
Novel mycophenolic acid precursor-based fluorescent probe for intracellular H2O2 detection in living cells and Daphnia magna and Zebrafish model systems†
Jongkeol
An
 a,
Sujeong
Park
b,
Neha
Jain
a,
Youngsam
Kim
*cd,
Satish Balasaheb
Nimse
*b and
David G.
Churchill
*ae
a,
Sujeong
Park
b,
Neha
Jain
a,
Youngsam
Kim
*cd,
Satish Balasaheb
Nimse
*b and
David G.
Churchill
*ae
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, 34141, Republic of Korea
bInstitute of Applied Chemistry and Department of Chemistry, Hallym University, Chuncheon, 24252, Republic of Korea
cEnvironmental Safety Group, KIST Europe Forschungsgesellschaft mbH, 66123 Saarbrücken, Germany
dDivision of Energy and Environment Technology, University of Science and Technology, Daejeon 34141, Republic of Korea
eKAIST Institute for Health Science and Technology (KIHST) (Therapeutic Bioengineering Section), Daejeon 34141, Republic of Korea
First published on 18th July 2024
Abstract
Innovative for the scientific community and attracting attention in the extensive biomedical field are novel compact organic chemosensing systems built upon unique core molecular frameworks. These systems may demonstrate customized responses and may be adaptable to analytes, showing promise for potential in vivo applications. Our recent investigation focuses on a precursor of Mycophenolic acid, resulting in the development of LBM (LOD = 13 nM) – a specialized probe selective for H2O2. This paper details the synthesis, characterization, and thorough biological assessments of LBM. Notably, we conducted experiments involving living cells, daphnia, and zebrafish models, utilizing microscopy techniques to determine probe nontoxicity and discern distinct patterns of probe localization. Localization involved the distribution of the probe in the Zebrafish model within the gut, esophagus, and muscles of the antennae.
1. Introduction
Reactive oxygen species (ROS) is terminology to indicate (partial or entirely consisting of) oxygen-containing substances with significant chemical reactivity. These include the superoxide anion (˙O2−), hydrogen peroxide (H2O2), hydroxyl radical (˙OH), ozone (O3), and singlet oxygen (1O2). Due to their marked chemical reactivity, and in some cases, their property of unpaired electrons, ROS play a vital role in a range of pathological, healthful, and physiological processes.1 Notably, among these species, H2O2 stands out as a significant molecule in nature and the environment. It arises as a result of active oxygen metabolism which is enzyme-catalyzed. In terms of serving as a messenger,2 H2O2 is a byproduct of essential cellular functions such as protein folding.3 H2O2 functions as a crucial modulator in numerous oxidative stress-related conditions as well. Humans are in contact with H2O2 through various common uses such as cosmetic and household products. H2O2 is processed within cells and tissue; excessive production and accumulation of hydrogen peroxide in the body, however, have been linked to various conditions/diseases, including various cancers, aging, asthma, cardiovascular, and neurodegenerative disorders such as Parkinson's disease and Dementia.4–7Despite the current scientific understanding, the full impact of H2O2 on human health and its involvement in diverse diseases, likely significant, is not fully understood. Hence, the development of a sensitive and effective method to detect H2O2 levels using small molecule design or devise design is critically important, and continued work in this direction is ongoing. Presently, primary techniques for detecting H2O2 encompass colorimetry, electrochemical assays, fluorescence probe methods, and spectrophotometry.8–10 However, sample preparation for spectrophotometry, electrochemistry, and colorimetry involves intricate processes and cannot dynamically capture changes in H2O2 levels or accurately measure H2O2 concentrations within live cells. In contrast, fluorescence probe methodologies offer a robust means of monitoring analyte (e.g., H2O2) levels within living systems.11–14
Fluorescent probes typically consist of fluorescent groups, detection units, and linking components. By combining different fluorescent and detection units, diverse fluorescent probes can be designed to address various detection requirements; the nature of the selected molecule may also have different distribution abilities in vivo. Thus, the utilization of fluorescence probes for detecting hydrogen peroxide associated with numerous human diseases continues to be a key technology; however scientific and technological optimization is still ongoing.
There are various foundational probe precedents. In 2003, the first boric acid-based H2O2 fluorescence probe was introduced,15 demonstrating high sensitivity and specificity in H2O2 detection. Notably, boric acid or borate esters are frequently employed as components in hydrogen peroxide reactions, with evidence showing the enhanced selectivity of probes based on borate ester oxidation reactions over other probe modalities that target ROS.16,17 The incorporation of recognition groups into probes has been validated for numerous classical fluorophores, including coumarin, naphthalimide, and fluorophores typically utilizing the AIE mechanism.18–21
Selenium or thioether-based fluorescent probes designed for the detection of H2O2 have been reported.22–25 Selenium is known to interact with ROS like hydrogen peroxide. In the design of fluorescent probes, the photoinduced electron transfer (PET) mechanism is frequently utilized. Typically, the PET process suppresses a fluorophore's fluorescence, which recovers when the PET is inhibited. These probes often involve selenium oxidation, which blocks the PET process. Thioether reacts with H2O2 to form sulfoxide or sulfone, during which fluorescence or color change occurs.
We formulated and synthesized a novel fluorescent probe named LBM to identify and characterize hydrogen peroxide's origins and roles as a transient redox messenger. This design combines the H2O2-reactive aryl boronic acid group with a specific recognition unit derived from mycophenolic acid (a precursor in the well-known synthesis thereof), allowing for the selective detection of hydrogen peroxide. The framework also includes a Br group for further conjugation and substitution.
The use of live cell imaging and organism models continues to be vitally important for validating the efficacy of the molecules and conditions necessary for analyte probing. We were able to use live cells of types A549 (lung cancer) and MRC-5 (lung normal).
Owing to their heightened susceptibility to toxicants, Daphnia magna (D. magna) have become indispensable model organisms for the explorations of ecotoxicology and other fields.26–29 Therefore herein, this animal model study was utilized. Additionally, we used Zebrafish models to extend our findings beyond those of closely related probes.21 Zebrafish serve as a versatile model for evaluating fluorescent dyes and conducting developmental toxicity studies. Their easy availability, small size, and transparent embryos make them ideal for early screening assays and high-throughput toxicity assessments. With its short life cycle, high fertility, and genetic similarity to humans, zebrafish are valuable scientific tools for analyzing the toxicity and bioactivity of various compounds30 which can later pave the way to help preserve the health and wellness of humans.
2. Experimental
2.1. Materials and apparatuses
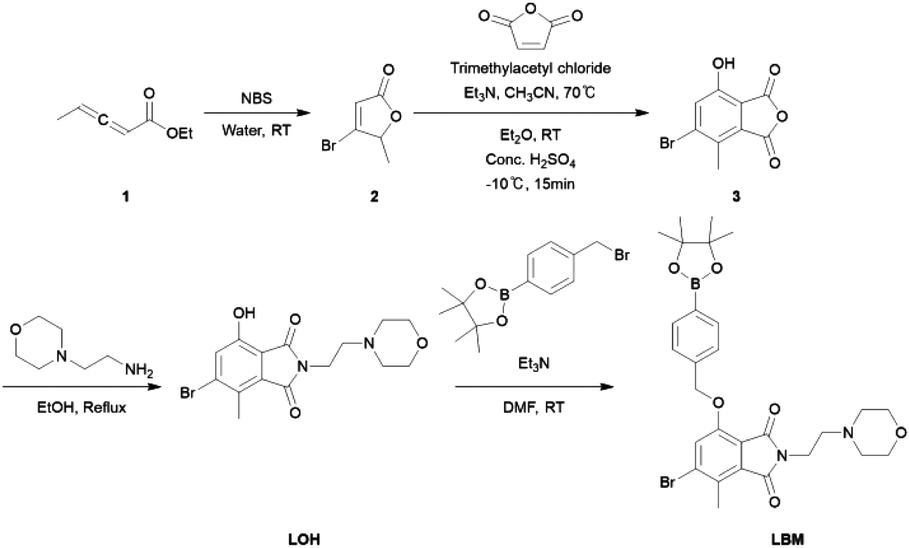
All chemicals were used as received from commercial suppliers (i.e., Aldrich, Tokyo Chemical Industry, etc.). 1H, 13C, COSY, HMBC, HSQC, and NOESY NMR spectra were acquired using a Bruker Avance 400 MHz spectrometer. Chemical shifts (δ) are reported in the standard notation of ppm relative to the residual solvent peak either at 7.26 (CDCl3) for 1H or 77.16 (CDCl3) for 13C as an internal reference. The following abbreviations are used for splitting patterns: br, broad; s, singlet; d, doublet; dd, doublet of doublets; t, triplet; td, triplet of doublets; q, quartet; quint, quintet; m, multiplet; ESI-mass spectrometry was performed on a Waters Corporation XEVO G2-XS QT of by the research support staff at KARA (KAIST Analysis center for Research Advancement). Absorption spectra were measured using a Shimadzu UV-1900i spectrophotometer. Fluorescence measurements were carried out with a Shimadzu RF-5301pc spectrofluorophotometer. A549 cells were obtained from the Korea Cell Line Bank and used as received. The confocal laser-scanning microscopy (CLSM) images were obtained with a Scope A1 confocal microscope (ZEISS).2.2. Synthesis of compound 3 and LOH
The synthetic routes of the probes are shown in Scheme 1. Compound 3 was synthesized according to a reported literature method.31 Compound 3 (0.257 g, 1.00 mmol) was dissolved in EtOH (10.0 mL) under a nitrogen atmosphere. Subsequently, the compound was treated dropwise with 4-(2-aminoethyl) morpholine (0.262 mL, 2.00 mmol) and refluxed at 78 °C for 24 hours. The reaction was monitored by TLC. After the reaction was determined to be finished by TLC, the solvent was evaporated and dried. The residue was purified by silica gel flash column chromatography using CH2Cl2/MeOH as the eluent (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume) to help afford the formation of compound LOH (0.217 g, 59%) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ = 7.37 (s, 1H, H1), 3.76 (t, 3JH–H = 6.5 Hz, 2H, H14), 3.66 (t, 3JH–H = 4.6 Hz, 4H, H19, 21), 2.64 (t, 3JH–H = 6.5 Hz, 2H, H16), 2.62 (s, 3H, H8) 2.54 (t, 3JH–H = 4.6 Hz, 4H, H18, 22); 13C NMR (100 MHz, CDCl3): δ = 169.49 (C10, 12), 167.84 (C6), 152.78 (C4), 134.27 (C5), 130.47 (C2), 128.82 (C3), 126.35 (C1), 66.82 (C19, 21), 56.08 (C18, 22), 53.46 (C14), 34.67 (C16), 16.31 (C8). MS/EI m/z: calculated for C15H17BrN2O4 + H, 369.04 found 369.0452.
:1 by volume) to help afford the formation of compound LOH (0.217 g, 59%) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ = 7.37 (s, 1H, H1), 3.76 (t, 3JH–H = 6.5 Hz, 2H, H14), 3.66 (t, 3JH–H = 4.6 Hz, 4H, H19, 21), 2.64 (t, 3JH–H = 6.5 Hz, 2H, H16), 2.62 (s, 3H, H8) 2.54 (t, 3JH–H = 4.6 Hz, 4H, H18, 22); 13C NMR (100 MHz, CDCl3): δ = 169.49 (C10, 12), 167.84 (C6), 152.78 (C4), 134.27 (C5), 130.47 (C2), 128.82 (C3), 126.35 (C1), 66.82 (C19, 21), 56.08 (C18, 22), 53.46 (C14), 34.67 (C16), 16.31 (C8). MS/EI m/z: calculated for C15H17BrN2O4 + H, 369.04 found 369.0452.
 | ||
| Scheme 1 Synthetic pathways of LBM. | ||
A sample of compound LOH (0.184 mg, 0.500 mmol) was dissolved in DMF (10.0 mL) under a nitrogen atmosphere. Subsequently, the mixture was treated with triethylamine (2.08 mL, 1.50 mmol), added with 4-bromomethyl phenylboronic acid pinacol ester (0.297 g, 1.00 mmol), and stirred at room temperature for 24 hours. The reaction was monitored by TLC. After the reaction was determined to be finished by TLC, the mixture was diluted with ethyl acetate (∼50 mL), washed with brine, dried with sodium sulfate, and dried as an isolated material to remove any remaining volatile solvent. The residue was purified by silica gel flash column chromatography with CH2Cl2/MeOH as the eluent (100:1 by volume) to help afford LBM (0.140 g, 24%) as a white/yellow solid. 1H NMR (400 MHz, CDCl3): δ = 7.83 (m, 2H, H26, 28), 7.46 (m, 2H, H25, 29), 7.38 (s, 1H, H1), 5.32 (s, 2H, H23), 3.77 (t, 3JH–H = 6.5 Hz, 2H, H14), 3.65 (t, 3JH–H = 4.6 Hz, 4H, H19, 21), 2.66 (s, 3H, H12), 2.61 (t, 3JH–H = 6.5 Hz, 2H, H16), 2.52 (t, 3JH–H = 4.6 Hz, 4H, H18, 22), 1.34 (s, 12H, H36–37); 13C NMR (100 MHz, CDCl3): δ = 168.02 (C7, 9), 165.85 (C6), 153.49 (C4), 138.48 (C5), 135.25 (C26, 28), 133.83 (C24), 131.09 (C27), 130.54 (C2), 126.08 (C25, 29), 123.43 (C1), 117.65 (C3), 83.92 (C32, 33), 71.17 (C23), 67.01 (C16), 56.11 (C22), 53.51 (C21), 34.80 (C14), 24.88 (C36–38), 16.33 (C12); MS/EI m/z: calculated for C28H34BBrN2O6 + H, 585.1693; found 585.1796.
2.3. Determination of the limit of detection
The experimentally determined limit of detection was calculated from fluorescence titration measurements. The fluorescence titration intensity of LBM (517 nm) was used to obtain the value for the slope in the x, y plot. The detection limit was calculated with the following equation of the graph:| Detection limit = 3σ/k |
In which σ is the standard deviation of the value obtained for 10 control measurements and k is the absolute value of the slope between the fluorescence intensity versus the H2O2 concentration.
2.4. Cell culture preparation and cellular imaging acquisition
The A549 (lung cancer) and MRC-5 (lung normal) cells, obtained from the Korea Cell Line Bank in Seoul, South Korea, were used to assess the cytotoxicity of LBM and its ability to detect intracellular H2O2 concentrations. These cells were cultured in a cell culture dish containing DMEM with 10% FBS and 1% penicillin/streptomycin in the presence of 5% CO2 at 37 °C until they reached about 80–90% confluency. The cells were then further subcultured in culture media after washing them with PBS (pH 7.4) followed by treatment with 0.25% trypsin–2.65 mM EDTA (EDTA = ethylenediamine tetra-acetic acid). The medium was replaced every two days. A549 cells were seeded and incubated for 24 h on poly-L-lysine-coated 14 mm coverslips in the 6-well plates for cell imaging experiments. In a control experiment, the culture medium was replaced with a fresh medium containing only Lyso-tracker dye (“Dye”, 2.0 μM), and the cells were incubated for 1 h at 37 °C to identify the lysosomes. In subsequent experiments, cells were treated with LBM (15.0 μM, final concentration) for 2 h followed by a 1 h treatment with “Dye” (2.0 μM). The LBM (15.0 μM) and “Dye” treated cells were further incubated with PMA (1.0 μg mL−1, final concentration) or H2O2 (100 μM, final concentration) or PMA and NAC (1.0 mM, final concentration) or NaOCl (100.0 μM, final concentration) at 37 °C for 1 hour. The cells were then prepared for cell-imaging analysis using a fluorescence microscope (Zeiss Axio Scope A1, Germany) by being washed with a PBS buffer solution maintained at physiological pH (pH = 7.4), fixing them with paraformaldehyde (2%), and mounting them onto glass slides.2.5. Cell viability assay
The cytotoxicity of LBM (0, 5.0, 10.0, 25.0, and 50.0 μM) against A549 (lung cancer) and MRC-5 (lung normal) cell lines was evaluated by MTT assay analysis. In brief, about 7000 cells per well were seeded into wells using the 96-well plate format and were allowed to incubate in the presence of 5% CO2 at 37 °C for 24 h. The LBM (10.0 mM) stock solution in DMSO was diluted in a culture medium to obtain the working solutions of 0, 5.0, 10.0, 25.0, and 50.0 μM concentrations. Respective solutions were then loaded in the wells containing cells and allowed to incubate for 24 h, followed by treatment with MTT for 2 h in the dark. The cytotoxicity of LBM was then determined by measuring the absorbance in each well at 490 nm using a spectrophotometer (SpectraMAX Plus, Molecular Devices Inc., Sunnyvale, CA, USA). All experiments were performed in triplicate, and the results were presented as values of mean ± SD (standard deviation).2.6. D. magna culture and fluorescence imaging of LBM in D. magna model systems
Ephippia of D. magna (Micro Biotests Inc., Belgium) were hatched under a light/dark cycle of 16/8 hours with a light intensity of 7000 lux for 72 hours and maintained at a temperature of 20.0 ± 1.0 °C. Fifteen adult daphnids were cultured in a 2.0 L beaker with a 1.5 L Elendt M4 medium. D. magna were fed daily with Chlorella vulgaris (0.1 mg C (individual per day, ∼1.5 × 108 cells per mL)). Regular maintenance included thrice replacement of beakers and medium in a week for water quality and daily removal of new neonates to prevent overcrowding. Before each medium replacement, the pH and dissolved oxygen levels were routinely checked. The interlaboratory test for verification of the test condition reliability was regularly performed according to guideline ISO 6341 with potassium dichromate as a reference chemical.Newborn neonates (<24 h) from the third brood were placed in six-well culture plates containing 10 mL ISO medium to concentrations of 10.0, 7.5, 5.0, 4.5, 4.0, 3.5, 3.0, 2.0, 1.0 μg L−1 (parts per billion, ppb) [Ag+], along with control series for effective concentration (EC) determinations according to OECD test guideline 202.32 Silver ion stock solution was prepared weekly by dissolving AgNO3 (Sigma-Aldrich, St Louis, MO, USA) to a concentration of 100 mg L−1 [Ag+]. Groups were divided into eight replicates with five daphnids each. Similarly, newborn neonates from the fourth brood also were placed in a six-well culture plates with a 10.0 mL ISO medium for fluorescence imaging. The experiment comprised three main groups: a control group with no treatment, a group treated with only probe LBM, and a group exposed to both LBM and 3.0 ppb [Ag+]. Each group was subjected to two distinct exposure durations, 6 hours and 24 hours. Only the groups involving Ag+ ion concentration were exposed to 3.0 ppb [Ag+]. After this initial exposure of 3 hours to Ag+, LBM treatment commenced according to the relevant groups. This was followed by a 30 minute incubation period before proceeding with the imaging. This sequence of exposure and treatment was the same for both the 6 hour and 24 hour exposure periods. Fluorescent imaging was then conducted using a ZEISS SteREO Discovery V8 microscope with Zeiss filters set 09 (band pass = 450–490 nm, long pass = 520 nm). The images were processed with ImageJ software for merging and relative fluorescence intensities. Briefly, fluorescence (green channel) from each sample was extracted, and the intensity was quantified. The obtained values were normalized by dividing them by the average value of the control group, allowing for the comparison of relative fluorescence differences.
2.7. Fluorescence imaging of zebrafish larvae
Zebrafish (ZF) wildtype AB embryos were obtained from the Helmholtz Institute for Pharmaceutical Research Saarland. The maintenance and experiments with ZF larvae (ZFL) were conducted according to the guidelines of the German Animal Welfare Act (§11 Abs. 1 TierSchG) and EU Directive 2010/63/EU.33,34 The received embryos were hatched in 0.3 × Danieau's medium, consisting of 17 mM NaCl, 2 mM KCl, 0.12 mM MgSO4, 1.8 mM Ca(NO3)2, 1.5 mM HEPES, and 1.2 μM methylene blue at 28 °C. Following the hatching of the organisms, ZFL was transferred to E3 egg water (E3), composed of 5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, and 0.33 mM MgSO4, and the resulting solution was incubated at 28 °C.ZFL at 96 hours post-fertilization (hpf) was utilized, aligning with the EU Directive 2010/63/EU, which does not consider embryos within the first 120 hpf as subjects of animal experimentation. The experiment was organized into three distinct groups: a control group with no treatment, a group treated solely with LBM, and a dual treatment of LBM and 5 mM H2O2. For the LBM-only group, the larvae were exposed to a 15 μM LBM solution in an E3 medium for 30 minutes. In contrast, the dual-treatment group first was exposed to 5 mM H2O2 for one hour, followed by a transfer to the 15.0 μM LBM solution in E3 medium for an additional 30 minutes.
For the LBM-only group, the larvae were exposed to a 15 μM LBM solution in an E3 medium for 30 minutes. In contrast, the dual-treatment group was first exposed to 5 mM H2O2 for one hour, followed by a transfer to the 15 μM LBM solution in E3 medium for an additional 30 minutes. The prepared zebrafish larvae were immobilized using tricaine (ethyl 3-amino-benzoate methanesulfonate salt) as an anesthetic. Then, for lateral observation under the microscope, the larvae were transferred to a 6% methylcellulose solution for fixation.35,36 The imaging process, consistent with the D. magna study, was performed using a ZEISS SteREO Discovery V8 microscope with Zeiss filter set 09 (band pass = 450–490 nm, long pass = 520 nm). The acquired images were then processed using ImageJ software, focusing on merging and analyzing the relative fluorescence intensities. Briefly, fluorescence (green channel) from each sample was extracted, and the intensity was quantified. The obtained values were normalized by dividing by the average value of the control group, allowing for the comparison of relative fluorescence differences.
3. Results and discussion
3.1. LBM probe design and synthesis
Our laboratory prepares chemosensors using standard synthetic organic chemistry methods, typically where starting materials are first involved in amounts on the gram scale and using many common separation, purification, and characterization techniques to help confirm the successful preparation of the intermediates and product molecules. The synthesis of LBM was achieved with satisfactory yields through a straightforward single-step nucleophilic substitution reaction (illustrated in Scheme 1), wherein LOH in DMF underwent a reaction with 4-bromomethylphenylboronic acid pinacol ester in the presence of triethylamine. The characterization of LBM was conducted using multinuclear NMR spectroscopy and mass spectral analysis (Fig. S1–S4†).Mycophenolic acid (MPA) and its analogs, along with related precursors, are currently under extensive investigation due to their significant medicinal and biological relevance. We have reported on these molecules in recent years.37–39 These compounds are applied in various research domains, thanks to their water solubility and compatibility for experiments in living cells. Despite dissimilarities, certain properties of MPA also apply to LOH and LBM. The core of LBM is extremely biocompatible, owing largely we believe to its similarity to MPA, a natural product that was first studied as an antibiotic and now as an immune suppressant (organ recipient antirejection medication). We thus were able to employ a 100% PBS buffer (10.0 mM, pH 7.4) without the use of cosolvent. The 4-bromomethylphenylboronic acid pinacol ester group was introduced at the fluorophore (LOH), although it did not appear to significantly impact the solubility of the derivative and allowed for quenching of the fluorescence of the fluorophore through a proposed intramolecular excited state intramolecular proton transfer (ESIPT); a “turn-on” fluorescence is enabled after the addition of H2O2 (Scheme S1†). Unlike our previous reports,37,40 this molecule bears, instead of an N–Me group, a full molecular pendant that includes a morpholino group [–CH2CH2N(CH2CH2)2O] as a suitable lysosome targeting unit.
3.2. Optical properties of LBM
After isolation and characterization, we analyzed the photophysical properties of LBM. The UV–vis absorption spectra were acquired and revealed a peak absorption at 354 nm. Notably, upon the introduction of 10 equiv. of H2O2 to LBM, a distinct and remarkable spectral red-shift occurred, centered at 417 nm. In contrast, the addition of 10 equiv. of other ROS does not yield any notable alteration in the absorption spectrum of LBM, as depicted in Fig. S5a.†To help further evaluate the fluorescence characteristics of LBM (15.0 μM) using the updated excitation spectra (417 nm), we performed a fluorescence emission selectivity analysis under physiological conditions relevant to human body physiology. This involved the usage of a 10.0 mM PBS solution at pH 7.4. When excited at 417 nm, an emission curve with nearly constant intensity is observed at 517 nm in the absence of H2O2. Remarkably, the addition of H2O2 induces a distinct “turn-on” fluorescence response in LBM. No detectable response is derived from different ROS within the examined time frame. However, upon introducing H2O2 to LBM in solution, a noticeable increase in the emission maximum at 517 nm is observed. The intensity increased by 13.4 times compared to the responses generated by the absence of H2O2 (Fig. 1a and b, Fig. S5b†). To validate the fluorescence enhancement attributed to H2O2, supplementary H2O2 is introduced into the samples. The addition of H2O2 results in increasing fluorescence intensity, indicating the probe's interaction with H2O2 (Fig. 1c) and the establishment of a “turn-on” mechanism (Fig. S9–S12†). To help confirm the mechanism of H2O2 detection with LBM, the reaction mixture of LBM with 5.0 equivalents of H2O2 was analyzed by high resolution mass spectrometry (HRMS). The obtained HRMS mass value matches the calculated value determined for LOH (calc.: 369.04, obtained: 369.0447, Fig. S9 and S10†). The 1H NMR spectrum study also helps explain the formation of LOH from the reaction between LBM and H2O2 (Fig. S12†). Upon reaction with H2O2, the doublets at δ 7.83 ppm and δ 7.46 ppm corresponding to two protons each at o- add m-positions to the boronate moiety, and a singlet corresponding to the two benzylic protons at δ 5.32 ppm, disappear. The comparison of the 1H NMR spectra obtained for LBM + H2O2 reaction and the LOH appear identical indicating the successful formation of the latter upon reaction of LBM with H2O2. This observation underscores the mechanism for the detection of H2O2 using LBM.
 | ||
| Fig. 1 (a) Fluorescence emission spectra of LBM in the presence of H2O2 (10.0 eq.) or in the absence of H2O2 in PBS (pH 7.4) (b) bar graph depiction of fluorescence emission of LBM (15.0 μM) with H2O2 (other ROS) (10.0 eq.) in solution (10.0 mM, PBS buffer solution, pH 7.4) (c) bar graph depiction of fluorescence emission of LBM (15.0 μM) with other ROS (10.0 eq.) + H2O2 (10.0 eq.) in solution (10.0 mM, PBS pH 7.4) (d) titrimetric fluorescence study reflecting the emission of the product from LBM (15.0 μM) with various concentrations of H2O2 (0–5.0 equiv.) in PBS solution (pH 7.4) (e) time-dependent emission spectra of LBM (15.0 μM) in solution (10.0 mM PBS, pH 7.4) with H2O2 (f) changes of the emission spectrum of LBM (15.0 μM) with H2O2 (10.0 eq.) under various pH values in solution (10.0 mM PBS buffer solution, pH 3–11); λex = 417 nm, λem = 517 nm; slit width = 5.0 nm/5.0 nm at RT. All spectra were recorded after 45 min. Error bars represent mean value ± SD (n = 3) (SD = standard deviation). | ||
To assess the sensitivity of LBM, a titration experiment was conducted by subjecting the probe to varying concentrations of H2O2 (ranging from 0.0 to 5.0 equiv.). Fluorometric titration results show emission intensity gradually increased upon the addition of H2O2 from 0 to 5.0 equiv. and was found to be linearly proportional to the concentration of H2O2 (Fig. 1d). Further, to determine the limit of detection, a linearity curve was plotted for H2O2 addition from 0.0 to 5.0 equiv. The limit of detection for LBM (3σ/k, in which σ stands for the standard deviation of 10 blank sample measurements and where k is the slope of the linear equation) was determined to be 13 nM (Fig. S13, Table S1†).
3.3. Time dependent detection studies
The changes in probe concentration over time, as well as a large fluorescence signal, are essential requirements for the proper function of a rapidly responsive sensor. A time-dependent investigation of LBM was conducted simultaneously with the addition of 10.0 equiv. of H2O2. Upon the addition of H2O2, LBM promptly exhibits a “turn-on” fluorescence change in the reaction mixture, with its signal maintaining stability for at least 120 minutes (Fig. 1e). These findings demonstarte a potential utility of LBM to be implemented as a real-time sensing agent within living cells.3.4. Studies of the effect that pH has on probing
For the probe to handle the analyte levels present in the cellular environment, LBM was investigated for its stability (or inertness) in a slightly acidic environment without undergoing decomposition (as determined by loss of the fluorescence signal intensity), including the occurrence of photobleaching. Hence, we tested the response of LBM towards H2O2 at various pH levels (Fig. 1f). The results reveal that the pH value of the solution influences the fluorescence response to H2O2. As shown in Fig. 1f, the fluorescence intensity at 517 nm of the LBM response to H2O2 gradually was enhanced when the pH increased in the range of 5.0 to 6.0; it reached the maximum intensity near the value of 7.4. However, in the pH range of 3.0 to 5.0, LBM did not give any response due to the pH effect on LBM, thus inhibiting its ability to carry out H2O2 detection in this regime. The trend of fluorescence intensity alterations with LBM at the given fluorescence maximum of 517 nm demonstrates that the probe can indeed tolerate a slightly acidic environment.3.5. Cell imaging studies
We investigated the potential of LBM in real biological systems by assessing its cytotoxicity using live normal lung cells (MRC-5) and lung cancer cells (A549) by way of running MTT assays. Remarkably, when exposed to LBM concentrations of up to 5–25 μM for 24 h, the cell viability in MRC-5 and A549 cells was found to be over 90% (Fig. 2a). The cell viability for both cell lines was over 85%, even at a very high concentration of 50 μM, indicating that the LBM shows very low cytotoxicity for cancer and normal cells. Thus, the outcome of the cytotoxicity assay affirms the favorable biocompatibility profile of LBM and assures its high applicability for imaging the intracellular H2O2. Subsequently, confocal fluorescence analyses were performed to validate the capability of LBM for detecting H2O2 in lysosomes using the lung cancer cells (A549). The cells treated with Lyso-tracker dye (red fluorescence signals) and LBM showed a very low intensity green signal, indicating the presence of H2O2 (Fig. 2b and c). However, upon induction of endogenous H2O2 production by treatment with PMA, the green fluorescence signals were significantly increased with a high co-localization of Lyso-tracker dye and LBM in the lysosomes indicated by the distinct orange color (bright field merge image). The co-localization (Pearson's Coefficient, r2 = 0.934) and overlap (r2 = 0.949) coefficients for Lyso-tracker dye and LBM indicate that the LBM effectively detects the H2O2 in the lysosomes (Fig. S14a†). Notably, PMA induces ROS production in cells, subsequently leading to the enhanced production of endogenous H2O2. To validate the detection of endogenous H2O2 using LBM, we treated the cells with NAC (N-acetylcysteine), a known suppressor of endogenous H2O2 production, before treating them with PMA (phorbol 12-myristate 13-acetate). As a result of the addition of NAC, the green fluorescence signals were significantly decreased (Fig. 2b and c). These findings indicate that LBM effectively detects endogenous H2O2 in living cells. Furthermore, to establish that LBM can exclusively discriminate H2O2 from other ROS, we treated the cells with H2O2 or NaOCl, followed by staining and imaging with the Lyso-tracker dye and LBM. The cells treated with H2O2 at a concentration of 100 μM demonstrated a further increase in the detected fluorescence signals in these samples. The values found for co-localization (Pearson's Coefficient, r2 = 0.943) and overlap (r2 = 0.941) coefficients for Lyso-tracker dye and LBM indicate that the LBM effectively detects the H2O2 in the lysosomes (Fig. S14b†). These results establish that LBM allows for the detection of both endogenous and exogenous H2O2. In contrast, when cells were exposed to NaOCl as the lone analyte, fluorescence intensity showed no notable increase. Consequently, these findings reaffirm that LBM specifically interacts with intracellular H2O2 among various ROS and exhibits a high degree of selectivity towards H2O2.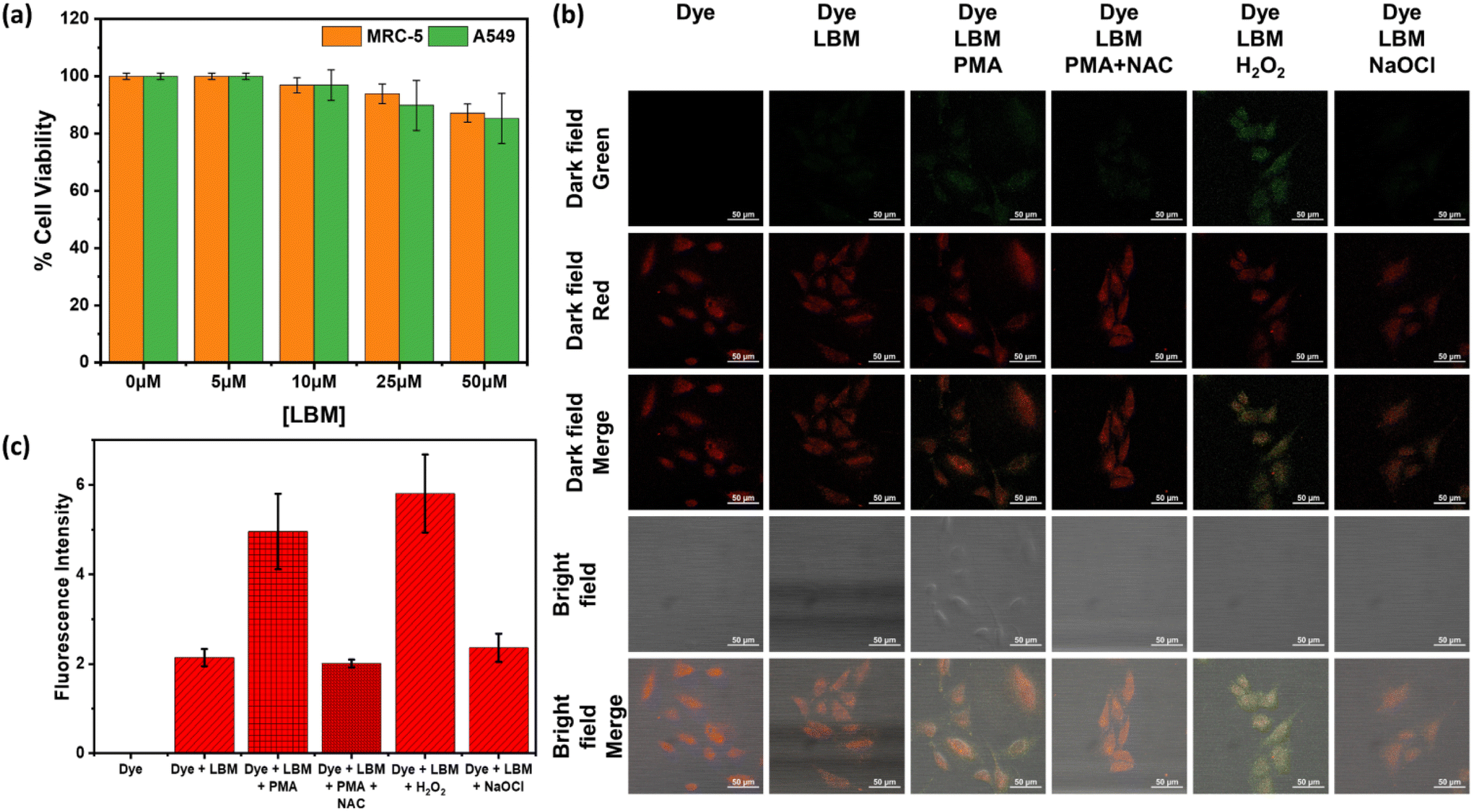 | ||
| Fig. 2 Results with LBM support that it allows the detection of intracellular H2O2. (a) Bar graphs representing the results of MTT assay upon 24 h treatment of MRC-5 and A549 cells with LBM (0, 5.0, 10.0, 25.0, 50.0 μM), (b) confocal microscopic images upon treatment of A549 cells with “Dye” (2.0 μM), “Dye” (2.0 μM) + LBM (15.0 μM), “Dye” + LBM + PMA (1.0 μg mL−1), “Dye” + LBM + PMA (1.0 μg mL−1) + NAC (1.0 mM), “Dye” + LBM + H2O2 (100.0 μM), “Dye” + LBM + NaOCl (100.0 μM), (c) fluorescence signal intensity of green (LBM) and red (Lyso-tracker “Dye”) channels measured for the dark field images. (scale bar: 50 μm) (LBM, λex: 410 nm, λem: 535 nm; Lyso-tracker dye, λex: 555 nm, λem: 592 nm). | ||
We explored the suitability of LBM for bioimaging H2O2 in A549 cells and found that LBM can easily cross both the cell and lysosomal membranes. LBM exhibited notable sensitivity and specificity in detecting H2O2, engaging with exogenous and endogenous H2O2 within the cell. The enhanced fluorescence signal of LBM upon interaction with endogenously produced H2O2 underscores its efficacy in facilitating the bioimaging of H2O2. Treatment of cells with PMA and NAC demonstrated the effectiveness of LBM in detecting endogenous H2O2. The presence of a morpholine group on LBM allows it to target the lysosomes explicitly. The preliminary investigation presented here establishes the potential of LBM in detecting H2O2 in the lysosome.
3.6. In vivo visualization and detection of H2O2 in D. magna
D. magna as a test organism in the biological sciences has many advantages as a visualization and test subject such as a short lifespan for monitoring and transparency. Herein, we have selected D. magna as the animal in vivo model for the visualization and validation study. In this test, newborn neonates (<24 hours old) from the third and fourth brood were utilized due to their relatively stable conditions compared to the first and second brood. Preliminary tests were conducted with silver ion concentrations ranging from 1.0 to 10.0 ppb over 48 hours. The 48 h EC50 of Ag ions in D. magna was found to be 4.15 ppb (Fig. S15†). Consequently, a concentration of 3.0 ppb below EC5 was selected for the experiment to avoid immobilization and induce oxidative stress. According to previous research,41 Ag+ is known to act as a xenobiotic within D. magna, increasing ROS levels.This study was thus designed to increase ROS within D. magna using silver ions and to verify their detection using the LBM probe. As illustrated in Fig. 3, after a 3 hour incubation of D. magna with a concentration of 3.0 ppb of Ag+ ions, the gastrointestinal tract and carapace of D. magna exhibit a discernibly augmented green fluorescent signal attributed to the presence of the probe reacting with the intrinsic hydrogen peroxide (H2O2) present (carapace = “shield extending from the head region and enveloping a smaller or larger part of the body” internet source, academic.oup.com (https://academic.oup.com/book/4028/chapter/145671887)). Furthermore, following a 24 hour exposure, the aforementioned anatomical structures of D. magna manifest a pronounced intensified green fluorescent signal (Fig. 3). In the 24 hour old specimens, particularly within the gastrointestinal tract, midgut and muscles of antennae exhibit strong fluorescence, where an increase in mitochondria activity and consequent ROS production.42 This observation indicates an increase in ROS production in areas dense with mitochondria, as well as a reaction with the LBM probe. These findings elucidate the predominant distribution of the probe within the gut, esophagus, and muscle tissues (antennae), affirming its interaction with endogenous H2O2. Additionally, to understand the relative enhancement in fluorescence, the fluorescence intensities were quantified and normalized against the control group for each set. With only LBM treatment, an increase of approximately 1.7-fold (3 h) and 2.5-fold (24 h) was observed. However, the ANOVA revealed no differences in a comparison test. On the other hand, the ROS-induced groups showed statistically significant differences (p < 0.01 for 3 h and p < 0.001 for 24 h) compared to other groups at both time points. These results suggest that LBM has the potential to detect endogenous induced-ROS within aquatic organisms.
 | ||
| Fig. 3 (a) Representative fluorescent images of D. magna; control series of (a) 3 h and (d) 24 h, incubation with only 15.0 μM LBM for 30 min at (b) 3 h and (e) 24 h and exposed to 3 ppb Ag+ for (c) 3 h and (f) 24 h, followed by incubation with 15.0 μM LBM for 30 min. Band pass = 450–490 nm, long pass = 520 nm. Scale bar = 100 μm. (g) Relative intensity of fluorescence normalized by control series (n = 3). Statistical differences were analyzed by one-way analysis of variance (ANOVA) and Bonferroni multiple comparison tests. Error bars indicate standard deviations of the values; *, ** and *** indicate p < 0.05, <0.05, and <0.001, respectively. | ||
3.7. In vivo visualization and detection of H2O2 in zebrafish larvae
In further in vivo visualization experiments utilizing the zebrafish larvae (ZFL) model (Fig. 4), ZFL at 96 hpf was exposed to 5 mM H2O2, aiming to induce endogenous ROS levels, aligning with methodologies from previous studies.43,44 In the ZFL group treated solely with LBM, detectable fluorescence was observed in the stomach and around the eyes. This suggests that LBM was absorbed into an organism, potentially due to naturally occurring endogenous ROS. In contrast, autofluorescence was observed around the yolk sac in the control group, exhibiting a broader and more diffuse signal compared to the distinct fluorescence in the LBM-treated group. These observations suggest effective absorption of the LBM probe in ZFL organisms. Remarkably, in dual-treated ZFL, a significant increase in fluorescence was observed from the eyes to the yolk sac and intestines. Additionally, quantitative analyses of average fluorescence intensities in each experimental group were conducted using the same methods as those in the experiments for daphnids. In the ZFL model, an approximately 1.7-fold increase in fluorescence was observed between the control and only the LBM treatment group. However, ANOVA testing revealed no significant differences in the comparison test. On the contrary, the dual-treatment group showed statistically significant differences when compared to the LBM-only treated group (p < 0.05) and the control group (p < 0.01). Combined with the results of D. magna, LBM is a promising tool for detecting ROS within aquatic organisms. | ||
| Fig. 4 Representative fluorescent images of 96 hpf zebrafish larvae; (a) control series, (b) incubation with only 15.0 μM LBM for 30 min, and (c) exposure to 5 mM H2O2 for 1 h, followed by incubation with 15.0 μM LBM for 30 min. Band pass = 450–490 nm, long pass = 520 nm. Scale bar = 250 μm. (d) Relative intensity of fluorescence normalized by control series (n = 5). Statistical differences were analyzed by one-way analysis of variance (ANOVA) and Bonferroni multiple comparison tests. Error bars indicate standard deviations of the values; * and ** indicate p < 0.05 and <0.01, respectively. | ||
4. Conclusions
A fluorescent probe, LBM, was synthesized based on a mycophenolic acid core, featuring a detachable aryl–boronic acid group aimed at enhancing its sensitivity and specificity towards hydrogen peroxide. LBM allows for the detection of both endogenous and exogenous H2O2. Compared to closely related probes, LBM features (i) lower cytotoxicity (cell viability is higher at higher concentrations), (ii) selective detection in organelles (lysosome tracker checking), (iii) a lower limit of detection (LOD), and (iv) more specific bio-testing (both Daphnia magna and Zebrafish).21Furthermore, experimental findings confirm the probe's (LBM's) effective detection of H2O2 with high selectivity. As a result, this novel fluorescent probe presents a promising avenue for monitoring H2O2, and in so doing, helps begin identifying a range of disorders caused by excessive H2O2 concentration. LBM demonstrated a distinct absorption peak at 417 nm, exclusively attributable to LOH. The reaction with H2O2 with LBM involves the analyte attacking the boronic acid center and allowing for the loss of the phenol protecting group. After H2O2 has worked its effect, LOH floats freely and its ESIPT spectroscopic signature is picked up by the spectrofluorometer. H2O2 is reactive as opposed to other reactive oxygen species (ROS) tested under these conditions. (Caution: sometimes peroxynitrite is picked up by boronate probes.45) Following its reaction with H2O2, LBM exhibited strong fluorescence emission at 517 nm, manifesting a substantial Stokes’ shift of 110 nm. Additionally, the limit of detection for LBM was quantified at 13 nM.
According to our findings from cell viability assessments and confocal fluorescence microscopy investigations, LBM displayed exceptional biocompatibility. Notably, LBM exhibited the capacity for selective H2O2 detection in preference to NaOCl. Furthermore, LBM proved to be a valuable tool for monitoring H2O2 levels within the A549 cell line. Consequently, LBM holds significant promise for the specific visualization of endogenous H2O2 and the real-time monitoring of intracellular redox state in living cells.
The application of LBM proved efficacious in elucidating alterations in H2O2 levels within the D. magna and zebrafish larvae models. In both trusted and well-understood animal models, distinct and strong fluorescence was observed in major organelles due to LBM when ROS was induced. Utilizing two crucial models in a freshwater environment, the LBM probe has demonstrated its potential even under aquatic exposure conditions. The entirety of the findings substantiates the potential of LBM as an innovative platform for investigating the chemical–biological underpinnings of H2O2 and its associations within model systems and thus complex biosystems, including possibly disease “models” of relevance.
Author contributions
Jongkeol An: Conceptualization, methodology, chemical and photophysical characterization, manuscript writing; Sujeong Park: Cell imaging studies, manuscript writing; Neha Jain: Formal analysis; Youngsam Kim: Zebrafish studies, manuscript writing; Satish Balasaheb Nimse, David G. Churchill: Project supervision; specifically, critical review, commentary or revision.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Molecular Logic Gate Laboratory operated by Prof. David G. Churchill acknowledges support from KAIST (N11240029) and the National Research Foundation of Korea (NRF-2021R1F1A104657613). The research support staff at KARA (KAIST Analysis Center for Research Advancement) facilitated the acquisition of MS data. Prof. S. B. Nimse was supported by the National Research Foundation of Korea (NRF-2021R1C1C1008480). Prof. Y. Kim was supported by the Nanomaterial Technology Development Program (NRF-2017M3A7B6052455) funded by the South Korean Ministry of Science.References
- E. C. Cheung and K. H. Vousden, Nat. Rev. Cancer, 2022, 22, 280–297 CrossRef CAS.
- E. W. Miller, O. Tulyathan, E. Y. Isacoff and C. J. Chang, Nat. Chem. Biol., 2007, 3, 263–267 CrossRef CAS PubMed.
- H. Sies, J. Biol. Chem., 2014, 289, 8735–8741 CrossRef CAS PubMed.
- M. López-Lázaro, Cancer Lett., 2007, 252, 1–8 CrossRef.
- P. Kirkham and I. Rahman, Pharmacol. Ther., 2006, 111, 476–494 CrossRef PubMed.
- E. Schröder and P. Eaton, Curr. Opin. Pharmacol., 2008, 8, 153–159 CrossRef.
- C. H.-L. Hung, S. S.-Y. Cheng, Y.-T. Cheung, S. Wuwongse, N. Q. Zhang, Y.-S. Ho, S. M.-Y. Lee and R. C.-C. Chang, Redox Biol., 2018, 14, 7–19 CrossRef CAS PubMed.
- K. B. R. Teodoro, F. L. Migliorini, W. A. Christinelli and D. S. Correa, Carbohydr. Polym., 2019, 212, 235–241 CrossRef CAS PubMed.
- F. Xie, X. Cao, F. Qu, A. M. Asiri and X. Sun, Sens. Actuators, B, 2018, 255, 1254–1261 CrossRef CAS.
- R. F. P. Nogueira, M. C. Oliveira and W. C. Paterlini, Talanta, 2005, 66, 86–91 CrossRef CAS PubMed.
- D. Kim, G. Kim, S.-J. Nam, J. Yin and J. Yoon, Sci. Rep., 2015, 5, 8488 CrossRef CAS PubMed.
- J. Liu, S. Zhou, J. Ren, C. Wu and Y. Zhao, Analyst, 2017, 142, 4522–4528 RSC.
- G. Feng, P. Zhai, Z. Li, M. Fan, Y. Jiang, N. Qiao, R. Chen, S. Tang, Z. Xu, X. Wang, G. Lin, C. Yang, M. Ying, B. Dong, Y. Shao and G. Xu, Bioorg. Chem., 2023, 130, 106199 CrossRef CAS PubMed.
- Y. Liu, C. Jiao, W. Lu, P. Zhang and Y. Wang, RSC Adv., 2019, 9, 18027–18041 RSC.
- L.-C. Lo and C.-Y. Chu, Chem. Commun., 2003, 2728–2729, 10.1039/B309393J.
- P. M. Sonawane, T. Yudhistira, M. B. Halle, A. Roychaudhury, Y. Kim, S. S. Surwase, V. K. Bhosale, J. Kim, H.-S. Park, Y.-C. Kim, C.-H. Kim and D. G. Churchill, Dyes Pigm., 2021, 191, 109371 CrossRef CAS.
- W.-L. Cui, M.-H. Wang, Y.-H. Yang, J.-Y. Wang, X. Zhu, H. Zhang and X. Ji, Coord. Chem. Rev., 2023, 474, 214848 CrossRef CAS.
- Y. Cheng, J. Dai, C. Sun, R. Liu, T. Zhai, X. Lou and F. Xia, Angew. Chem., Int. Ed., 2018, 57, 3123–3127 CrossRef CAS PubMed.
- Q. Wu, Y. Li, Y. Li, D. Wang and B. Z. Tang, Mater. Chem. Front., 2021, 5, 3489–3496 RSC.
- W.-X. Wang, W.-L. Jiang, G.-J. Mao, M. Tan, J. Fei, Y. Li and C.-Y. Li, Anal. Chem., 2021, 93, 3301–3307 CrossRef CAS.
- Y. Wu, Z. Li and Y. Shen, ACS Omega, 2019, 4, 16242–16246 CrossRef CAS.
- Y.-X. Liao, K. Li, M.-Y. Wu, T. Wu and X.-Q. Yu, Org. Biomol. Chem., 2014, 12, 3004–3008 RSC.
- R. Mamgain and F. V. Singh, ACS Org. Inorg. Au, 2022, 2, 262–288 CrossRef CAS PubMed.
- A. G. Griesbeck, B. Öngel and M. Atar, J. Phys. Org. Chem., 2017, 30, e3741 CrossRef.
- M. Świerczyńska, D. Słowiński, R. Michalski, J. Romański and R. Podsiadły, Spectrochim. Acta, Part A, 2023, 289, 122193 CrossRef PubMed.
- S. B. Lovern and R. Klaper, Environ. Toxicol. Chem., 2006, 25, 1132–1137 CrossRef CAS PubMed.
- Z. Zhang, J. Fan, Y. Zhao, Y. Kang, J. Du and X. Peng, ACS Sens., 2018, 3, 735–741 CrossRef CAS PubMed.
- T. Gomes, Y. Song, D. A. Brede, L. Xie, K. B. Gutzkow, B. Salbu and K. E. Tollefsen, Sci. Total Environ., 2018, 628–629, 206–216 CrossRef CAS PubMed.
- H. Franquet-Griell, C. Gómez-Canela, F. Ventura and S. Lacorte, Environ. Res., 2015, 138, 161–172 CrossRef CAS.
- T.-Y. Choi, T.-I. Choi, Y.-R. Lee, S.-K. Choe and C.-H. Kim, Exp. Mol. Med., 2021, 53, 310–317 CrossRef CAS PubMed.
- M. B. Halle, T. Yudhistira, K. J. Lee, J. H. Choi, Y. Kim, H.-S. Park and D. G. Churchill, ACS Omega, 2018, 3, 13474–13483 CrossRef CAS.
- OECD, Test No. 202: Daphnia sp. Acute Immobilisation Test, 2004 Search PubMed.
- U. Strähle, S. Scholz, R. Geisler, P. Greiner, H. Hollert, S. Rastegar, A. Schumacher, I. Selderslaghs, C. Weiss, H. Witters and T. Braunbeck, Reprod. Toxicol., 2012, 33, 128–132 CrossRef.
- M. Westerfield, The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio Rerio), University of Oregon Press, 2000 Search PubMed.
- G. Lanzarin, C. Venâncio, L. M. Félix and S. Monteiro, Journal, 2021, 9, 1784 CAS.
- Y. Sim, H.-J. Cho, J.-S. Lee, W. S. Lee, H. Kim and J. Jeong, Chemosphere, 2023, 330, 138723 CrossRef CAS.
- P. M. Sonawane, N. Jain, A. Roychaudhury, S. J. Park, V. K. Bhosale, M. B. Halle, C.-H. Kim, S. B. Nimse and D. G. Churchill, Analyst, 2023, 148, 5203–5209 RSC.
- N. Jain, P. M. Sonawane, H. Liu, A. Roychaudhury, Y. Lee, J. An, D. Kim, D. Kim, Y. Kim, Y.-C. Kim, K.-B. Cho, H.-S. Park, C.-H. Kim and D. G. Churchill, Analyst, 2023, 148, 2609–2615 RSC.
- M. B. Halle, T. Yudhistira, W.-H. Lee, S. V. Mulay and D. G. Churchill, Org. Lett., 2018, 20, 3557–3561 CrossRef CAS PubMed.
- W. Lee, T. Yudhistira, W. Youn, S. Han, M. B. Halle, J. H. Choi, Y. Kim, I. S. Choi and D. G. Churchill, Analyst, 2021, 146, 2212–2220 RSC.
- R. Miller, Y. Kim, C. G. Park, C. Torres, B. Kim, J. Lee, D. Flaherty, H.-S. Han, Y. J. Kim and H. Kong, ACS Appl. Mater. Interfaces, 2022, 14, 39759–39774 CrossRef CAS.
- B. Mittmann, P. Ungerer, M. Klann, A. Stollewerk and C. Wolff, EvoDevo, 2014, 5, 12 CrossRef.
- L. Wang, X. Fu, J. Hyun, J. Xu, X. Gao and Y.-J. Jeon, Polymers, 2023, 15, 1612 CrossRef CAS.
- P. Niu, J. Liu, F. Xu, L. Yang, Y. Li, A. Sun, L. Wei, X. Liu and X. Song, ACS Appl. Bio Mater., 2022, 5, 1683–1691 CrossRef CAS.
- J. Zielonka, A. Sikora, R. Podsiadly, M. Hardy and B. Kalyanaraman, in Mitochondrial Medicine: Volume 1: Targeting Mitochondria, ed. V. Weissig and M. Edeas, Springer US, New York, NY, 2021, pp. 315–327. DOI:10.1007/978-1-0716-1262-0_20.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an00742e |
| This journal is © The Royal Society of Chemistry 2024 |