
A method for the quantitative analysis of polar anionic pesticides in milk/infant formula, cereals and fruit and vegetables using ion chromatography coupled to tandem mass spectrometry†
Jim
Garvey
 *,
Anatte
Margalit
,
Michael
Kelly
,
Eoin
Geoghegan
and
Stephen
Burrell
*,
Anatte
Margalit
,
Michael
Kelly
,
Eoin
Geoghegan
and
Stephen
Burrell
The Food Chemistry Laboratories, The Department of Agriculture, Food and the Marine, Celbridge, Co. Kildare, Ireland. E-mail: Jim.Garvey@agriculture.gov.ie
First published on 23rd May 2024
Abstract
Polar pesticides such as anionic or ionisable compounds have always provided a challenge for analytical chemists. Methods of analysis have been developed using a range of techniques including normal phase chromatography, ion-pairing, derivatisation and HILIC or multi-mode chromatography. These work well with some of these compounds but, except for HILIC, all of them have their limitations and none of them cover the range required by legislation. Some of these compounds, glyphosate, chlorate and phosphonic acid, are found regularly in a range of food matrices, and therefore reliable methods of analysis are essential. This study describes an ion chromatography method with tandem mass spectrometry detection which not only covers the full range of compounds required by legislation but also can be expanded to include other anionic or ionisable pesticides and metabolites. These include glyphosate and its metabolites, glufosinate and its metabolites, ethephon and its metabolites as well as fosetyl aluminium, chlorate and perchlorate. The method is fully validated according to the performance criteria from the SANTE guidelines for the analysis of pesticides in food and feed over a wide range of matrices, including milk, infant formula, cereals and fruits and vegetables. Over 300 food samples have analysed as part of our routine monitoring program.
1. Introduction
In general, methods for the analysis of pesticide residues in food commodities tend to be multi-residue methods because they are the most efficient use of resources. They also allow for the analysis of a large number of pesticides in one analytical run. These methods usually involve a combination of Gas chromatography (GC) and liquid chromatography (LC) or ultra-high performance liquid chromatography (UHPLC) combined with mass spectrometry (MS) in tandem mode. They can cover more than 90% of the scope required for pesticide residue analysis. There are a small number of compounds which do not fit well into multi-residue methods either because they do not extract under the method conditions, or they are not amenable to reverse phase chromatography normally used. These pesticide residues require specific methods for their accurate identification and quantification. One such group of pesticides is the highly polar pesticides which exist as anions or can be ionised to give anions.There are a number of important compounds in this group of highly polar anionic pesticides. The most well-known of these is glyphosate which has become the most used herbicide in the world due to its effectiveness in clearing vegetation to allow for planting of a new crop.1 It is also the most well-known among the group due to the controversy over its toxicity.2–4 A recent review of this toxicity by the European Food Safety Authority (EFSA) suggests that there are no areas of concern for this pesticide allowing its continued use within the European Union.5 There are other important pesticides in this group such as the herbicides glufosinate and fosetyl aluminium, which are used on a wide range of leafy vegetables and brassicas and the growth regulator ethephon, which is used on a range of cereals and soft fruit. Also included in the scope is the disinfectant chlorate and the food contaminant perchlorate. These pesticides have traditionally been considered difficult to analyse and single residue methods or methods for a small group of compounds have been applied to their analysis. Also included are a range of metabolites of glyphosate, glufosinate and ethephon. Currently these are not part of the residue definitions of these compounds for maximum residue level (MRL) assignment, but they are useful as confirmatory tools for inferring the use of these molecules. These molecules tend to be small, highly polar and in some cases acidic. They are difficult to extract and analyse by more conventional methods such as normal-phase or reverse phase chromatography where many of the compounds are not retained on the column.6
Methods used for their analysis include activated carbon black columns, ion-pairing, derivatisation or HILIC methods.7,8 All of these methods have major disadvantages: the use of activated carbon columns requires a great deal of deactivation of active sites and can only handle a relatively small number of samples before the deactivation process needs to be repeated. Derivatisation works well with glyphosate and AMPA but is untested over the scope we require.9–11 The use of ion-pairing reagents has been used successfully for the analysis of fosetyl aluminium which allowed this pesticide to be retained on a C18 reversed phase column.12 The most successful of these methods so far has been HILIC but again these methods require frequent passivation, the use of a chelating agent to remove metals and the columns require significant conditioning. HILIC methods are also prone to very pronounced matrix effects.13–16 and chlorate and perchlorate do not work well with this method. Ion chromatography with various detectors has been used to determine active substances in formulated products.17,18 Rajski et al. developed an ion chromatography method for some of these pesticides and metabolites in fruit and vegetables using a high-resolution accurate mass system for detection.19 Because a lot of these compounds fragment to give common ions, PO3 (78.9) and PO2 (62.9) the use of a triple quadrupole mass spectrometry system seemed to us to be a more appropriate detection system and such a method has been used to analyse fruits and vegetables including grapes and cereals.20–25
For the work described here the extraction and clean-up for this method is based on a modification of the QuEChERS for polar pesticides method (QuPPE) developed by the European Union Reference Laboratory in Stuttgart and published on their website.9 Depending on the matrix the extraction is carried out with methanol or methanol–water, in some cases acidified. The samples are then cleaned-up by solid phase extraction (SPE) prior to analysis by ion chromatography with tandem MS detection. The use of ion chromatography has a number of advantages over all previous methods in that we use dedicated systems which are metal free. This means a chelating agent is not required and there is also no requirement to passivate the systems. The columns used are robust and require minimal conditioning and only need to be reactivated after thousands of injections. Matrix effects14–16 are still a major issue and need to be dealt with by one of a number of techniques available to us. Another problem reported with ion chromatography has been retention time drift which has been attributed to the matrix type.19,20 In the work described here the sensitivity of the mass spectrometer allows for dilution of the samples to levels where the matrix effects are greatly reduced, although we cannot eliminate them entirely. This also results in much improved retention time stability.
2. Experimental
2.1 Chemicals and reagents
Wheat and infant formula for this study were obtained from local producers. Organic tomatoes, oranges and carrots were obtained from local supermarkets. Only polypropylene vials and volumetric flasks were used for the preparation of standards and samples as some of these compounds are known to be adsorbed onto glass surfaces.9 High purity water with a resistivity of 18 MΩ was produced in the laboratory from deionised water using an Adrona water polisher. Methanol, formic acid and acetic acid (LC/MS grade) were purchased from Fisher Scientific, Ireland. Centrifuge tubes (50 mL) were purchased from Fisher Scientific, Ireland. Titan 3 0.2 μm pesticide membrane filters were purchased from Fisher Scientific, Ireland. Dionex OnGuard II RP ICC cartridges were purchased from ThermoFisher Scientific. Analytical standards were purchased from LGC, U.K. For all 16 analytes tested, pure standards were made up in either methanol–water (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) or pure water at a concentration of 300–600 mg L−1. A spiking mixture containing all 16 pesticides and metabolites at a final concentration of ∼3 mg L−1 was made up in water. This mixture was used to spike the blank samples for recovery studies and to make up a range of calibration standards at twice the required concentration. For recovery studies these standards were matrix matched with the commodity being studied to give an eight-point calibration curve between 0.1 and 250 μg L−1. The matrix was diluted to give the same final concentration in the standards and samples. During each run samples were bracketed by two sets of calibration standards and a calibration curve was constructed from both sets of standards.
:50) or pure water at a concentration of 300–600 mg L−1. A spiking mixture containing all 16 pesticides and metabolites at a final concentration of ∼3 mg L−1 was made up in water. This mixture was used to spike the blank samples for recovery studies and to make up a range of calibration standards at twice the required concentration. For recovery studies these standards were matrix matched with the commodity being studied to give an eight-point calibration curve between 0.1 and 250 μg L−1. The matrix was diluted to give the same final concentration in the standards and samples. During each run samples were bracketed by two sets of calibration standards and a calibration curve was constructed from both sets of standards.
2.2 Sample preparation and extraction
Wheat samples were milled prior to analysis. Infant formula samples were prepared by adding water according to the manufacturer's instructions. Fruit and vegetable samples were first chopped and then frozen at −80 °C. The frozen samples were then comminuted26,27 using a Stephan industrial blender. Samples were stored in a freezer at −20 °C prior to extraction. Validation was carried out according to the SANTE document28 “Guidance document on analytical quality control and method validation procedures for pesticides residues analysis in food and feed”.The variations in the extraction procedure for each matrix is summarised in Table S1.†
2.3 Ion chromatography method
The samples were analysed using a Dionex ICS 5000+ Ion Chromatograph with a dual pump system. One pump delivers high purity water to the eluent generator and the second pump delivers high purity water to the CTC regeneration cartridge. A dynamically regenerated suppressor is placed between the analytical column and the conductivity detector, and this removes potassium hydroxide from the mobile phase and replaced it with water to protect the ion source. A third, auxiliary pump, delivers acetonitrile post column and before the eluent gets to the mass spectrometer. The organic solvent helps with the desolvation of the sample in the ion source of the Mass Spectrometer. Rajski et al.19 have shown that acetonitrile is better than methanol for this purpose with an average increase in signal intensity of 300% with acetonitrile. The signal increase was more moderate with methanol with most compounds giving an improvement of 120–160%. Fosetyl was higher at 242%. Separation was achieved using a Thermo Scientific™ Dionex™ IonPac™ AS19-4 μm analytical column, 2 × 250 mm, held at 40 °C with elution of the polar anionic analytes using a potassium hydroxide gradient at a flow rate of 0.35 mL min−1. The gradient was optimised to give the best chromatographic separation of the analytes. The gradient is given in Fig. 1. The current of the in-line AERS suppressor was set to 52 mÅ.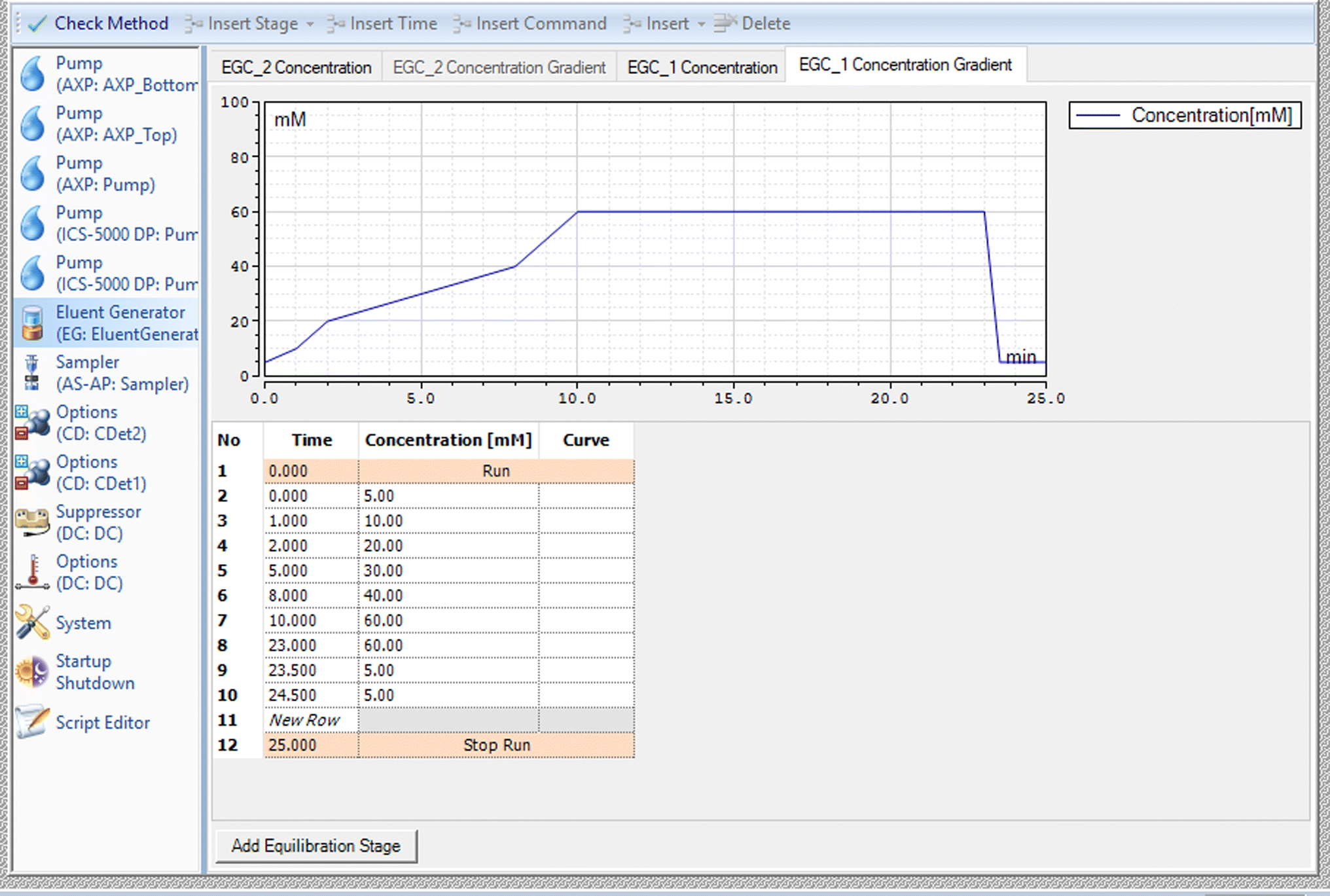 | ||
| Fig. 1 Summary of the Ionic Strength gradient used in the ion chromatography part of the method. | ||
2.4 Mass Spectrometry method
The Mass Spectrometer used for this method was the Thermofisher TSQ Altis+ and the ion source parameters are given in Table S2.† Each of the analytes in the method were optimised and the transitions used in the method are given in Table S3 and Fig. S1a† for wheat and Fig. S1b† for infant formula.The Mass Spectrometer was operated in selected reaction monitoring mode (SRM) and each compound was optimised individually. In most cases the most abundant transition was chosen for quantitation and at least two other transitions were used for confirmation of identity.
3. Results and discussion
3.1 Quantification
One transition was used for quantitation and further transitions were used for conformation of structure. An eight-point calibration curve was constructed using the two sets of bracketing standards included in each run. The standards were matrix matched so that the matrix concentration was the same as the samples when the appropriate dilution was taken into account. In the initial work a comparison was done between the quantifications with and without isotopically labelled internal standards (ILIS's). The results are given in Table 1 for some of the analytes. Based on of these results there was no advantage to using ILIS's so all subsequent work was done with external, matrix matched, standards.| No ILIS | ILIS | |
|---|---|---|
| Maleic hydrazide | 81.8 | 85.3 |
| Glufosinate | 120.3 | 70.7 |
| Glyphosate | 85.3 | 61.8 |
Very good accuracy (trueness) was obtained without the use of isotopically labelled internal standards and quantifications from this method also compared very well with other methods as evaluated by the performance in proficiency tests.
3.2 Linearity
Linearity was assessed using matrix matched external standards. A weighting of 1/x was used. The calibration range was between 0.5 μg L−1 (0.1 μg L−1 for cereals) and 250 μg L−1. Taking the dilution into account this is equivalent to 10 μg L−1 to 5 mg L−1 in the samples. The co-efficient of variation (R2) was used to evaluate the linearity with a value of 0.95 or better considered acceptable. Fig. 2 shows an example of transition data and a calibration curve for glyphosate in wheat at 10 μg kg−1. The data is summarised in Fig. 3, and from this figure it can be seen that the linearity criteria are met for all analytes, except for perchlorate in wheat The difference between the actual standard concentration and the back calculated standard concentrations are all less than 20% which means the linearity is acceptable – Table S4.† All the analytes gave acceptable linearity in all matrices except for perchlorate in wheat. Cyanuric acid was not evaluated for wheat and infant formula so no data is available for these matrices.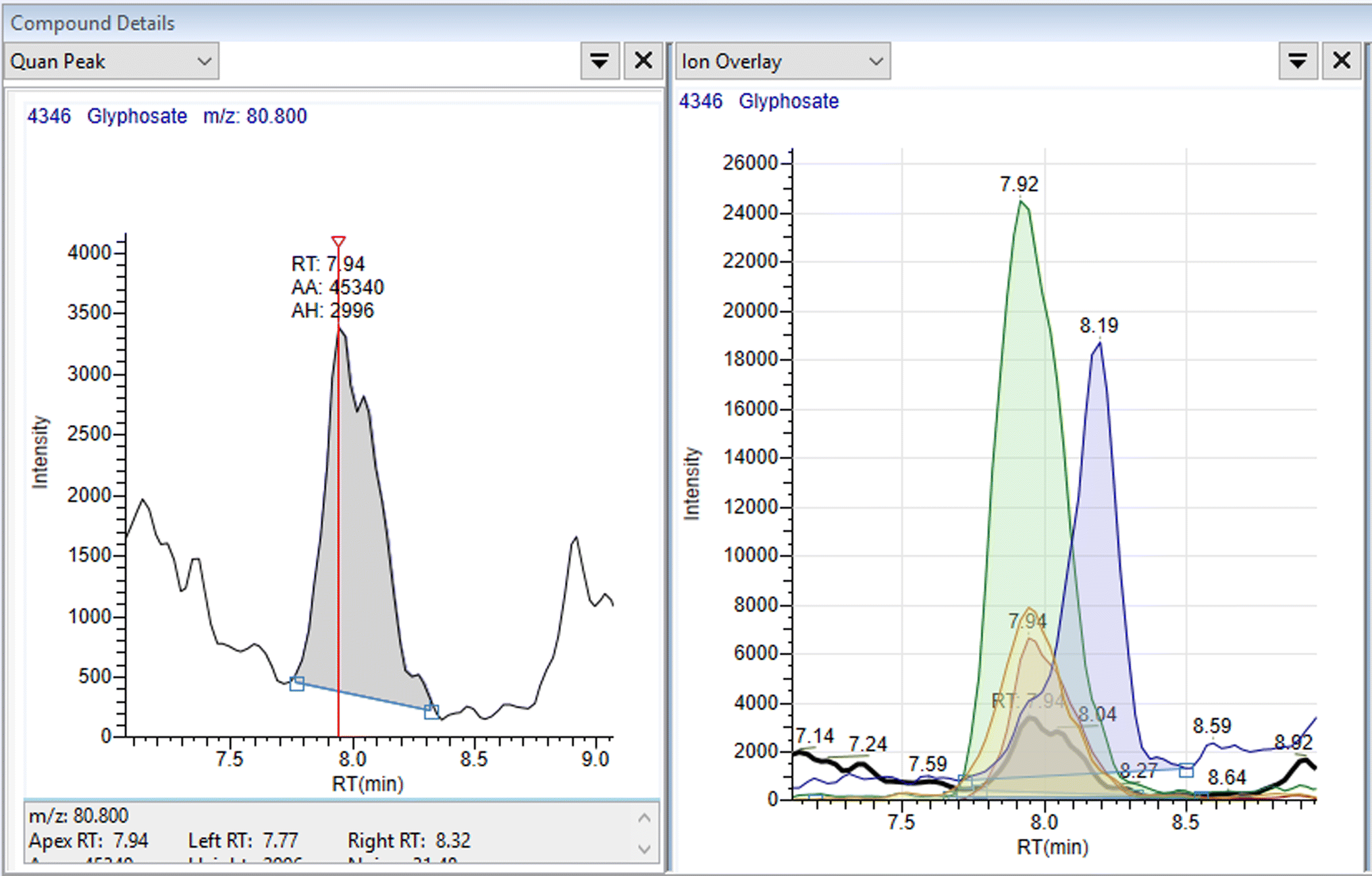 | ||
| Fig. 2 Example chromatograms for glyphosate in wheat. | ||
 | ||
| Fig. 3 Summary of the linearity data. | ||
3.3 Recovery data
The recovery data is used to estimate accuracy and precision. Acceptable accuracy is defined as average recovery for a repeatability experiment between 70% and 120%. Precision or repeatability is defined as a % CV of 20% or less. Acceptable accuracy for reproducibility data is defined as % recovery between 60% and 140% and acceptable precision is defined as a % CV of 30% or less.Repeatability studies were carried out at four concentration levels between 10 mg L−1 and 250 mg L−1 by spiking blank samples with a mix containing all the compounds in the method. Six replicates were carried out at each concentration level by two different analysts to give 60 different recovery experiments per commodity. The average recovery standard deviation and relative standard deviation were calculated for each set of repeatability experiments. The means from each repeatability study were then combined to calculate the within laboratory reproducibility.
The repeatability data is summarised in Table S5a and Fig. S5b.† For cereals, the criteria were not met at the lowest levels for glufosinate or at any level for fosetyl aluminium or phosphonic acid. The data meets the validation criteria for all other analytes. All analytes met the validation criteria for infant formula. In fruit and vegetables cyanuric acid and dicamba were problematic at low levels in carrots. All other fruit and vegetable data met the validation criteria.
Fig. 4a and b shows a summary of the reproducibility data and from this it is evident that all the compounds meet the criteria for accuracy with the exception of fosetyl aluminium and phosphonic acid in wheat.
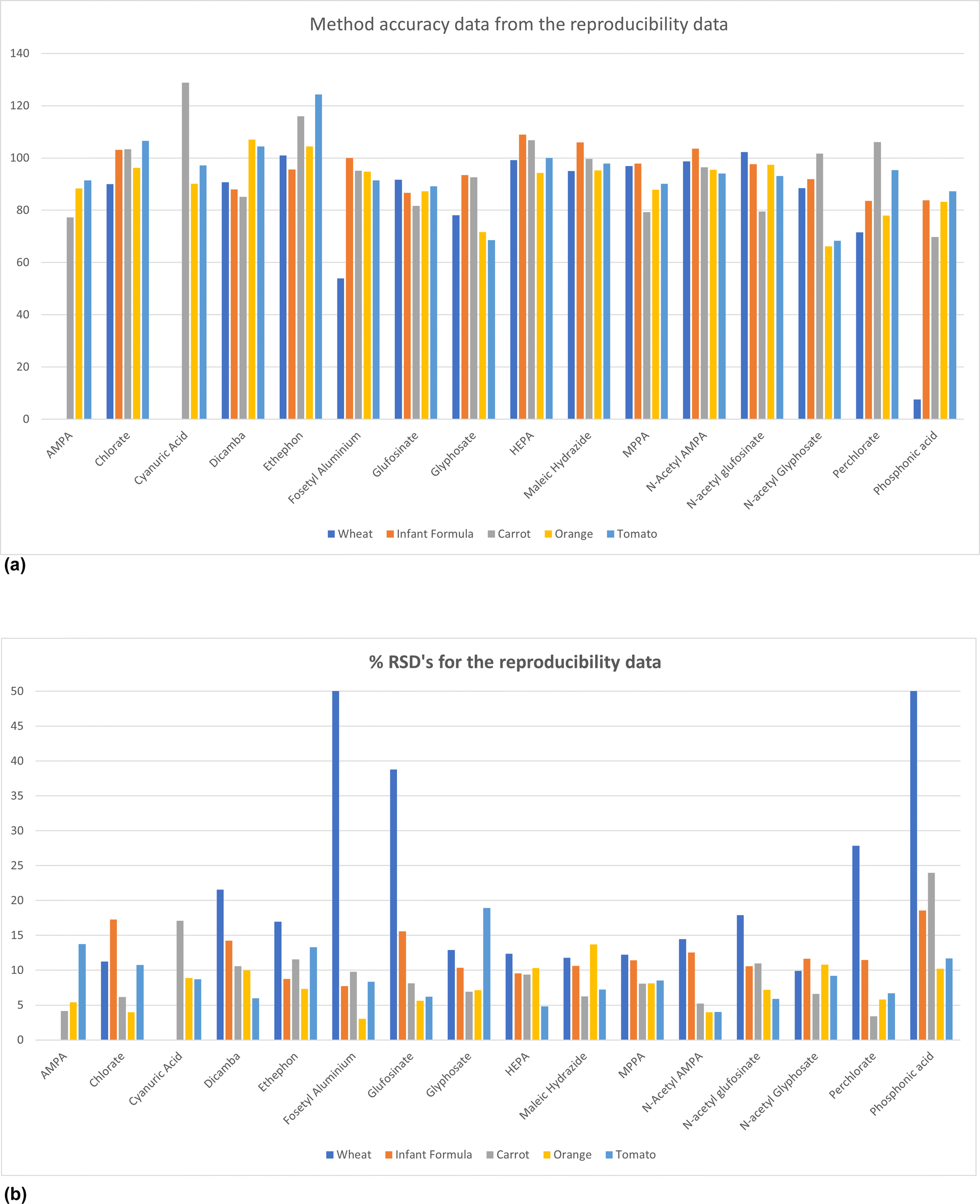 | ||
| Fig. 4 Summary of the within lab reproducibility data (a) accuracy and (b) precision. | ||
3.4 Ion ratio data
Normal practice in pesticide residue analysis is to use one transition for quantification and a second transition for confirmation of identity. The ratio of the area of the confirmation transition to the quantification transition (referred to as ion ratio) is then used as a criterion for confirmation of the identity of the analyte. A value of 30% or less is considered acceptable. The ion ratio data is summarised in Fig. S2.† From this data it is clear that dicamba and maleic hydrazide failed to meet the criteria for all matrices, except for infant formula in the case of dicamba. HEPA and N-acetyl glyphosate dd not meet the criteria in oranges while phosphonic acid failed for carrots. For all other analytes the criteria are met for all matrices and concentration levels. Where the ion ratio criteria are not met screening of the samples for these analytes is still possible however reliable confirmation of the analyte identity requires the use of another analytical method or the presence of metabolites.3.5 Matrix effect data
To ensure accurate quantification matrix effects are important to understand and control. These effects can result in very significant suppression or enhancement of the signal in the mass spectrometer, and this can lead to poor quantification accuracy. The matrix effect is defined as the slope of the calibration curve measured with matrix matched standards compared to the slope measured with solvent standards. It can be quantified using the equation:| Matrix effects = ((Slopematrix − Slopesolvent)/Slopesolvent) × 100 |
A negative matrix effect occurs with suppression of the analyte signal compared to solvent standards and a positive matrix effect occurs when enhancement of the signal occurs. Matrix effects within the range ±20% are considered manageable and anything outside this range is considered significant. The easiest way to deal with significant matrix effects is dilution of the sample which often reduces the effects to a manageable level. If this doesn't work, then matrix matching of the standards or the use of isotopically labelled internal standards should be considered.
For wheat, the matrix effects were only significant for AMPA, fosetyl aluminum and ethephon at a dilution level of 1/100. For infant formula, the matrix effects were significant for AMPA, fosetyl aluminum ethephon, and glufosinate at a dilution level of 1/20. In these cases, the use of matrix matched standards with the matrix diluted to the same level as the samples was used for quantification. The situation was not so straightforward for fruit and vegetables. In this case dilution reduced the matrix effect but for more than half the compounds the effect is still significant. This is a problem for routine work where batches of mixed matrices are run, and an exact matrix match is rarely possible. Additional dilution does not bring down the matrix effect any further. However, for each analyte which displayed a significant matrix effect the effect did not seem to be dependent on the matrix e.g. for ethephon 80% suppression was obtained regardless of the matrix. Similar effects were observed for other analytes such as cyanuric acid, dicamba and fosetyl aluminium. This suggests that matrix matching with any matrix should be used for quantitation. The matrix effect data is summarized in Fig. S3† for wheat and infant formula and Fig. S4† for fruit and vegetables.
3.6 Proficiency test and real sample data
This method has been used to analyse proficiency tests from the European Union Reference Laboratory for the last three years and the results are given in Table 2. Acceptable z-scores have been achieved for all analytes found and no false positives or false negatives were reported.| Scheme | Year | Matrix | Pesticide | z-score |
|---|---|---|---|---|
| EUPT SRM 16 | 2021 | Sesame seeds | Ethephon | −0.3 |
| Glufosinate | −0.1 | |||
| Glyphosate | 0.0 | |||
| Phosphonic acid | −0.7 | |||
| Chlorate | 0.2 | |||
| EUPT SRM 17 | 2022 | Tomato | Maleic hydrazide | 0.8 |
| EUPT SRM 18 | 2023 | Honey | Chlorate | −0.4 |
| Glyphosate | −1.0 | |||
| Phosphonic acid | 0.3 | |||
| Perchlorate | −0.3 |
This method, or a slight variation of it was used to analyse 300 routine samples in our laboratory. Of the 16 pesticides and metabolites covered by the scope nine were found in real samples. The most detected was the fosetyl aluminium metabolite, phosphonic acid. There is some debate as to whether this is from the use of fosetyl aluminium or whether it is coming from the type of fertiliser used.29 This metabolite was found 49 times in total, fruit and vegetables – 34, Cereals/feed – 11, milk/baby food – 2 and Meat/fat 2. The disinfectant chlorate was found 15 times, milk/baby food – 7, meat/fat – 4, fruit and veg – 3, and cereals/feed 1. The ethephon metabolite HEPA was found 14 times, 12 in bovine liver and 2 in cereals. Glyphosate was found 13 times, 9 in fruit and vegetables and 4 in cereals. Of the others maleic hydrazide was found 4 times in fruit and vegetables, perchlorate was found 3 times (cereals/feed – 1 and milk/babyfood 2), cyanuric acid was found twice (fruit and veg. – 2 and cereals – 2), glyphosate metabolites were found twice AMPA once in fruit and vegetables and N-acetyl AMPA was detected once in cereals. The data is summarised in Fig. 5.
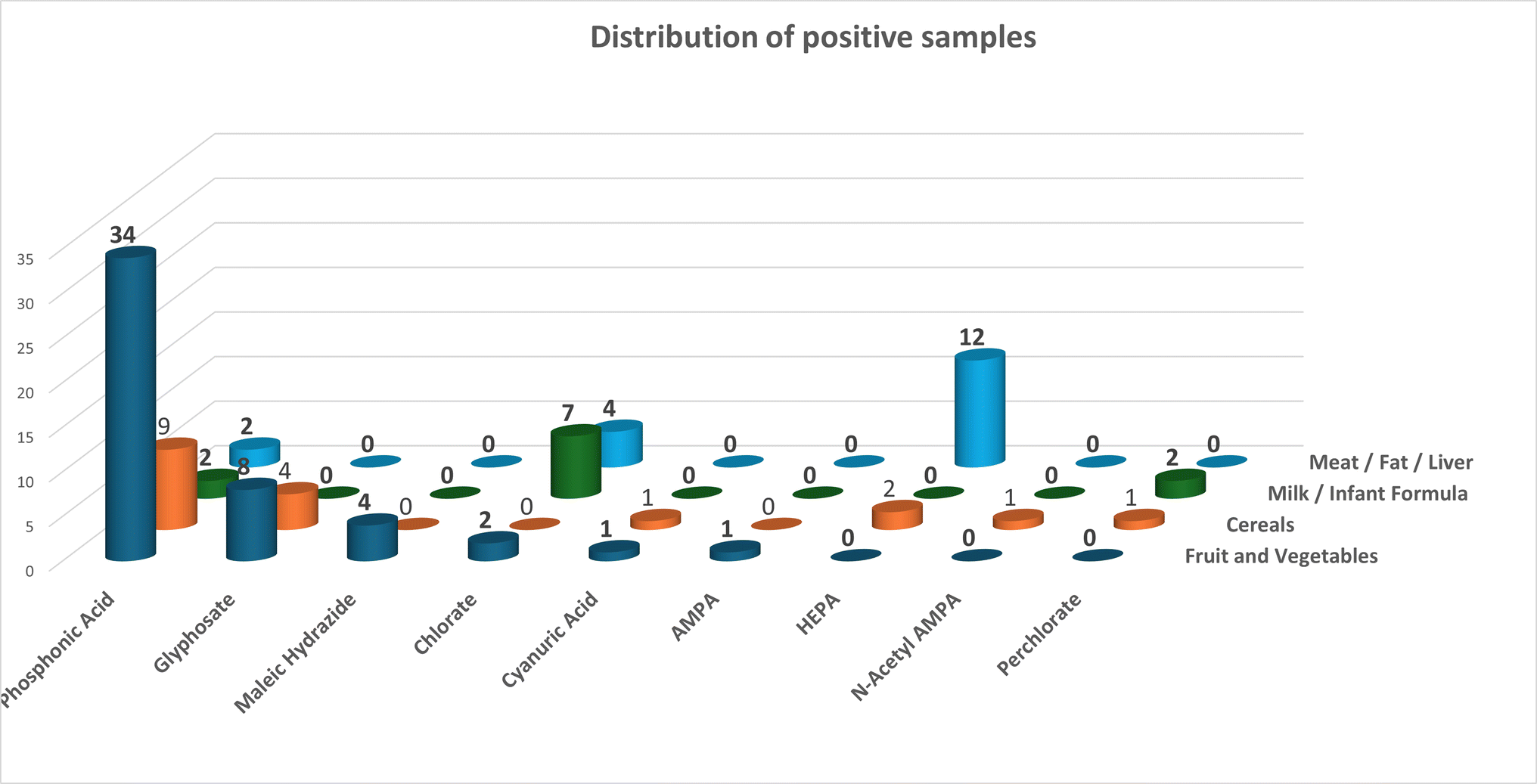 | ||
| Fig. 5 Summary of positive results from routine monitoring samples using this method. | ||
Conclusions
The method described in this study allows for the analysis of fruit and vegetables, infant formula/milk and cereals with a minor modification of the extraction procedure, for a range of 16 polar pesticides and metabolites. Validation has been carried out according to the EU SANTE guidance document and has been verified by using the method to analyse over 300 routine samples in the laboratory. Robustness has been demonstrated by also being used to analyse samples of liver and chicken meat though not yet fully validated for these matrices.Good precision and accuracy was achieved without the need to use isotopically labelled internal standards which makes the analysis straightforward and cost effective. The use of suppressed ion chromatography provides us with a stable chromatographic system with minimal need for conditioning and no requirement for passivation in the system. The sensitivity of the mass spectrometer used allows the use of dilution to solve some of the problems experienced with other methods such as severe matrix effects and unstable retention times in different commodities.
The analytes which proved problematic were AMPA which gave poor data generally in cereals and Infant Formula. Reliable confirmatory data could not be obtained for dicamba and maleic hydrazide. Matrix effects were high in fruit and vegetables, but these were controlled by matrix matching without the need to use isotopically labelled internal standards as demonstrated by comparison with other methods through the proficiency testing program.
This study demonstrates that ion chromatography with tandem mass spectrometry provides a stable and robust method for the analysis of polar pesticides in a wide range of matrices which meets the legislative requirements within the EU and has proven its value with the analysis of a large number of routine samples. These analytes have always proven difficult to analyse however we are satisfied that this method provides us with a reliable and robust means of analysing polar pesticides in a wide variety of matrices.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. E. Richmond, Glyphosate: A review of its global use, environmental impact, and potential health effects on humans and other species, J. Environ. Sci. Stud., 2018, 8, 416–434, DOI:10.1007/s13412-018-0517-2.
- International Agency for Research on Cancer World Health Organization, Q&A on Glyphosate, 2016, last accessed 2024 January 29, https://www.iarc.fr/en/mediacentre/iarcnews/pdf/Q%26A_Glyphosate.pdf.
- R. Mesnage and M. N. Antoniou, Facts and Fallacies in the Debate on Glyphosate Toxicity, Front. Public Health, 2017, 316, DOI:10.3389/fpubh.2017.00316 , https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5705608/.
- L. Zhang, I. Rana, R. M. Shaffer, E. Taioli and L. Sheppard, Exposure to glyphosate-based herbicides and risk for non-Hodgkin lymphoma: A meta-analysis and supporting evidence, Mutat. Res., Rev. Mutat. Res., 2019, 781, 186–206, DOI:10.1016/j.mrrev.2019.02.001.
- EFSA Journal, [Internet], 2023, last accessed 2024 Jan 26th, DOI:10.2903/j.efsa.2023.8164, https://www.efsa.onlinelibrary.wiley.com.
- H. G. Mol and R. C. van Dam, Rapid detection of pesticides not amenable to multi-residue methods by flow injection–tandem mass spectrometry, Anal. Bioanal. Chem., 2014, 406, 6817–6825, DOI:10.1007/s00216-014-7644-8.
- A. Vass, J. Robles-Molina, P. Perez-Ortega, B. Gilbert-Lopez, M. Dernovics, A. Molina-Diaz and J. F. Garcia-Reyes, Study of different HILIC, mixed-mode, and other aqueous normal-phase approaches for the liquid chromatography/mass spectrometry-based determination of challenging polar pesticides, Anal. Bioanal. Chem., 2016, 408, 4857–4869, DOI:10.1007/s00216-016-9589-6.
- R. Domingos Alves, R. Romero-Gonzalez, R. Lopez-Ruiz, M. L. Jimenez-Medina and A. Garrido Frenich, Fast determination of four polar contaminants in soy nutraceutical products by liquid chromatography coupled to tandem mass spectrometry, Anal. Bioanal. Chem., 2016, 408, 8089–8098, DOI:10.1007/s00216-016-9912-2.
- European Union Reference Laboratories for Residues of Pesticides Quick Method for the Analysis of Highly Polar Pesticides in Food Involving Extraction with Acidified Methanol and LC- or IC-MS/MS Measurement (QuPPe-Method), 2024, Version 12.2: 2023 Dec 21, last accessed May 17, https://www.eurl-pesticides.eu/userfiles/file/EurlSRM/EurlSrm_meth_QuPPe_PO_V12_2.pdf Search PubMed.
- S. Goscinny, H. Unterluggauer, J. Aldrian, V. Hanot and S. Masselter, Determination of Glyphosate and Its Metabolite AMPA (Aminomethylphosphonic Acid) in Cereals After Derivatization by Isotope Dilution and UPLC-MS/MS, Food Anal. Methods, 2012, 5, 1177–1185, DOI:10.1007/s12161-011-9361-7.
- T. Poiger, I. J. Buerge, A. Bachli, M. D. Muller and M. E. Balmer, Occurrence of the herbicide glyphosate and its metabolite AMPA in surface waters in Switzerland determined with on-line solid phase extraction LC-MS/MS, Environ. Sci. Pollut. Res. Int., 2016, 24, 1588–1596, DOI:10.1007/s11356-016-7835-2.
- A. M. Botero-Coy, M. Ibanez, J. V. Sancho and F. Hernandez, Direct liquid chromatography–tandem mass spectrometry determination of underivatized glyphosate in rice, maize and soybean, J. Chromatogr. A, 2013, 1313, 157–165, DOI:10.1016/j.chroma.2013.07.037.
- R. Nortes-Méndez, J. Robles-Molina, R. Lopez-Blanco, A. Vass, A. Molina-Díaz and J. F. Garcia-Reyes, Determination of polar pesticides in olive oil and olives by hydrophilic interaction liquid chromatography coupled to tandem mass spectrometry and high resolution mass spectrometry, Talanta, 2016, 158, 222–228, DOI:10.1016/j.talanta.2016.05.058.
- S. Herrera-Lopez, J. Dias and A. de Kok, Analysis of highly polar pesticides and their main metabolites in animal origin matrices by hydrophilic interaction liquid chromatography and mass spectrometry, Food Control, 2020, 115, 107289, DOI:10.1016/j.foodcont.2020.107289.
- S. Herrera-Lopez, J. Dias, H. Mol and A. de Kok, Selective multiresidue determination of highly polar anionic pesticides in plant-based milk, wine and beer using hydrophilic interaction liquid chromatography combined with tandem mass spectrometry, J. Chromatogr. A, 2020, 1625, 461216, DOI:10.1016/j.chroma.2020.461226.
- S. Herrera-Lopez, J. Scholten, B. Kiedrowska and A. De Kok, Method validation and application of a selective multiresidue analysis of highly polar pesticides in food matrices using hydrophilic interaction liquid chromatography and mass spectrometry, J. Chromatogr. A, 2019, 1594, 93–104, DOI:10.1016/j.chroma.2019.02.024.
- N. D. Gangal, S. S. Bandre and P. S. Ramanathon, Determination of some pesticides and intermediates by ion chromatography, J. Chromatogr. A, 2000, 884, 243–249, DOI:10.1016/S0021-9673(00)00305-8.
- N. C. Maragou and G. Balayiannis, Determination of Ethephon in Pesticide Formulations by Ion Exchange Chromatography with Indirect Spectrophotometric Detection, Anal. Lett., 2019, 53(5), 795–806, DOI:10.1080/00032719.2019.1677700.
- L. Rajski, F. J. Diaz Galliano, V. Cutillas and A. R. Fernandes-Alba, Coupling Ion Chromatography to Q-Orbitrap for the Fast and Robust Analysis of Anionic Pesticides in Fruits and Vegetables, J. AOAC Int., 2018, 101, 352–359, DOI:10.5740/jaoacint.17-0410.
- S. Adams, J. Guest, M. Dickinson, R. J. Fussell, J. Beck and F. Shoutsen, Development and Validation of Ion Chromatography–Tandem Mass Spectrometry-Based Method for the Multiresidue Determination of Polar Ionic Pesticides in Food, J. Agric. Food Chem., 2017, 65, 7294–7304, DOI:10.1021/acs.jafc.7b00476.
- L. Melton, M. J. Taylor and E. E. Flynn, The utilisation of ion chromatography and tandem mass spectrometry (IC-MS/MS) for the multi-residue simultaneous determination of highly polar anionic pesticides in fruit and vegetables, Food Chem., 2019, 299, 125028, DOI:10.1016/j.foodchem.2019.125028.
- A. Bauer, J. Luetjohann, S. Rohn, J. Kuballa and E. Jantzen, Ion chromatography tandem mass spectrometry (IC-MS/MS) multimethod for the determination of highly polar pesticides in plant-derived commodities, Food Control, 2018, 86, 71–76, DOI:10.1016/j.foodcont.2017.11.007.
- J. B. Ngere, K. H. Ebrahimi, R. Williams, E. Pire, J. Walsby-Tickle and J. S. McCullagh, Ion-Exchange Chromatography Coupled to Mass Spectrometry in Life Science, Environmental, and Medical Research, Anal. Chem., 2023, 95(1), 152–166, DOI:10.1021/acs.analchem.2c04298.
- A. J. Krynitsky, R. A. Niemann and D. A. Nortrup, Determination of perchlorate anion in foods by ion chromatography-tandem mass spectrometry, Anal. Chem., 2004, 76, 5518–5522, DOI:10.1021/ac049281+.
- K. Granby, S. Johannesen and M. Vahl, Analysis of glyphosate residues in cereals using liquid chromatography-mass spectrometry (LC-MS/MS), Food Addit. Contam., 2003, 20, 692–698, DOI:10.1080/0265203031000109477.
- R. J. Fussell, M. T. Hetmanski, A. Colyer, M. Caldow, F. Smith and D. Findlay, Assessment of the stability of pesticides during the cryogenic processing of fruits and vegetables, Food Addit. Contam., 2007, 24(11), 1247–1256 CrossRef CAS PubMed.
- S. J. Lehotay, N. Michlig and A. R. Lightfield, Assessment of Test Portion Sizes after Sample Comminution with Liquid Nitrogen in an Improved High-Throughput Method for Analysis of Pesticide Residues in Fruits and Vegetables, J. Agric. Food Chem., 2020, 68(5), 1468–1479, DOI:10.1021/acs.jafc.9b07685.
- SANTE/11312/2021, Guidance document on analytical quality control and method validation procedures for pesticides residues analysis in food and feed, 2024, last accessed January 26, https://food.ec.europa.eu/system/files/2023-11/pesticides_mrl_guidelines_wrkdoc_2021-11312.pdf.
- W. Nader, A. Zahn and J. Jaschik, Phosphonic acid in plant-based food and feed products – Where does it come from?, Food Control, 2023, 150, 109701 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ay00183d |
| This journal is © The Royal Society of Chemistry 2024 |