased at +1.2 V. The corresponding pH was calculated, exhibiting a gradual decrease of the initial pH of the solution (5.6) to reach pH 1 when the acidifier is biased at +1.2 V. These results confirm that the application of an anodic potential to the acidifier leads to a change in the local pH at the sensor surface electrode, as a greater flux of protons is produced.
Experiments showed that the pH at the surface of the sensor electrode can be dropped by 4.6 pH unit by applying a potential of +1.2 V to the acidifier. This local acidification of the electrolyte solution is sufficient for sensitive detection of Pb2+. However, applying potentials greater than +1.2 V to the acidifier gave very noisy CVs due to the generation of oxygen bubbles at the acidifier. For these reasons, a potential of +1.2 V for the acidifier was selected in the following experiments.
Cyclic voltammetry at Au-SPE was then carried out in pH 5.6 after applying the optimized acidified potential (Eacidifier = +1.2 V) for different times (tacidifier of 5 s and 80 s) to determine the optimum acidification time to reach pH 1 in the vicinity of sensing electrode. The obtained cyclic voltammograms are presented in Fig. S3,† with the acidification process tested three times under both tacidifier conditions. As it can be seen, the position of reduction peak potential confirms the pH change from 5.6 to 1 when applying Eacidifier in tacidifier 5 s and 80 s. However, tacidifier of 5 s gave an average of reduction peak potential of 0.45 V ± 0.02 (N = 3), while the average of reduction peak potential obtained when acidifier was biased at +1.2 V during tacidifier of 80 s, is 0.46 V ± 0.01 (N = 3). Consequently, the acidification process is more reproductible when tacidifier is 80 s, which agrees with previous results.
3.2. Electrochemical behaviour of Pb2+ under sample acidification
The cyclic voltammetry analysis of 100 ppm Pb2+ was investigated on gold screen-printed electrode at pH 1 and at pH 5.6 when the acidifier was either switched on or off (Fig. 3). At pH 1, the peaks observed are attributed to the reduction of lead ions Pb2+(aq.) to its zero-valence metallic state Pb(s) and then the dissolution of the Pb(s) back to lead ions Pb2+(aq.), as indicated in the Pourbaix diagram for lead.28 The first two cathodic peaks (C1 and C2) observed at −0.25 V and −0.51 V, are attributed to the formation of a Pb monolayer on the Au-SPE surface at potentials more positive than the reversible Nernst potential for bulk metal formation.29 This electrochemical phenomenon is called underpotential deposition (UPD),30–32 which takes place when a metal is deposited on a sensing electrode surface of different nature. The deposition of Pb multilayers is observed with another cathodic peak at −0.77 V (C3). This bulk deposition is defined as overpotential deposition (OPD), which takes place at potential greater than the Nernst potential (−0126 V/SHE33,34). This can be explained by the fact that the UPD of Pb takes place at a potential less negative than its equilibrium potential due to a difference in affinity between Pb and the gold electrode surface compared with the Pb–Pb interaction. Overall, the interactions between Pb and the electrode surface (adatom-substrate bond) are stronger than the Pb–Pb interactions (adatom–adatom bond). Thus, the energy required to reduce the first layer of Pb metal is lower than that required for multilayer formation.35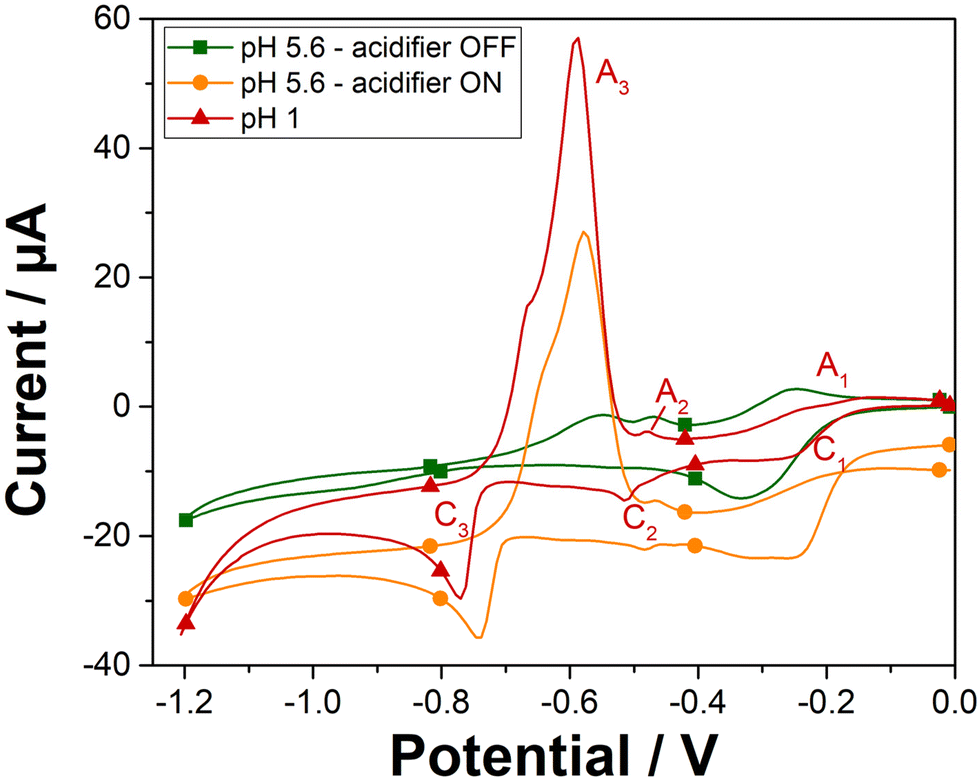 | ||
| Fig. 3 Cyclic voltammograms of gold electrode obtained in 100 ppm Pb2+ at pH 1 (red triangle curve), and at pH 5.6 with the acidifier switched off (green square curve), and with the acidifier switched on (orange circle curve). Scan rate 50 mV s−1. | ||
On the reverse scan, the bulk deposit is first stripped from the sensing electrode surface and dissolved into the solution. Thus, a well sharped anodic peak (A3) corresponded to the bulk stripping of lead, is observed at a potential of −0.59 V. Afterwards, the last monolayer of lead underpotential deposit, is then stripped, with the two other peaks (A2 and A1) attributed to the UPD stripping observed at potentials of −0.48 V and −0.24 V, respectively. Solution pH has a significant impact on the electrochemical deposition and stripping behavior of lead ions. Specifically, the pH influences the availability of protons, which are crucial for the stripping process. Cyclic voltammetry on Au-SPE in the presence of 100 ppm of lead were also carried out at different pH solutions 2; 3 and 5.6 (Fig. S4†). The resulted voltammograms at different pH values have shown that lower pH levels are more favorable for lead ion deposition and stripping using our sensor system, resulting in more defined and higher peaks. In the case of pH 5.6 with the acidifier switched off, only one cathodic peak is observed at potential of −0.33 V, which can be attributed to an overlapping of the two cathodic peaks (C1 and C2) of lead UPD. The disappearance of the cathodic C3 peak confirms that the bulk deposition of lead appears only when peaks of lead UPD reach their maximal charge.36 The significant change in the electrochemical behaviour of lead as a function of pH, proves that in less acidic medium, the proton deficiency makes difficult the redox reaction of lead. However, lower pH values lead to higher currents that boost the sensor response's sensitivity. Consequently, these results confirm the need to acidify the pH of the electrolyte solution to detect lead in neutral medium.
Cyclic voltammetry was then recorded in the presence of lead and with the acidifier biased at +1.2 V (Fig. 3). As expected, a similar electrochemical behaviour for 100 ppm Pb2+ is obtained when the acidifier is switched on as compared to the case of pH 1. The chemically prepared pH 1 solution gives a bulk stripping A3 peak at +0.59 V with a current magnitude of 63.7 μA, while the anodic A3 peak is observed at +0.58 V with the acidifier switched on with a peak magnitude of 43.2 μA (68% of the signal compared to the magnitude of A3 obtained for the untreated pH 1). Hence, these results confirm the possibility to reach pH conditions to perform Pb2+ electroanalysis through acidification by in situ electrochemical method.
3.3. Pb2+ ASV under sample acidification
ASV of 100 ppb of Pb2+ was repeated three times under the same conditions (Fig. S5†). The resulting square wave voltammograms shows a lack of repeatability in the measurements. Stripping peak potential and current are dependent on the oxidation state of the electrode surface. Cyclic voltammetry of the platinum grid was performed in 0.1 M NaNO3 (pH 5.6). The current shoulder around +0.70 V is assigned to oxidation of platinum on the forward scan and a peak at −0.07 V (on the reverse scan) is assigned to the oxide reduction (Fig. S6†). Water oxidation reaction at a platinum electrode is a multiple step reaction.37 It proceeds through an adsorbate evolution mechanism. Hydroxide ions, resulting from H2O dissociation, are adsorbed onto the Pt electroactive sites (eqn (3)). Then a deprotonation process undergoes to form PtO intermediates (eqn (4)). A nucleophilic attack by solvent water molecules leads to the formation of PtOOH at the electrode surface (eqn (5)). Finally, PtOOH goes through a deprotonation process to release H+ and O2 (eqn (6)).| Pt + H2O → PtOH + H+ + e− | (3) |
| PtOH → PtO + H+ + e− | (4) |
| PtO + H2O → PtOOH + H+ + e− | (5) |
| PtOOH → Pt + O2 + H+ + e− | (6) |
The electro-generation of H+ leads to the formation of Pt oxide layers, disturbing the surface atom arrangements, to which electrode reactions are sensitive to. Iizuka et al. have shown that oxygen evolution reaction activity of Pt electrodes dropped when an anodic potential of +1.4 V (vs. RHE, pH = 1) was held for 100 s. OER activity could be restored when a potential sufficiently low to reduce the Pt oxide was applied.38
An electrochemical regeneration of the surface of the platinum grid is performed before each detection experiment to restore the electrode activity degraded by the prolonged oxidation of the platinum grid. The full analytical procedure involves a three-step process: (i) sensor cleaning and acidifier regeneration, (ii) deposition & acidification, (iii) detection. The cleaning step starts by applying an anodic potential of +0.1 V to the sensor to dissolve any metal deposits left over from a previous experiment. A potential of 0 V is applied to the acidifier to reduce the Pt oxide layer that was formed during the acidification procedure. After 60 s, the deposition step begins by the application of −0.7 V to the sensing electrode. This step involves the reduction of lead ions Pb2+(aq.) to its zero-valence metallic state Pb(s) on the Au-SPE surface. The acidification step starts by the application of +1.2 V to the acidifier for the suitable optimized time. During this step, water is oxidised and generates protons, which diffuse to the surface of the sensor. Deposition and acidification processes are then followed by the stripping step. The electrolyte solution is stirred to ensure the homogenization of the solution during the cleaning step, to improve mass transport during the deposition step and to avoid the accumulation of oxygen bubbles during the acidification step. Stirring is stopped before the start of the stripping. Fig. 4A displays the anodic stripping voltammograms recorded in the presence of 100 ppb of Pb2+, at pH 1 and pH 5.6 solution, without and with the acidifier switched on. At pH 1, an anodic stripping peak is observed at −0.27 V with a peak magnitude of 14.1 ± 1.4 μA. At pH 5.6, the application of the acidification process leads to the improvement of the anodic stripping current of Pb. Without acidification a stripping peak located at −0.28 V with a current magnitude of 9.4 ± 0.8 μA is recorded. When the acidifier is switched on, a sharp anodic stripping peak is obtained at −0.28 V with a peak magnitude of 15.9 ± 0.7 μA (N = 3). Thus, the peak current intensity increased by 59% compared to the initial obtained current when the pH of detection is dropped from 5.6 to 1. This behaviour gives direct evidence that lower pH values provide a higher current, which offers a higher sensitivity for SWV detection. These results prove the efficiency of the optimized acidification procedure. In our experimental conditions, the sample volume is 0.5 mL for a concentration of 100 ppb Pb2+. If 100% of Pb2+ contained in the sample is deposited on a 4 mm diameter disc electrode (Aelectrode = 0.126 cm2), 0.397 μg cm−2 of Pb will be deposited. Given that Pb has an atomic radius of 1.86 × 10−8 cm and an atomic mass of 207.2 g mol−1, a full monolayer of Pb would correspond to 0.316 μg cm−2. It should be noted that for the simplicity of calculations, geometric surface area was considered. These results suggest that, in our experimental conditions, a single monolayer of Pb is formed during the preconcentration process. Such calculation agrees with our decision to use underpotential deposition instead of the bulk deposition for the lead detection. Considering the shapes of the Pb ASV peaks obtained in pH 1 and in pH 5.6 electrochemically acidified samples, the differences observed can be attributed to several factors, including variations in the local pH environment, the presence of different species already present in the electrolytes or formed during electrochemical acidification, and different kinetics of the stripping process under these conditions.
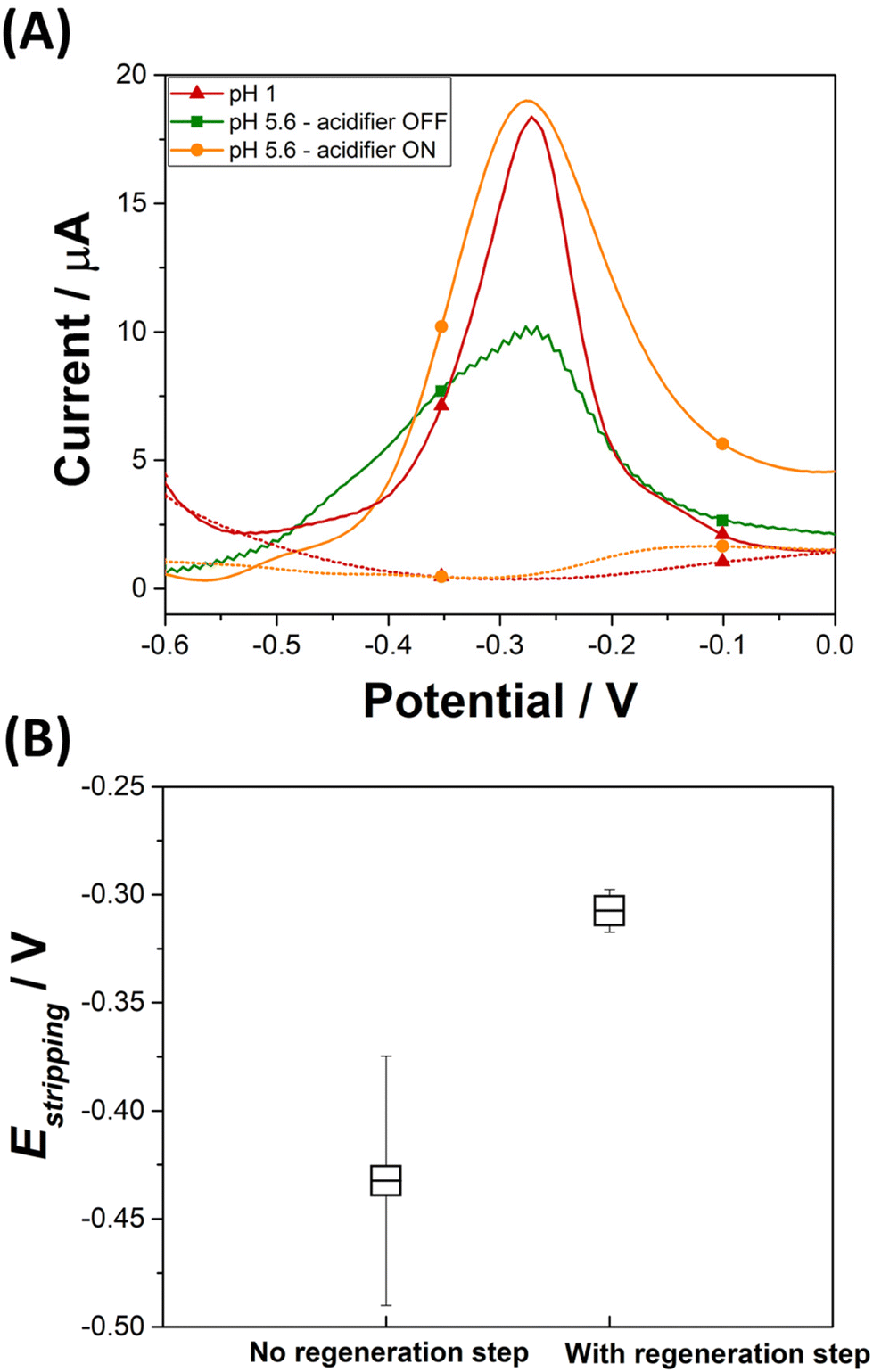 | ||
| Fig. 4 (A) SWV of 100 ppb Pb2+ on a gold electrode in HNO3 (pH 1) (red triangle curve) and in 0.1 M NaNO3 (pH 5.6) with the acidifier switch off (green square curve) and on (orange circle curve). (B) Average of stripping peak potentials values obtained with (N = 20) and without (N = 80) regeneration step of the acidifier. | ||
The average and the standard deviation of the stripping peak potential Estripping from a hundred measurements are estimated before and after applying the optimized SWV procedure (Fig. 4B). In the absence of the regeneration of the acidifier surface, the average potential value for the stripping of Pb(s) to Pb2+(aq.) is −0.43 ± 0.06 V. This wide distribution of Estripping values can be corrected by the application of a reductive potential that regenerates the acidifier surface. Once a regeneration step is included in the analytical procedure, the average Estripping value is −0.31 ± 0.01 V. This shows that applying the optimized SWV procedure, which includes the cleaning step, results in achieving stability in the stripping peak potential.
3.4. Analytical sensing of Pb2+ with in situ acidification
SWV analysis were performed in the presence of various concentrations of Pb2+ in 0.1 M NaNO3 (pH 5.6) with in situ acidification, using to the optimal procedure to build a calibration curve (Fig. 5A). In the presence of Pb2+, the peak current indicative of the anodic stripping of lead is observed at −0.28 V. A proportional increase of the stripping current response is observed when the concentration of Pb2+ increased from 10 to 100 ppb. The Pb stripping peaks shift to higher potentials when increasing Pb2+ concentration and consequently increasing the Pb2+/Pb ratio, which is in agreement with the Nernst equation. Furthermore, it is obvious that when the lead concentration reaches 100 ppb, the SWV stripping response shows the appearance of a small shoulder around −0.48 V. This can be explained by the fact that from this concentration, the second UPD stripping peak of Pb monolayer starts to be visible. The variation of the shape of the stripping peaks for various Pb2+ concentrations may be due to the several factors that may include the surface coverage of the electrode by Pb2+, the co-adsorption of nitrate anions and the variation of dissolved oxygen concentration linked to the acidification process. The dependency of the stripping peak current with Pb2+ concentration is linear as shown in Fig. 5B. All analyses were performed in triplicate to obtain standard deviation (shown as error bars). The correlation equation is given by istripping = (0.086 ± 0.003) [Pb2+] + (4.24 ± 0.164). This variation shows a wide range of linearity which extends from 10 ppb to 60 ppb with a correlation coefficient of R2 = 0.996 and a LOD of 6 ppb, which is below the WHO recommendation for Pb2+ concentration in drinking waters. As a comparison, the calibration curve when the acidifier is OFF gives a correlation equation of istripping = (0.034 ± 0.002) [Pb2+] − (0.138 ± 0.055). The equation for the calibration curve at pH 1 is istripping = (0.188 ± 0.017) [Pb2+] − (0.662 ± 0.436). The sensitivity of the Pb2+ stripping when the acidifier is switched ON is higher than when it is OFF. However, it does not achieve the same sensitivity as when the pH is close to one. The high value of the intercept found when the acidifier is ON can be explained by the high dissolved oxygen concentrations liked to the acidification process discussed beforehand.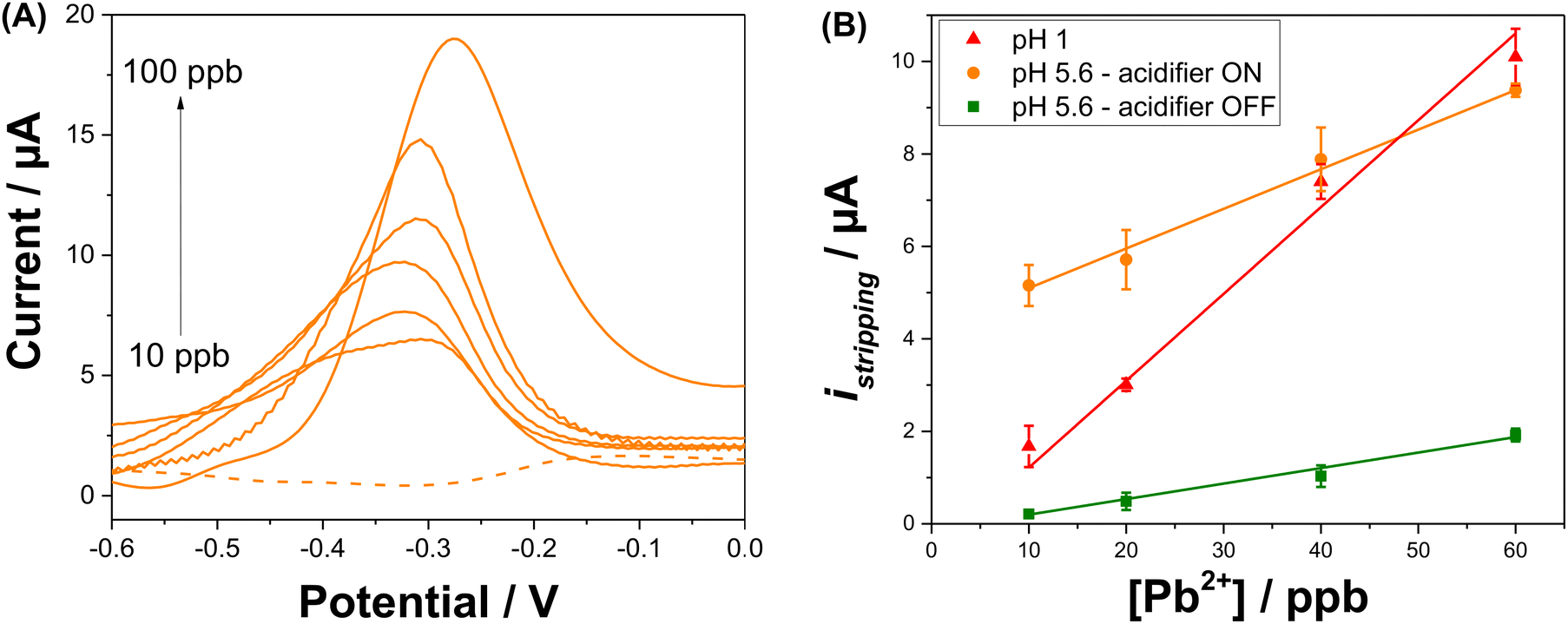 | ||
Fig. 5 (A) SWV of different Pb2+ concentrations (10, 20, 40, 60, 80, 100 ppb) recorded in 0.1 M NaNO3 (pH 5.6) with in situ acidification, according to the optimized procedure. (B) Calibration curves for anodic stripping peak current vs. Pb2+ concentration at pH 1 ( ), and at pH 5.6 when the acidifier is ON ( ), and at pH 5.6 when the acidifier is ON ( ) and OFF ( ) and OFF ( ). ). | ||
To study the impact of interference such as Cu2+ on the detection of Pb2+, SWV analysis was then undertaken with in situ acidification in the presence of a fixed concentration of 100 ppb (0.48 μM) of Pb2+ and an increasing concentrations of Cu2+: 200 ppb (3.2 μM), 500 ppb (7.9 μM), and 1000 ppb (15.7 μM). The average percentage of the stripping Pb2+ signal (N = 3) in the presence of Cu2+ is presented in Fig. S7.† Despite the presence of high concentrations of the interferent, Cu2+ slightly affects the stripping signal of Pb2+, resulting in a low decrease in the Pb2+ signal by approximately 22%, 15%, and 19% in the presence of 200, 500, and 1000 ppb of Cu2+, respectively.
4. Conclusions
We have proceeded to local electrochemical acidification involving water electrolysis for the detection of Pb2+ traces. An electrochemical cell was designed to integrate an acidifier close to a gold sensor. By applying an optimal positive potential to the acidifier, water oxidation leads to the electro-generation of protons, which diffuse to the vicinity of the sensing electrode and locally acidify the sample. Under the optimized conditions, the pH in the vicinity of the sensor surface was dropped from the initial value (pH 5.6) to the optimal detection value (pH 1). Anodic stripping voltammetry was then carried out, to successfully detect concentrations down to 10 ppb of Pb2+, in agreement with the WHO guidelines. The developed approach proves that the control of pH in situ paves the way for on-field analysis, whether for lead detection or other pH-dependent analytes. In this sense, experiments are underway to develop an innovative electrochemical device for real-time lead detection in water utility systems using the in situ pH control method developed in this work.Data availability
Data for this article, including raw voltammograms used to draw Fig. 2–5 and Fig. S1–S7† will be available, when the manuscript is accepted for publication at Recherche Data Gouv (https://entrepot.recherche.data.gouv.fr/) repository at https://doi.org/10.57745/F82FDN.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors extend their appreciation to SATT Sayens, ERDF and the National Research Agency (ANR) for providing funding to this study as a part of aQuaLead project (ANR-22-CE42-0028-01).References
- A. J. Borrill, N. E. Reily and J. V. Macpherson, Addressing the Practicalities of Anodic Stripping Voltammetry for Heavy Metal Detection: A Tutorial Review, Analyst, 2019, 144(23), 6834–6849, 10.1039/C9AN01437C.
- C. Ariño, C. E. Banks, A. Bobrowski, R. D. Crapnell, A. Economou, A. Królicka, C. Pérez-Ràfols, D. Soulis and J. Wang, Electrochemical Stripping Analysis, Nat. Rev. Methods Primers, 2022, 2(1), 1–18, DOI:10.1038/s43586-022-00143-5.
- G. Herzog and V. Beni, Stripping Voltammetry at Micro-Interface Arrays: A Review, Anal. Chim. Acta, 2013, 769, 10–21, DOI:10.1016/j.aca.2012.12.031.
- W. Chen, M.-K. Zhang, B.-Y. Liu, J. Cai and Y.-X. Chen, Challenges and Recent Progress in Unraveling the Intrinsic pH Effect in Electrocatalysis, Curr. Opin. Electrochem., 2022, 34, 101003, DOI:10.1016/j.coelec.2022.101003.
- X. Strakosas, J. Selberg, P. Pansodtee, N. Yonas, P. Manapongpun, M. Teodorescu and M. Rolandi, A Non-Enzymatic Glucose Sensor Enabled by Bioelectronic pH Control, Sci. Rep., 2019, 9(1), 10844, DOI:10.1038/s41598-019-46302-9.
- A. Wiorek, M. Cuartero, R. De Marco and G. A. Crespo, Polyaniline Films as Electrochemical-Proton Pump for Acidification of Thin Layer Samples, Anal. Chem., 2019, 91(23), 14951–14959, DOI:10.1021/acs.analchem.9b03402.
- A. Wiorek, G. Hussain, A. F. Molina-Osorio, M. Cuartero and G. A. Crespo, Reagentless Acid–Base Titration for Alkalinity Detection in Seawater, Anal. Chem., 2021, 93(42), 14130–14137, DOI:10.1021/acs.analchem.1c02545.
- F. Steininger, A. Wiorek, G. A. Crespo, K. Koren and M. Cuartero, Imaging Sample Acidification Triggered by Electrochemically Activated Polyaniline, Anal. Chem., 2022, 94(40), 13647–13651, DOI:10.1021/acs.analchem.2c03409.
- N. Fomina, C. A. Johnson, A. Maruniak, S. Bahrampour, C. Lang, R. W. Davis, S. Kavusi and H. Ahmad, An Electrochemical Platform for Localized pH Control on Demand, Lab Chip, 2016, 16(12), 2236–2244, 10.1039/C6LC00421K.
- H. Zhou, G. Li and S. Yao, A Droplet-Based pH Regulator in Microfluidics, Lab Chip, 2014, 14(11), 1917–1922, 10.1039/C3LC51442K.
- T. L. Read, E. Bitziou, M. B. Joseph and J. V. Macpherson, In Situ Control of Local pH Using a Boron Doped Diamond Ring Disk Electrode: Optimizing Heavy Metal (Mercury) Detection, Anal. Chem., 2014, 86(1), 367–371, DOI:10.1021/ac403519p.
- I. Seymour, B. O'Sullivan, P. Lovera, J. F. Rohan and A. O'Riordan, Electrochemical Detection of Free-Chlorine in Water Samples Facilitated by in situ pH Control Using Interdigitated Microelectrodes, Sens. Actuators, B, 2020, 325, 128774, DOI:10.1016/j.snb.2020.128774.
- L. Wasiewska, I. Seymour, B. Patella, R. Inguanta, C. Burgess, G. Duffy and A. O'Riordan, Reagent Free Electrochemical-Based Detection of Silver Ions at Interdigitated Microelectrodes Using in situ pH Control, Sens. Actuators, B, 2021, 333, 129531, DOI:10.1016/j.snb.2021.129531.
- A. Huseinov, A. Hoque, K. A. Ruble, B. J. Dee and N. T. Alvarez, Reagentless Dissolution and Quantification of Particulate Lead in Tap Water via Membrane Electrolysis, Anal. Chem., 2023, 95(24), 9297–9303, DOI:10.1021/acs.analchem.3c01201.
- D. Zhao, D. Siebold, N. T. Alvarez, V. N. Shanov and W. R. Heineman, Carbon Nanotube Thread Electrochemical Cell: Detection of Heavy Metals, Anal. Chem., 2017, 89(18), 9654–9663, DOI:10.1021/acs.analchem.6b04724.
- W. Kang, X. Pei, C. A. Rusinek, A. Bange, E. N. Haynes, W. R. Heineman and I. Papautsky, Determination of Lead with a Copper-Based Electrochemical Sensor, Anal. Chem., 2017, 89(6), 3345–3352, DOI:10.1021/acs.analchem.6b03894.
- Y. Zhang, H. Jiang and X. Wang, Cytidine-Stabilized Gold Nanocluster as a Fluorescence Turn-on and Turn-off Probe for Dual Functional Detection of Ag+ and Hg2+, Anal. Chim. Acta, 2015, 870, 1–7, DOI:10.1016/j.aca.2015.01.016.
- C. Wang, X. Cui, Y. Li, H. Li, L. Huang, J. Bi, J. Luo, L. Q. Ma, W. Zhou, Y. Cao, B. Wang and F. Miao, A Label-Free and Portable Graphene FET Aptasensor for Children Blood Lead Detection, Sci. Rep., 2016, 6(1), 21711, DOI:10.1038/srep21711.
- D. Dragoe, N. Spătaru, R. Kawasaki, A. Manivannan, T. Spătaru, D. A. Tryk and A. Fujishima, Detection of Trace Levels of Pb2+ in Tap Water at Boron-Doped Diamond Electrodes with Anodic Stripping Voltammetry, Electrochim. Acta, 2006, 51(12), 2437–2441, DOI:10.1016/j.electacta.2005.07.022.
- H. Needleman, Lead Poisoning, Annu. Rev. Med., 2004, 55(1), 209–222, DOI:10.1146/annurev.med.55.091902.103653.
- J. Strandberg. Lead in Drinking-Water, Health Risks, Monitoring and Corrective Actions – Technical Brief, 2022 Search PubMed.
- Background documents for development of WHO Guidelines for drinking-water quality and Guidelines for safe recreational water environments. https://www.who.int/publications/m/item/background-documents-for-development-of-who-guidelines-for-drinking-water-quality-and-guidelines-for-safe-recreational-water-environments (accessed 2023-12-01).
- K. Kulthanan, P. Nuchkull and S. Varothai, The pH of Water from Various Sources: An Overview for Recommendation for Patients with Atopic Dermatitis, Asian Pac. Allergy, 2013, 3(3), 155–160, DOI:10.5415/apallergy.2013.3.3.155.
- J. Gajdár, G. Herzog and M. Etienne, Amperometric Sensor for Selective On-Site Analysis of Free Sulfite in Wines, ACS Sens., 2022, 7(8), 2209–2217, DOI:10.1021/acssensors.2c00611.
- M. Etienne, T. X. H. Le, T. Nasir and G. Herzog, Electrochemical Filter To Remove Oxygen Interference Locally, Rapidly, and Temporarily for Sensing Applications, Anal. Chem., 2020, 92(11), 7425–7429, DOI:10.1021/acs.analchem.0c00395.
- S. Yang and D. G. H. Hetterscheid, Redefinition of the Active Species and the Mechanism of the Oxygen Evolution Reaction on Gold Oxide, ACS Catal., 2020, 10(21), 12582–12589, DOI:10.1021/acscatal.0c03548.
- J. P. Hoare, A Cyclic Voltammetric Study of the Gold–Oxygen System, J. Electrochem. Soc., 1984, 131(8), 1808, DOI:10.1149/1.2115966.
- M. Pourbaix and J. Burbank, Atlas D-Equilibres Electrochimiques, J. Electrochem. Soc., 1964, 111(1), 14C, DOI:10.1149/1.2426051.
- D. W. M. Arrigan, L. L. Bihan and M. J. Pickup, Apparent Enhanced Underpotential Voltammetry of Lead(II) at a Spontaneously Adsorbed Monolayer-Coated Gold Electrode, Analyst, 1999, 124(12), 1797–1802, 10.1039/A906125H.
- I. Mazerie and F. Geneste, Coupling of Anodic Stripping Voltammetry with Sampled-Current Voltammetry on an Electrode Array: Application to Lead Detection, Sensors, 2020, 20(5), 1327, DOI:10.3390/s20051327.
- G. Herzog and D. W. M. Arrigan, Underpotential Deposition and Stripping of Lead at Disorganized Monolayer-Modified Gold Electrodes, Electroanalysis, 2005, 17(20), 1816–1821, DOI:10.1002/elan.200503318.
- M. Brand, I. Eshkenazi and E. Kirowa-Eisner, The Silver Electrode in Square-Wave Anodic Stripping Voltammetry. Determination of Pb2+ without Removal of Oxygen, Anal. Chem., 1997, 69(22), 4660–4664, DOI:10.1021/ac970420f.
- A. J. Bard, Standard Potentials in Aqueous Solution, Routledge, New York, 2017. DOI:10.1201/9780203738764.
- P. Atkins, Physical Chemistry for the Life Sciences, Oxford University Press, Oxford, 2010 Search PubMed.
- G. Herzog and D. W. M. Arrigan, Determination of Trace Metals by Underpotential Deposition–Stripping Voltammetry at Solid Electrodes, TrAC, Trends Anal. Chem., 2005, 24(3), 208–217, DOI:10.1016/j.trac.2004.11.014.
- E. Kirowa-Eisner, Y. Bonfil, D. Tzur and E. Gileadi, Thermodynamics and Kinetics of Upd of Lead on Polycrystalline Silver and Gold, J. Electroanal. Chem., 2003, 552, 171–183, DOI:10.1016/S0022-0728(03)00181-5.
- B. E. Conway, Electrochemical Oxide Film Formation at Noble Metals as a Surface-Chemical Process, Prog. Surf. Sci., 1995, 49(4), 331–452, DOI:10.1016/0079-6816(95)00040-6.
- K. Iizuka, T. Kumeda, K. Suzuki, H. Tajiri, O. Sakata, N. Hoshi and M. Nakamura, Tailoring the Active Site for the Oxygen Evolution Reaction on a Pt Electrode, Commun. Chem., 2022, 5(1), 126, DOI:10.1038/s42004-022-00748-7.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an00647j |
| This journal is © The Royal Society of Chemistry 2024 |