DOI:
10.1039/D4AY00735B
(Paper)
Anal. Methods, 2024,
16, 5178-5190
Simultaneous determination of 78 pesticide residues and 16 mycotoxins in tsampa by an improved QuEChERS method coupled with ultra performance liquid chromatography-tandem mass spectrometry†
Received
22nd April 2024
, Accepted 6th June 2024
First published on 11th June 2024
Abstract
Tsampa may contain pesticide residues and mycotoxins, which may pose a risk to human health. Currently, pesticide detection and mycotoxin detection are two independent experiments. To improve the efficiency of the analysis, a method based on QuEChERS combined with ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) for the simultaneous determination of 78 pesticides and 16 mycotoxins in tsampa was developed. All the target compounds showed good linear correlation with correlation coefficients (R2) greater than 0.9990. The limits of detection (LODs) and limits of quantification (LOQs) were in the ranges of 0.10–3.00 μg kg−1 and 0.40–10.00 μg kg−1, respectively. The average recoveries of the pesticides and mycotoxins spiked at the 1, 2, and 10-fold LOQ were in the range of 73.0–115.2%, and the relative standard deviations (RSDs) were lower than 11.7%. This method was applied to 19 batches of real samples in which 32% of samples exceeded the maximum residue limits of the European Union involving aflatoxin G2, ochratoxin A, and hexaconazole. It proved to be excellent, efficient, greatly simplified, and highly applicable, which could reduce the workload and time significantly for the daily monitoring of the pesticides and mycotoxins in tsampa.
1. Introduction
Tsampa is one of the traditional staple foods of the Tibetan people,1 which comes from barley. After removing impurities, cleaning, drying, stir-frying, grinding, and other processes from barley, tsampa is produced, retaining more nutrients than traditional cereal products, such as proteins, vitamins, β-glucans, etc.2 In the process of barley cultivation, in order to effectively prevent the emergence of pests and plant diseases, the use of pesticides is inevitable, resulting in the presence of pesticide residues in barley.3 However, these pesticides may still be in the tsampa, after the barley is processed into tsampa, posing a potential threat to the health of consumers.4 This may lead to acute poisoning, chronic poisoning, cancer, neurological damage, etc.5 Mycotoxins, the toxic metabolites produced by fungi that grow in substrates like grain or feed, can affect the tsampa during cultivation, storage, or transport.6–8 Most of the mycotoxins are highly biotoxic, suppressing the immune system and causing adverse effects on human health, such as carcinogenicity, teratogenicity, and mutagenicity.9,10 Mycotoxins for instance aflatoxin,11 fumonisin B1,12 ochratoxin A13 and zearalenone14 and deoxynivalenol15 are classified as carcinogens by the International Agency for Research on Cancer (IARC). Therefore, more attention needs to be paid to monitoring pesticide residues as well as mycotoxins in tsampa to protect consumer health and safety.
Numerous studies have been conducted on the detection of pesticide residues or mycotoxins in whole-grain foods. The commonly used pretreatment techniques for pesticide residues include solid-phase extraction,16,17 dispersive liquid–liquid microextraction,18,19 and QuEChERS.20,21 Meanwhile, immunoaffinity columns,22,23 QuEChERS, and magnetic solid-phase extraction24 have been mainly used for the detection of mycotoxins. QuEChERS is the preferred method for extracting pesticides and mycotoxins due to its ability to significantly reduce the difficulty, time, and cost of sample processing compared to other pretreatment techniques. With regard to detection techniques, UPLC-MS/MS is a reliable and effective method for the detection of pesticides and mycotoxins in food. It has become the most commonly used method for multi-analyte analysis.25–27 In the previous study, pesticide residues and mycotoxins were detected separately. However, if a modified method was developed to detect both simultaneously, it would significantly simplify the pretreatment and improve the detection efficiency. Chau et al. used UPLC-MS/MS to simultaneously determine 31 pesticide residues and 6 mycotoxins in Pu-erh tea using UPLC-MS/MS after the extraction by QuEChERS.28 He et al. used the modified QuEChERS/UPLC-MS/MS to determine 49 pesticide residues and 17 mycotoxins in wine.29 However, there are few reported methods for the co-detection and analysis of pesticides and mycotoxins in cereal foods. Tsampa, as a food made from barley, is one of the main food sources for people in Tibet. Therefore, a method for simultaneously detecting pesticide residues and mycotoxins in tsampa is necessary to improve the efficiency of detecting target compounds and protect consumer health and safety.
The potential existence of pesticides and mycotoxins in tsampa increases the challenge and cost of daily monitoring. In order to enhance the efficiency of detecting pesticide residues and mycotoxins in tsampa, this study aimed to optimize the QuEChERS method to establish an effective method using UPLC-MS/MS for the simultaneous detection of 78 pesticides and 16 mycotoxins in tsampa, and to apply it to real samples. Therefore, the scientific basis and data support provided will contribute to the quality and safety of food in Tibet.
2. Materials and methods
2.1. Chemicals and reagents
78 standard solutions of pesticides (1000 μg mL−1), 14 standard solutions of mycotoxins (1000 μg mL−1) and 2 standard solutions of mycotoxins (100 μg mL−1) were purchased from Altascientific (Tianjin, China). LC-grade methanol (MeOH) and acetonitrile (ACN) were provided by Thermo Fisher (Waltham, USA); LC-grade acetic acid (CH3COOH), sodium chloride (NaCl), and anhydrous magnesium sulfate (MgSO4) were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China); MS-grade formic acid and ammonium formate were provided by Agilent (California, USA); graphitised carbon black (GCB), N-propylethylenediamine (PSA), and octadecylsilane (C18) were purchased from Bonna-Agela (Tianjin, China).
2.2. Apparatus
The Milli-Q ultrapure water dispenser was supplied by Millipore (Massachusetts, USA); and the N-EVAP112 nitrogen blowing machine was supplied by Millipore (Massachusetts, USA). The N-EVAP112 Nitrogen Blow Concentrator was provided by Organomation Associates (Massachusetts, USA); the SR-2DS Horizontal Oscillator was provided by TATEC (Koshigaya, Japan); the Allegra X-30R centrifuge was provided by BECAKMEN COULTER (California, USA); the PL602-L electronic balance was provided by Mettler-Toledo (Zurich, Switzerland).
2.3. Preparation of standard solutions
Preparation of pesticide standard intermediate: 0.1 mL of each pesticide standard solution (concentration of 1000 μg mL−1) was pipetted into a 10 mL volumetric flask and diluted with methanol. Then the concentration of the solution was 10 mg L−1; it was stored at 4 °C, protected from light.
Preparation of mycotoxin standard intermediate solution: 16 mycotoxins were divided into group I (1000 μg mL−1) and group II (100 μg mL−1). 0.1 mL of group I was pipetted into a 10 mL volumetric flask, then fixed with methanol. The standard intermediate solution of group I (10 mg L−1) was stored at 4 °C away from light.
Standard work solution preparation: 1 mL of the standard intermediate solution of group I (10 mg L−1) and pesticide standard intermediate solution (10 mg L−1), as well as 0.1 mL of group II, were pipetted into a 10 mL volumetric flask diluted with MeOH. The mixed standard working solution (1 mg L−1) was obtained and stored at 4 °C away from light.
2.4. Instrumentation
The separation was carried out using a UPLC-MS/MS system (ACQUITY UPLC-Xevo TQXS, Waters Corp., Milford, MA, USA) at 40 °C, chromatographic column: ACQUITY UPLC BEH C18 column (100 mm × 2.1 mm id, 1.7 μm, Waters, Milford, MA, USA). Mobile phase: phase A was 0.1% formic acid in water containing 5 mmol ammonium formate and phase B was MeOH. The gradient elution procedure was as follows: 0.0–0.2 min: 2% B; 0.2–1.5 min: 2–30% B; 1.5–3 min: 30–40% B; 3–4.5 min: 40–45% B; 4.5–7.5 min: 45–65% B; 7.5–9 min: 65–98% B; 9–11 min: 98% B; 11.01–15 min: 2% B; flow rate 0.3 mL min−1, injection volume: 2 μL. The mass spectrometers were set as follows: electrospray ion source (ESI); positive ion scanning, ion source temperature: 150 °C; electrospray voltage: 2.0 kV; desolvation gas temperature: 550 °C; desolvation gas flow rate: 800 L h−1; cone pore gas flow rate: 50 L h−1; multiple reaction monitoring (MRM) mode acquisition.
2.5. Sample preparation
Accurately 2.5 g tsampa was placed into a 50 mL centrifuge tube, 15 mL ultrapure water was added, the tube was vortexed for 30 s, and stood for 20 min. Ten mL of ACN (5% formic acid) and 4 zirconium beads were added, then 4 g MgSO4 and 1 g NaCl were added, vortexed for 5 min, and centrifuged for 5 min (4500 rpm). Three mL of supernatant was placed in an advance-equipped 15 mL purification tube (300 mg MgSO4 + 100 mg PSA + 150 mg C18), vortexed for 5 min, and centrifuged for 5 min (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm). One mL of supernatant was placed in a 10 mL glass tube, and nitrogen was blown at 40 °C to dryness. The residue was diluted to 0.5 mL MeOH–water solution (3:2, v/v), vortexed, and then passed through a 0.22 μm filter.
000 rpm). One mL of supernatant was placed in a 10 mL glass tube, and nitrogen was blown at 40 °C to dryness. The residue was diluted to 0.5 mL MeOH–water solution (3:2, v/v), vortexed, and then passed through a 0.22 μm filter.
2.6. Real samples
Thirty batches of tsampa samples (22 batches of white tsampa, 3 batches of black tsampa, 3 batches of pea tsampa, and 2 batches of water-milled tsampa) were purchased from Lhasa, Tibet, China, of which 8 batches of white tsampa, 2 batches of black tsampa, and 1 batch of water-milled tsampa were purchased online, and the rest of the tsampa samples were taken from the local shops. The purchased samples were stored in a refrigerator at 4 °C.
3. Results and discussion
3.1. Optimization of UPLC-MS/MS conditions
To ensure maximum sensitivity for accurate quantification of the target compounds, the mass spectrometry parameters were optimized for each pesticide and mycotoxin. In this study, the mass spectrometry conditions were optimized in positive ion electrospray mode, including the precursor ions, product ions, cone voltage, and collision energy. The precursor ion with the strongest response signal was determined by optimizing the cone voltage in MS scan mode. The precursor ions were cleaved in daughter scan mode, and two of the product ions with relatively higher response signals were selected as quantitative and qualitative ions by adjusting the collision energy. The retention time and mass spectrometry parameters of 78 pesticides and 16 mycotoxins are shown in Table 1.
Table 1 Retention time and mass spectrometry parameters of 78 pesticides and 16 mycotoxins
| No. |
Compound |
CAS |
Molecular formula |
Retention time (min) |
Ion pair (m/z) |
Collision energy (eV) |
Cone (V) |
|
Quantitative ion.
|
|
Pesticides (78)
|
| 1 |
Acetamiprid |
135410-20-7 |
C10H11ClN4 |
3.71 |
223.1/126.0a, 223.1/56.0 |
20, 15 |
30 |
| 2 |
Aldicarb |
116-06-3 |
C7H14N2O2S |
4.48 |
208.1/116.2a, 208.1/89.2 |
7, 16 |
10 |
| 3 |
Aldicarb sulfone |
1646-88-4 |
C7H14N2O4S |
6.67 |
223.1/86.0a, 223.1/148.1 |
13, 8 |
40 |
| 4 |
Aldicarb sulfoxide |
1646-87-3 |
C7H14N2O3S |
2.58 |
207.1/132.0a, 207.1/89.0 |
5, 15 |
20 |
| 5 |
Azoxystrobin |
131860-33-8 |
C22H17N3O5 |
8.16 |
404.1/372.1a, 404.1/329.1 |
16, 30 |
30 |
| 6 |
Bitertanol |
55179-31-2 |
C20H23N3O2 |
9.72 |
338.2/70.0a, 338.2/269.1 |
8, 8 |
30 |
| 7 |
Butralin |
33629-47-9 |
C14H21N3O4 |
10.32 |
296.2/240.1a, 296.2/222.1 |
13, 21 |
30 |
| 8 |
Cadusafos |
95465-99-9 |
C10H23O2PS2 |
9.80 |
271.1/159.0a, 271.1/131.0 |
16, 22 |
16 |
| 9 |
Carbendazim |
10605-21-7 |
C9H9N3O2 |
2.96 |
192.1/160.0a, 192.1/132.0 |
15, 30 |
10 |
| 10 |
Carbofuran |
1563-66-2 |
C12H15NO3 |
5.52 |
222.1/165.0a, 222.1/123.0 |
10, 20 |
25 |
| 11 |
Carbofuran-3-hydroxy |
16655-82-6 |
C12H15NO4 |
3.71 |
238.1/181.0a, 238.1/220.1 |
10, 4 |
30 |
| 12 |
Chlorantraniliprole |
500008-45-7 |
C18H14BrCl2N5O2 |
7.68 |
484.0/453.0a, 484.0/286.0 |
18, 20 |
30 |
| 13 |
Chlorbenzuron |
57160-47-1 |
C14H10Cl2N2O2 |
9.44 |
309.0/156.0a, 309.0/139.0 |
15, 30 |
30 |
| 14 |
Chlordimeform |
6164-98-3 |
C10H13ClN2 |
3.22 |
197.1/46.0a, 197.1/117.0 |
18, 29 |
20 |
| 15 |
Chlorfenvinphos |
18708-86-6 |
C12H14Cl3O4P |
9.61 |
359.0/155.0a, 359.0/99.0 |
12, 30 |
28 |
| 16 |
Chlorsulfuron |
64902-72-3 |
C12H12ClN5O4S |
6.15 |
358.0/141.0a, 358.0/167.0 |
16, 18 |
24 |
| 17 |
Chlortoluron |
15545-48-9 |
C10H13ClN2O |
6.46 |
213.1/72.0a, 213.1/46.0 |
15, 15 |
25 |
| 18 |
Clothianidin |
210880-92-5 |
C6H8ClN5O2S |
3.40 |
250.0/132.0a, 250.0/169.0 |
12, 8 |
30 |
| 19 |
Cymoxanil |
57966-95-7 |
C7H10N4O3 |
3.95 |
199.2/128.0a, 199.2/111.1 |
10, 18 |
25 |
| 20 |
Difenoconazole |
119446-68-3 |
C19H17Cl2N3O3 |
9.81 |
406.1/251.0a, 406.1/111.0 |
20, 60 |
35 |
| 21 |
Diflubenzuron |
35367-38-5 |
C14H9ClF2N2O2 |
9.29 |
311.0/158.0a, 311.0/141.0 |
12, 15 |
34 |
| 22 |
Ethametsulfuron |
111353-84-5 |
C14H16N6O6S |
4.44 |
397.0/196.1a, 397.0/170.1 |
15, 15 |
30 |
| 23 |
Ethoprophos |
13194-48-4 |
C8H19O2PS2 |
9.05 |
243.1/97.0a, 243.1/173.0 |
30, 12 |
20 |
| 24 |
Etoxazole |
153233-91-1 |
C21H23F2NO2 |
10.27 |
360.2/141.1a, 360.2/57.1 |
25, 25 |
30 |
| 25 |
Fenamiphos |
22224-92-6 |
C13H22NO3PS |
9.31 |
304.1/217.1a, 304.1/202.1 |
24, 36 |
27 |
| 26 |
Fenamiphos sulfone |
31972-44-8 |
C13H22NO5PS |
6.21 |
336.1/266.1a, 336.1/188.2 |
20, 28 |
34 |
| 27 |
Fenamiphos sulfoxide |
31972-43-7 |
C13H22NO4PS |
5.95 |
320.1/108.0a, 320.1/171.1 |
35, 22 |
30 |
| 28 |
Fenbuconazole |
114369-43-6 |
C19H17ClN4 |
9.28 |
337.1/125.0a, 337.1/70.0 |
30, 20 |
15 |
| 29 |
Fenoxanil |
115852-48-7 |
C15H18Cl2N2O2 |
9.38 |
329.1/86.0a, 329.1/302.0 |
22, 10 |
26 |
| 30 |
Fonofos |
944-22-9 |
C10H15OPS2 |
9.49 |
247.0/109.0a, 247.0/137.0 |
20, 10 |
24 |
| 31 |
Heptenophos |
23560-59-0 |
C9H12ClO4P |
7.37 |
251.0/127.0a, 251.0/89.0 |
14, 34 |
26 |
| 32 |
Hexaconazole |
79983-71-4 |
C14H17Cl2N3O |
9.67 |
314.1/70.0a, 314.1/159.0 |
20, 30 |
30 |
| 33 |
Hexaflumuron |
86479-06-3 |
C16H8Cl2F6N2O3 |
9.91 |
461.0/158.0a, 461.0/141.0 |
25, 59 |
19 |
| 34 |
Imidacloprid |
138261-41-3 |
C9H10ClN5O2 |
3.37 |
256.1/175.0a, 256.1/209.0 |
20, 12 |
25 |
| 35 |
Indanofan |
133220-30-1 |
C20H17ClO3 |
9.05 |
341.1/174.9a, 341.1/186.9 |
14, 12 |
21 |
| 36 |
Isofenphos-methyl |
99675-03-3 |
C14H22NO4PS |
9.45 |
332.2/230.9a, 332.2/121.0 |
15, 30 |
20 |
| 37 |
Isoprothiolane |
50512-35-1 |
C12H18O4S2 |
8.51 |
291.1/189.0a, 291.1/231.0 |
22, 12 |
17 |
| 38 |
Malathion |
121-75-5 |
C10H19O6PS2 |
8.53 |
331.0/127.0a, 331.0/99.0 |
12, 25 |
30 |
| 39 |
Metalaxyl |
57837-19-1 |
C15H21NO4 |
7.18 |
280.2/220.1a, 280.2/192.1 |
15, 20 |
30 |
| 40 |
Methamidophos |
10265-92-6 |
C2H8NO2PS |
1.47 |
142.0/94.0a, 142.0/125.0 |
12, 14 |
30 |
| 41 |
Methidathion |
950-37-8 |
C6H11N2O4PS3 |
7.30 |
303.0/145.0a, 303.0/85.0 |
10, 35 |
30 |
| 42 |
Methiocarb |
2032-65-7 |
C11H15NO2S |
8.13 |
226.1/169.0a, 226.1/121.0 |
10, 20 |
25 |
| 43 |
Methiocarb sulfone |
2179-25-1 |
C11H15NO4S |
3.89 |
258.1/122.0a, 258.1/201.0 |
16, 7 |
45 |
| 44 |
Methiocarb sulfoxide |
2635-10-1 |
C11H15NO3S |
3.55 |
242.1/185.0a, 242.1/122.0 |
14, 28 |
26 |
| 45 |
Methomyl |
16752-77-5 |
C5H10N2O2S |
2.88 |
163.1/88.0a, 163.1/106.0 |
10, 10 |
15 |
| 46 |
Metrafenone |
220899-03-6 |
C19H21BrO5 |
9.73 |
409.1/209.1a, 409.1/227.0 |
14, 16 |
19 |
| 47 |
Metsulfuron-methyl |
74223-64-6 |
C14H15N5O6S |
5.59 |
382.1/167.0a, 382.1/199.0 |
16, 22 |
28 |
| 48 |
Mevinphos |
7786-34-7 |
C7H13O6P |
3.70 |
225.1/127.0a, 225.1/193.0 |
15, 10 |
15 |
| 49 |
Myclobutanil |
88671-89-0 |
C15H17ClN4 |
8.80 |
289.1/70.1a, 289.1/125.1 |
15, 30 |
25 |
| 50 |
Monocrotophos |
6923-22-4 |
C7H14NO5P |
3.10 |
224.1/193.0a, 224.1/127.0 |
7, 15 |
20 |
| 51 |
Nitenpyram |
150824-47-8 |
C11H15ClN4O2 |
2.81 |
271.1/126.0a, 271.1/237.1 |
30, 17 |
30 |
| 52 |
Omethoate |
1113-02-6 |
C5H12NO4PS |
8.75 |
214.0/125.0a, 214.0/183.0 |
22, 10 |
25 |
| 53 |
Phorate |
298-02-2 |
C7H17O2PS3 |
9.64 |
261.0/75.0a, 261.0/47.0 |
12, 33 |
20 |
| 54 |
Phorate sulfone |
2588-04-1 |
C7H17O4PS3 |
6.85 |
293.0/171.0a, 293.0/97.0 |
10, 30 |
24 |
| 55 |
Phorate sulfoxide |
2588-05-8 |
C7H17O3PS3 |
6.64 |
277.0/97.0a, 277.0/143.0 |
32, 20 |
24 |
| 56 |
Pirimicarb |
23103-98-2 |
C11H18N4O2 |
4.75 |
239.2/72.0a, 239.2/182.1 |
20, 15 |
25 |
| 57 |
Profenofos |
41198-08-7 |
C11H15BrClO3PS |
9.99 |
372.9/303.0a, 372.9/345.0 |
20, 12 |
30 |
| 58 |
Propiconazole |
60207-90-1 |
C15H17Cl2N3O2 |
9.57 |
342.1/159.0a, 342.1/69.0 |
20, 30 |
35 |
| 59 |
Pyraclostrobin |
175013-18-0 |
C19H18ClN3O4 |
9.64 |
388.1/194.0a, 388.1/163.0 |
12, 25 |
25 |
| 60 |
Pyridaben |
96489-71-3 |
C19H25ClN2OS |
10.49 |
365.2/147.1a, 365.2/309.1 |
24, 12 |
10 |
| 61 |
Pyrimethanil |
53112-28-0 |
C12H13N3 |
7.15 |
200.1/107.0a, 200.1/82.0 |
24, 24 |
25 |
| 62 |
Quinoxyfen |
124495-18-7 |
C15H8Cl2FNO |
10.18 |
308.0/197.0a, 308.0/214.0 |
30, 32 |
20 |
| 63 |
Spirodiclofen |
148477-71-8 |
C21H24Cl2O4 |
10.36 |
411.1/71.0a, 411.1/313.0 |
15, 10 |
35 |
| 64 |
Tebuconazole |
107534-96-3 |
C16H22ClN3O |
9.52 |
308.2/70.0a, 308.2/125.0 |
24, 40 |
30 |
| 65 |
Terbufos |
13071-79-9 |
C9H21O2PS3 |
10.04 |
289.1/103.0a, 289.1/233.0 |
10, 4 |
15 |
| 66 |
Terbufos-sulfone |
56070-16-7 |
C9H21O4PS3 |
7.95 |
321.0/97.0a, 321.0/171.0 |
40, 12 |
20 |
| 67 |
Terbufos-sulfoxide |
10548-10-4 |
C9H21O3PS3 |
7.97 |
305.1/187.0a, 305.1/97.0 |
11, 40 |
20 |
| 68 |
Tetrachlorvinphos |
22248-79-9 |
C10H9Cl4O4P |
9.38 |
364.9/127.0a, 364.9/204.0 |
16, 35 |
32 |
| 69 |
Thiamethoxam |
153719-23-4 |
C8H10ClN5O3S |
2.99 |
292.0/211.0a, 292.0/181.0 |
10, 20 |
25 |
| 70 |
Triadimefon |
43121-43-3 |
C14H16ClN3O2 |
8.66 |
294.1/69.0a, 294.1/197.0 |
20, 14 |
30 |
| 71 |
Triadimenol |
55219-65-3 |
C14H18ClN3O2 |
9.54 |
296.1/70.0a, 296.1/99.0 |
10, 15 |
30 |
| 72 |
Triallate |
2303-17-5 |
C10H16Cl3NOS |
10.24 |
304.0/86.0a, 304.0/143.0 |
18, 28 |
32 |
| 73 |
Triazophos |
24017-47-8 |
C12H16N3O3PS |
8.85 |
314.1/162.0a, 314.1/119.0 |
18, 35 |
22 |
| 74 |
Tricyclazole |
41814-78-2 |
C9H7N3S |
4.29 |
190.0/163.0a, 190.0/136.0 |
20, 25 |
10 |
| 75 |
Phoxim |
14816-18-3 |
C12H15N2O3PS |
9.64 |
299.1/129.0a, 299.1/77.0 |
13, 20 |
25 |
| 76 |
Diazinon |
333-41-5 |
C12H21N2O3PS |
9.56 |
305.1/169.0a, 305.1/153.0 |
22, 20 |
20 |
| 77 |
Deltamethrin |
52918-63-5 |
C22H19Br2NO3 |
10.47 |
523.0/281.0a, 523.0/506.0 |
16, 10 |
10 |
| 78 |
Sedaxane |
874967-67-6 |
C18H19F2N3O |
8.84 |
332.2/159.1a, 332.2/292.2 |
17, 15 |
40 |
|
|
Mycotoxins (16)
|
| 1 |
15-O-Acetyl-4-deoxynivalenol |
88337-96-6 |
C17H22O7 |
3.90 |
339.1/137.1a, 339.1/261.1 |
9, 10 |
50 |
| 2 |
3-Acetyldeoxynivalenol |
50722-38-8 |
C17H22O7 |
3.88 |
339.1/231.1a, 339.1/203.1 |
10, 15 |
55 |
| 3 |
Aflatoxin B1 |
1162-65-8 |
C17H12O6 |
5.34 |
313.1/285.1a, 313.1/241.1 |
30, 50 |
25 |
| 4 |
Aflatoxin B2 |
7220-81-7 |
C17H14O6 |
4.95 |
315.1/259.0a, 315.1/287.1 |
40, 35 |
25 |
| 5 |
Aflatoxin G1 |
1165-39-5 |
C17H12O7 |
4.65 |
329.1/243.1a, 329.1/311.0 |
20, 25 |
20 |
| 6 |
Aflatoxin G2 |
7241-98-7 |
C17H14O7 |
4.33 |
331.1/313.2a, 331.1/245.1 |
25, 25 |
25 |
| 7 |
Deoxynivalenol |
51481-10-8 |
C15H20O6 |
2.77 |
297.1/249.0a, 297.1/203.0 |
10, 16 |
20 |
| 8 |
Fumonisin B1 |
116355-83-0 |
C34H59NO15 |
8.21 |
722.4/334.4a,722.4/352.3 |
36, 40 |
40 |
| 9 |
Fumonisin B2 |
116355-84-1 |
C34H59NO14 |
9.33 |
706.4/336.3a, 706.4/318.3 |
36, 38 |
40 |
| 10 |
HT-2 toxin |
26934-87-2 |
C22H32O8 |
7.27 |
442.3/215.1a, 442.3/263.1 |
10, 10 |
20 |
| 11 |
Neosolaniol |
36519-25-2 |
C19H26O8 |
3.36 |
400.2/305.1a, 400.2/184.9 |
12, 20 |
10 |
| 12 |
Sterigmatocystin |
10048-13-2 |
C18H12O6 |
8.79 |
325.1/310.0a, 325.1/281.1 |
25, 26 |
22 |
| 13 |
T-2 toxin |
21259-20-1 |
C24H34O9 |
8.19 |
484.2/305.1a, 484.2/215.1 |
15, 22 |
20 |
| 14 |
Zearalenone |
17924-92-4 |
C18H22O5 |
8.63 |
319.2/283.2a, 319.2/187.1 |
10, 19 |
20 |
| 15 |
Ochratoxin A |
303-47-9 |
C20H18ClNO6 |
8.80 |
404.1/239.1a, 404.1/221.0 |
19, 39 |
20 |
| 16 |
Stachybotrylactam |
163391-76-2 |
C23H31NO4 |
9.60 |
386.3/178.2a, 386.3/150.2 |
40, 30 |
80 |
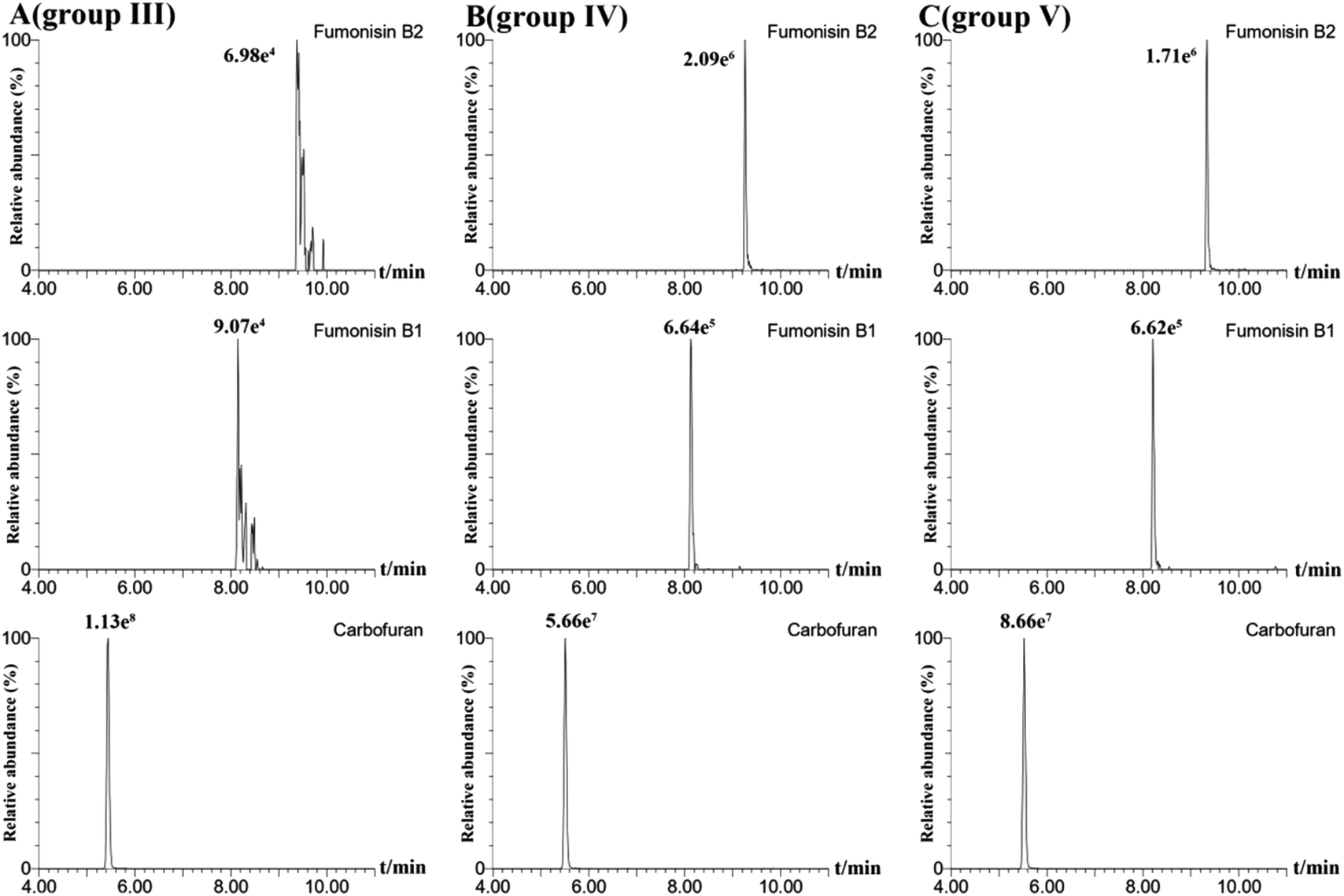
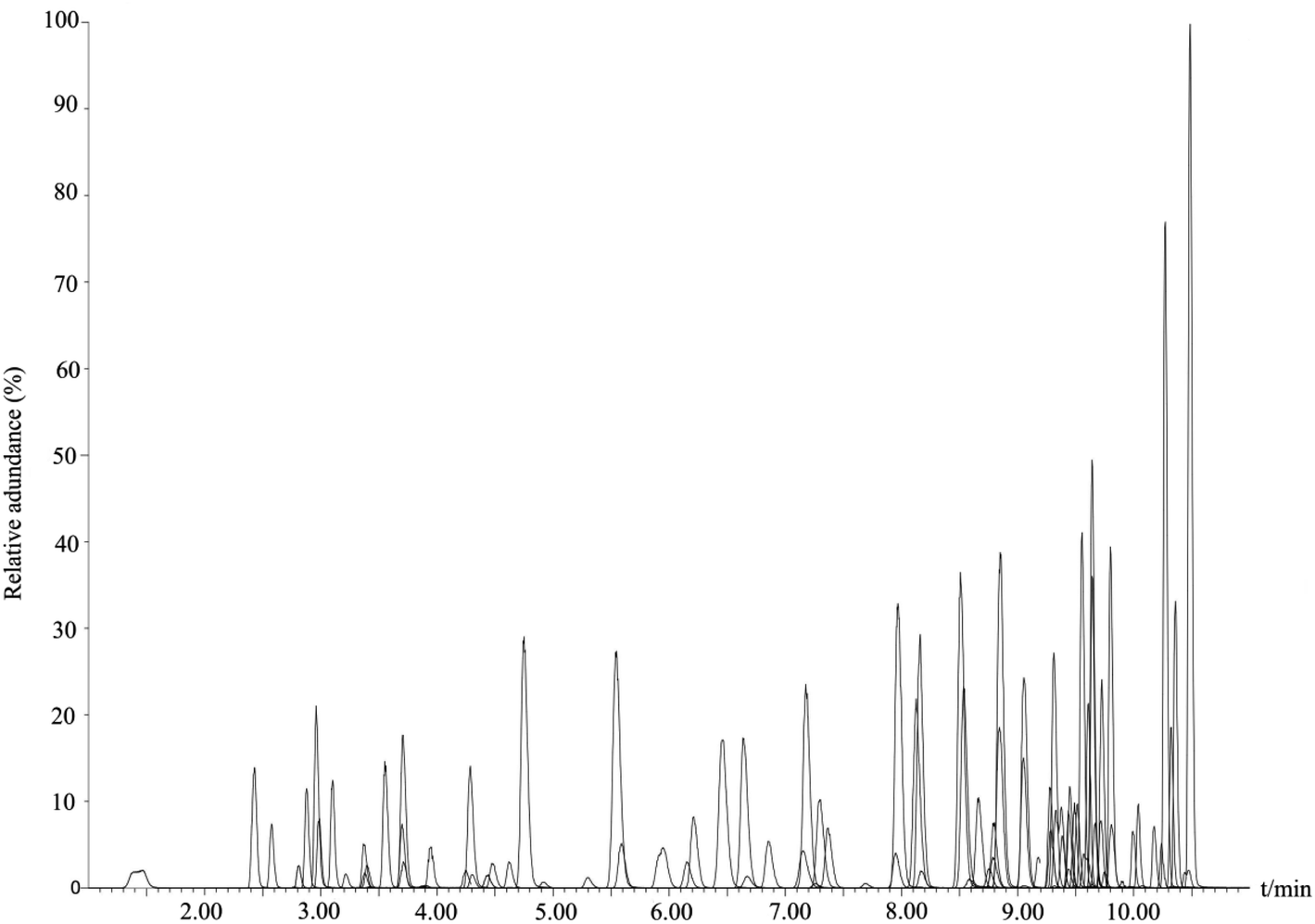
In order to obtain the optimal response and peak shapes for the target compounds, the chromatographic conditions were optimized. This study investigated the effects of five different mobile phases: (I) ACN–water, (II) MeOH–water, (III) MeOH–0.01% formic acid in water (with 2 mmol per L ammonium formate), (IV) MeOH–0.1% formic acid in water (with 2 mmol per L ammonium formate), and (V) MeOH–0.1% formic acid in water (with 5 mmol per L ammonium formate) on peak shapes and responses for 78 pesticides and 16 mycotoxins. The responses of the target compounds in group II were higher than those in group I. It is because the MeOH could give slight H+ to the analytes, which promotes hydrogenation peaks of the pesticide and mycotoxin with the response increased.30 Small amounts of formic acid and ammonium formate in the mobile phase could facilitate the formation of [M + H]+, increase the response of the compounds, and improve the peak shapes of the compounds.31 As shown in Fig. 1, most of the pesticides had an optimal response using group III as the mobile phase, however some mycotoxins, for instance, fumonisin B1 and fumonisin B2, appeared as more heterogeneous peaks. In group IV, as the acidity increased, the peak shape of the mycotoxins such as fumonisin B1 and fumonisin B2 improved significantly with the response increased, but the response of the pesticides such as carbofuran decreased. In group V, the response of pesticides such as carbofuran increased and there were no obvious changes in the response and peak shapes of the compounds. Therefore, in order to ensure that the 78 pesticides and 16 mycotoxins had better peak shapes and response, 0.1% formic acid in water (containing 5 mmol per L ammonium formate)–MeOH solution (group V) was selected as the mobile phase. The total ion chromatogram of the 78 pesticides and 16 mycotoxins in the mixed standard solution (100 μg mL−1) is shown in Fig. 2.
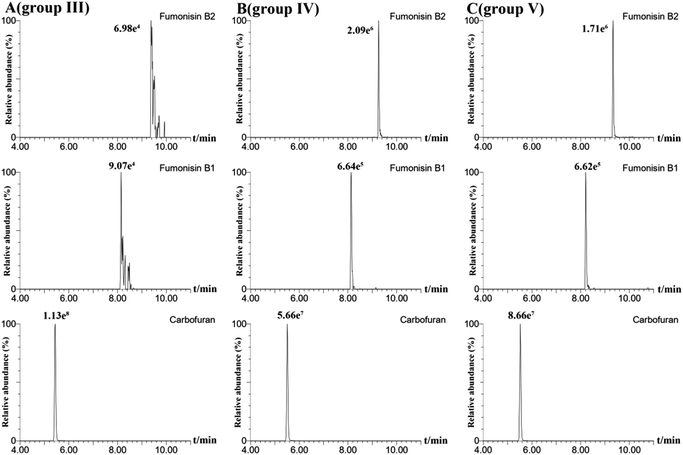 |
| | Fig. 1 Effect of addition of different levels of formic acid and ammonium formate on carbofuran, fumonisin B2 and fumonisin B1: 0.01% formic acid and 2 mmol ammonium formate (A); 0.1% formic acid and 2 mmol ammonium formate (B); 0.1% formic acid and 5 mmol ammonium formate (C). | |
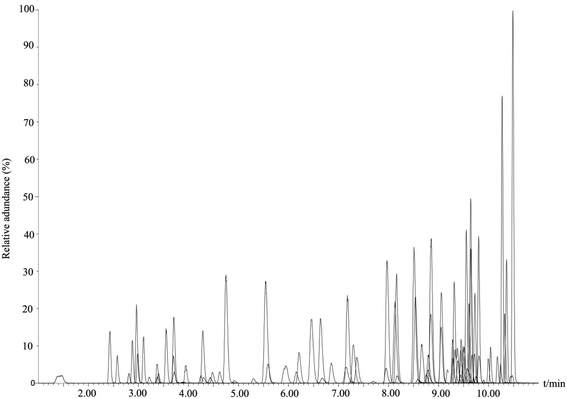 |
| | Fig. 2 TIC for 78 pesticides and 16 mycotoxins. | |
3.2. Optimization of pretreatment conditions
3.2.1. Optimization of hydration volume.
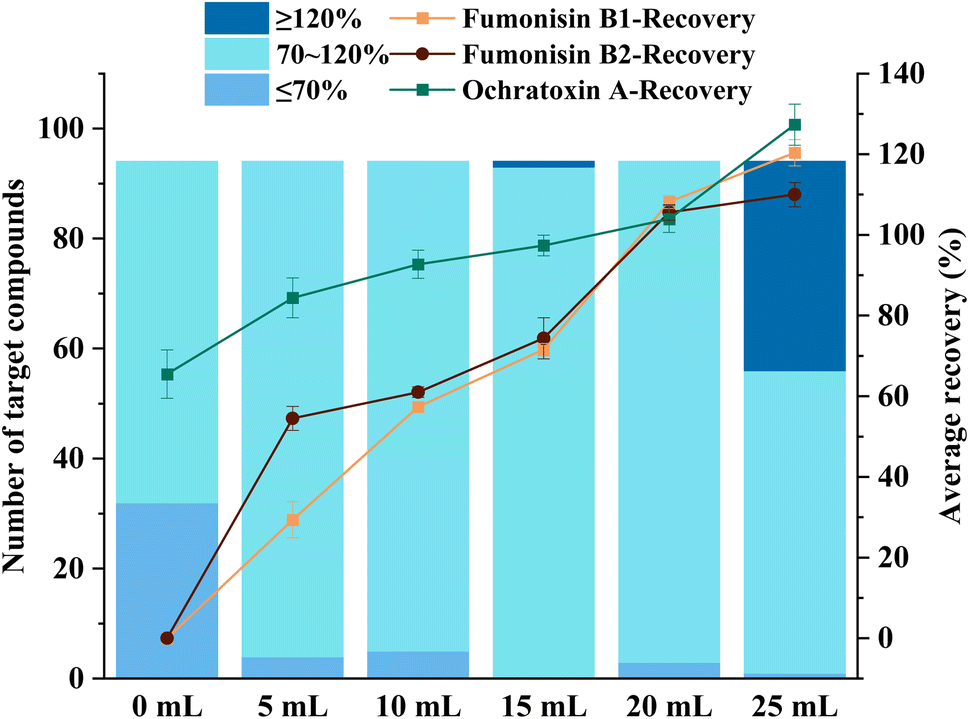
An appropriate amount of water added to the dried sample could improve the mass transfer efficiency between the sample and the extraction solvent, increasing the extraction efficiency of the target compounds.32,33 Therefore, a certain amount of water was required to ensure that the target compounds could be adequately extracted from the tsampa sample. In this study, the effects of 0 mL, 5 mL, 10 mL, 15 mL, 20 mL, and 25 mL of water on the recovery of target compounds at the spiked level of 40 μg kg−1 were investigated. The results showed that the recovery of fumonisin B1, fumonisin B2, and ochratoxin A increased with the addition of water (Fig. 3). This is because they have hydroxyl groups, which are easily soluble in water. When the water was added up to 15 mL, the number of target compounds meeting the criterion (recovery between 70 and 120%) reached the highest. When the water was greater than 15 mL, the number of target compounds meeting the criterion decreased. When the water was 25 mL, it appeared that the recovery of 40% of the target compounds exceeded 120%. Therefore, 15 mL water was confirmed for the subsequent experiments.
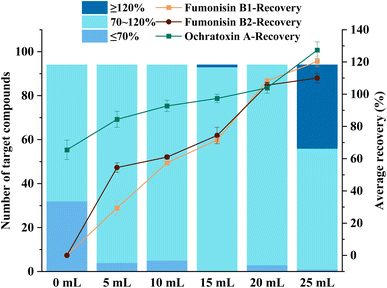 |
| | Fig. 3 Effect of different hydration volumes on the recovery of pesticides and mycotoxins (n = 3). | |
3.2.2. Optimisation of extraction solvents.
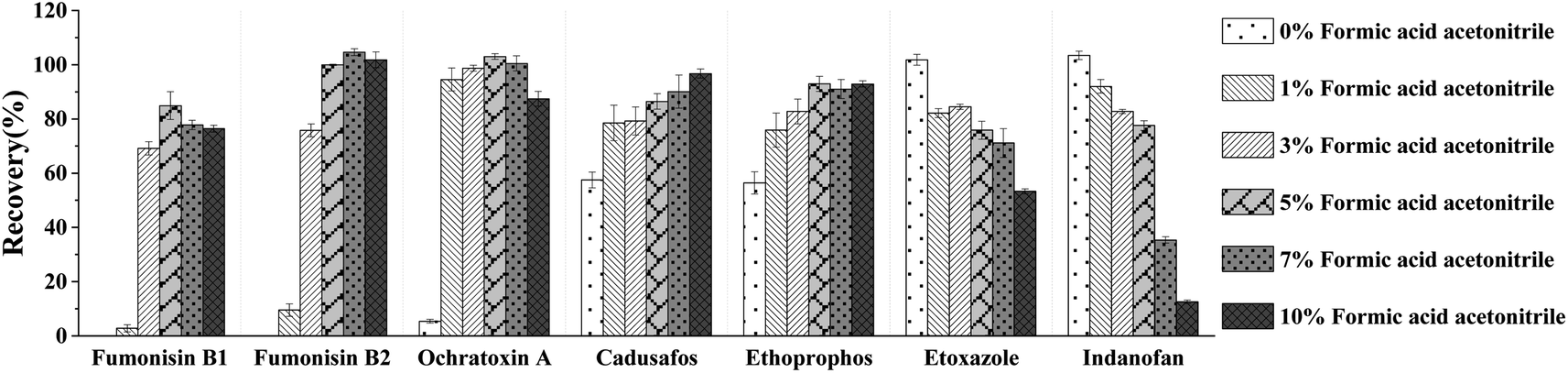
ACN has been widely used in the extraction of pesticides and mycotoxins. Compared with MeOH, ACN could improve the recovery of target compounds and effectively reduce the influence of impurities including fat, protein, and pigments during the extraction process.34 For some target compounds, fumonisin B1, fumonisin B2, ochratoxin A, ethoprophos and cadusafos were unstable under an alkaline environment resulting in low-extraction efficiency, so a suitable amount of acid was required to adjust the pH of the extraction solvents to improve the stability and extraction efficiency. In this study, we investigated the extraction efficiency of target compounds spiked at 40 μg kg−1 in six groups of ACN extraction solvents with varying levels of acidity. The solvents tested were ACN, 1% formic acid ACN, 3% formic acid ACN, 5% formic acid ACN, 7% formic acid ACN, and 10% formic acid ACN. The results showed that the number of target compounds with recovery rates in the range of 70–120% tended to increase and then decrease as the formic acid content increased (Fig. 4). When using ACN as the extraction solvent, the recoveries for fumonisin B1, fumonisin B2, and ochratoxin A were all low. However, when using 5% formic acid ACN as the extraction solvent, the recoveries for fumonisin B1 and ochratoxin A were optimal, and the number of target compounds with recovery in the range of 70–120% was the highest. For some acid-sensitive target compounds, such as cadusafos and ethoprophos, the recovery showed an upward trend with increasing acidity. However, for some acid-unstable target compounds, such as etoxazole and indanofan, the recovery showed an opposite trend with the increasing acidity. Therefore, 5% formic acid ACN was selected as the extraction solvent for subsequent optimization.
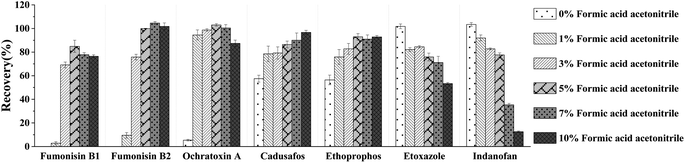 |
| | Fig. 4 Effect of ACN extraction solvents with different formic acid contents on the recovery of pesticides and mycotoxins (n = 3). | |
3.2.3. Optimization of extraction salt.
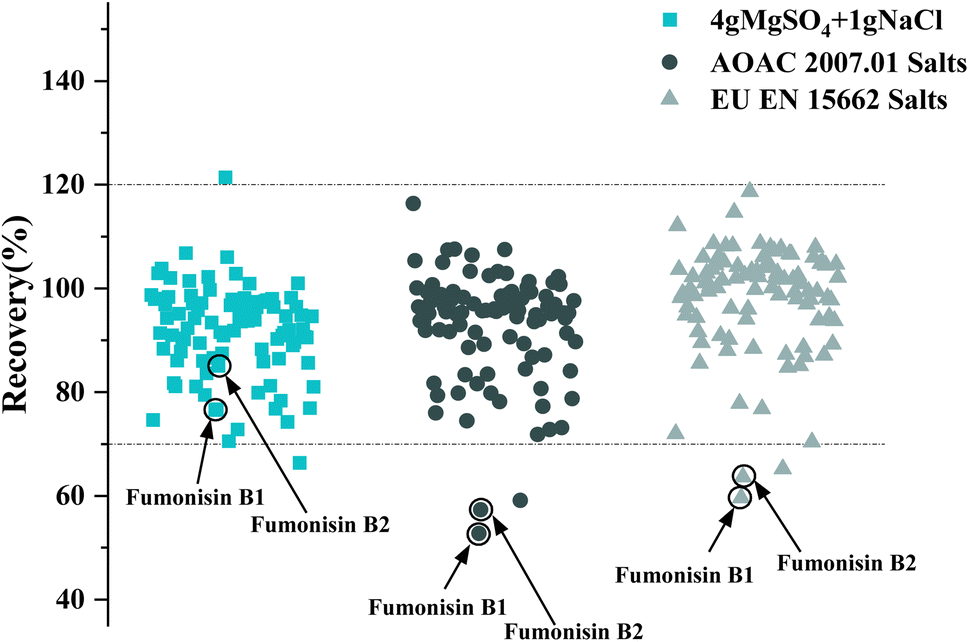
In this study, the extraction effects of three different extraction salts of QuEChERS (4 g MgSO4 + 1 g NaCl), AOAC 2007.01 (6 g MgSO4 + 1.5 g NaOAc),35 and EU EN 15662 (4 g MgSO4 + 1 g NaCl + 1 g sodium citrate + 0.5 g disodium citrate)32 were investigated on pesticides and mycotoxins spiked at 40 μg kg−1. The results showed that the number of the target compounds with recovery in the range of 70–120% were similar for the three buffer salts. Of these, these buffer salts have less effect on pesticides, however, the variations of fumonisin B1 and fumonisin B2 were more pronounced (Fig. 5). Compared to 4 g MgSO4 + 1 g NaCl, the recoveries of fumonisin B1 and B2 decreased from 76.8% to 52.8% and 85.1% to 57.3%, respectively, as the pH of the solution increased from 1.63 to 4.49 when using the AOAC 2007.01 extraction salt. Similarly, the recoveries of fumonisin B1 and B2 decreased from 76.8% to 59.6% and 85.1% to 63.6%, respectively, as the pH of the solution increased from 1.63 to 2.56, after the addition of the EU EN 15662 extraction salt. It was indicated that the addition of three different extraction salts in tsampa results in different pH ranges of the extraction, among which the traditional extraction salts could provide the lowest pH environment. Acid-sensitive compounds such as fumonisin B1 and fumonisin B2 could be fully extracted in acidic environments, therefore, 4 g MgSO4 and 1 g NaCl were finally selected as the extraction salts.
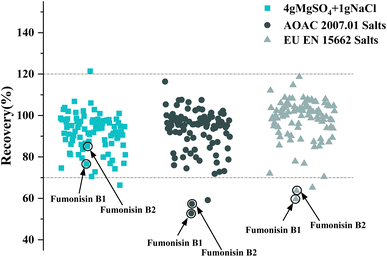 |
| | Fig. 5 Effect of different extraction salts on the recovery of pesticides and mycotoxins (n = 3). | |
3.2.4. Optimization of purification materials.
The composition of tsampa is more complex, containing rich carbohydrates, proteins, lipids, and other components. Therefore, the co-extracts will be extracted together with target compounds from tsampa, which can contaminate the chromatographic column and reduce the sensitivity of the instrument. PSA, C18, and GCB were commonly used as adsorbents. PSA is a weak anion exchanger adsorbent, which is capable of removing fatty acids, polar pigments, and other hydrogen-bonding compounds. C18 could effectively remove lipids, sterols and other nonpolar compounds. GCB could effectively remove chlorophyll and other pigments, but it has strong adsorption on compounds with a planar structure.
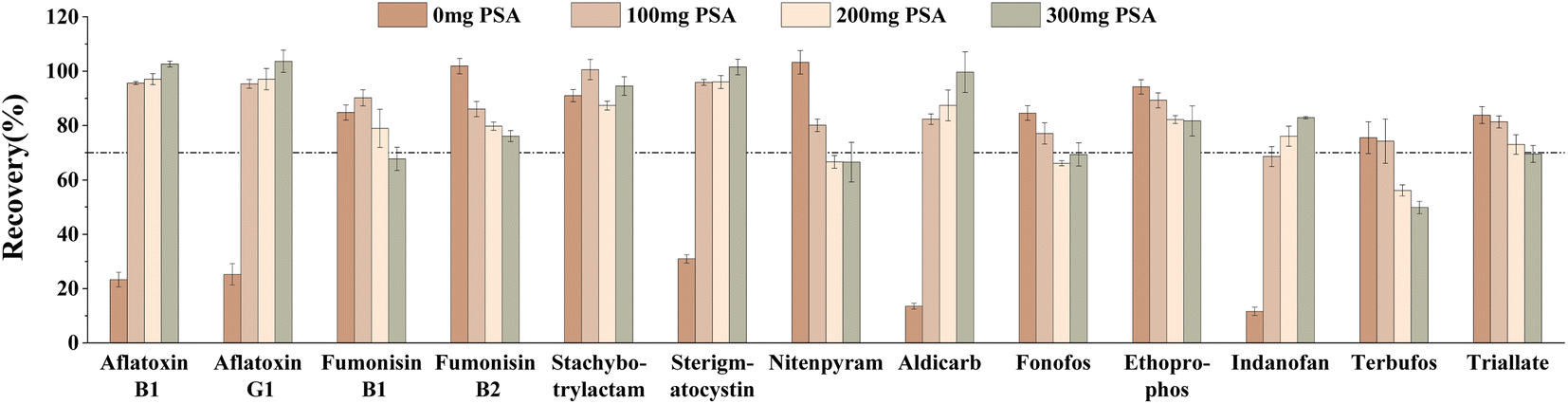
In this study, the purification effects of different amounts of PSA, C18, and GCB on target compounds spiked at 40 μg kg−1 were investigated. Firstly, the effects of different contents of PSA (0 mg, 100 mg, 200 mg, 300 mg) on the target compounds were compared. The results showed that the recoveries of several target compounds, including aflatoxin B1, aflatoxin G1, sterigmatocystin, aldicarb and indanofan, were less than 40% when the PSA was 0 mg (Fig. 6). However, as the amount of PSA increased, the recoveries gradually improved. Conversely, for other target compounds, such as fumonisin B2, nitenpyram, ethoprophos, terbufos, and triallate, the recoveries decreased with the increase in PSA amount. When the PSA was 100 mg, the number of target compounds meeting the recovery range (70–120%) was maximum and the recoveries of fumonisin B1 and stachybotrylactam were optimal. Therefore, the final PSA was determined to be 100 mg.
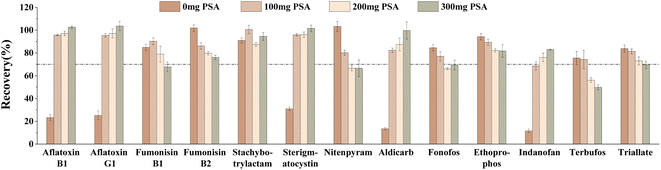 |
| | Fig. 6 Effect of different contents of PSA on the recovery of pesticides and mycotoxins (n = 3). | |
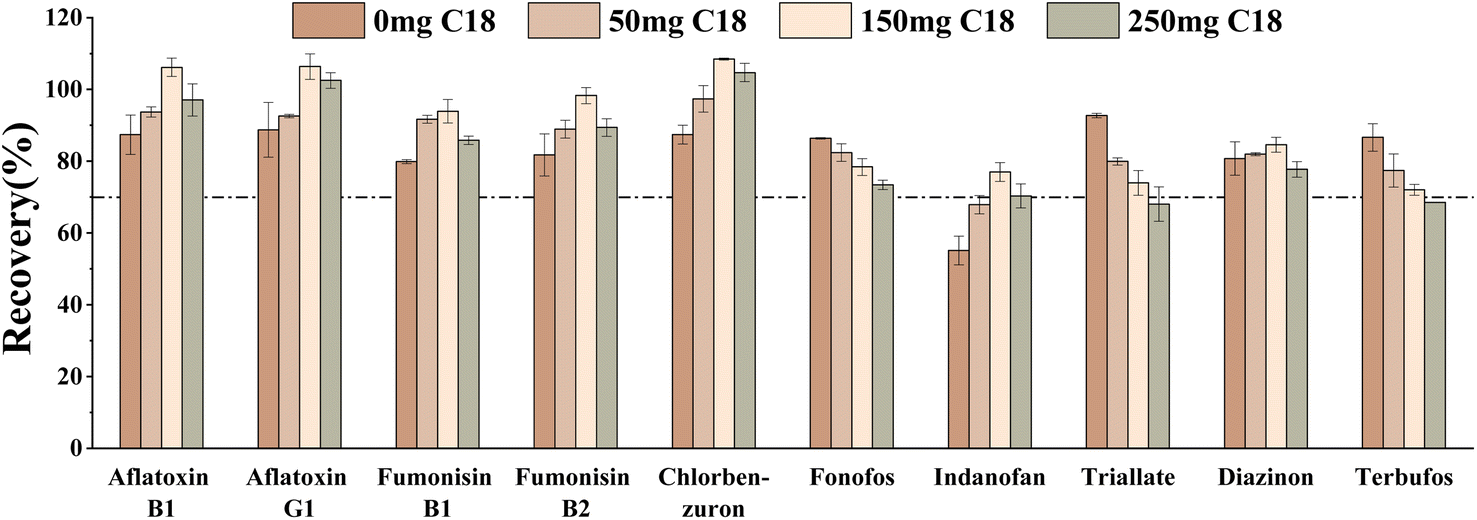
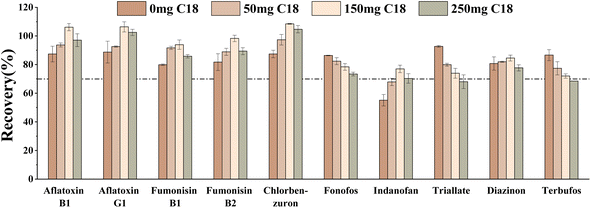
Secondly, different amounts of C18 (0 mg, 50 mg, 150 mg, and 250 mg) on the target compounds were studied. The results showed that the recovery of fonofos, terbufos, and triallate decreased gradually with increasing C18 amount (Fig. 7). When the C18 was 150 mg, the recoveries of aflatoxin B1, aflatoxin B2, fumonisin B1, fumonisin B2, indanofan, and chlorbenzuron were optimal with the largest number of target compounds meeting the criterion (70–120%). Therefore, 150 mg C18 was determined to be the optimal condition.
 |
| | Fig. 7 Effect of different contents of C18 on the recovery of pesticides and mycotoxins (n = 3). | |
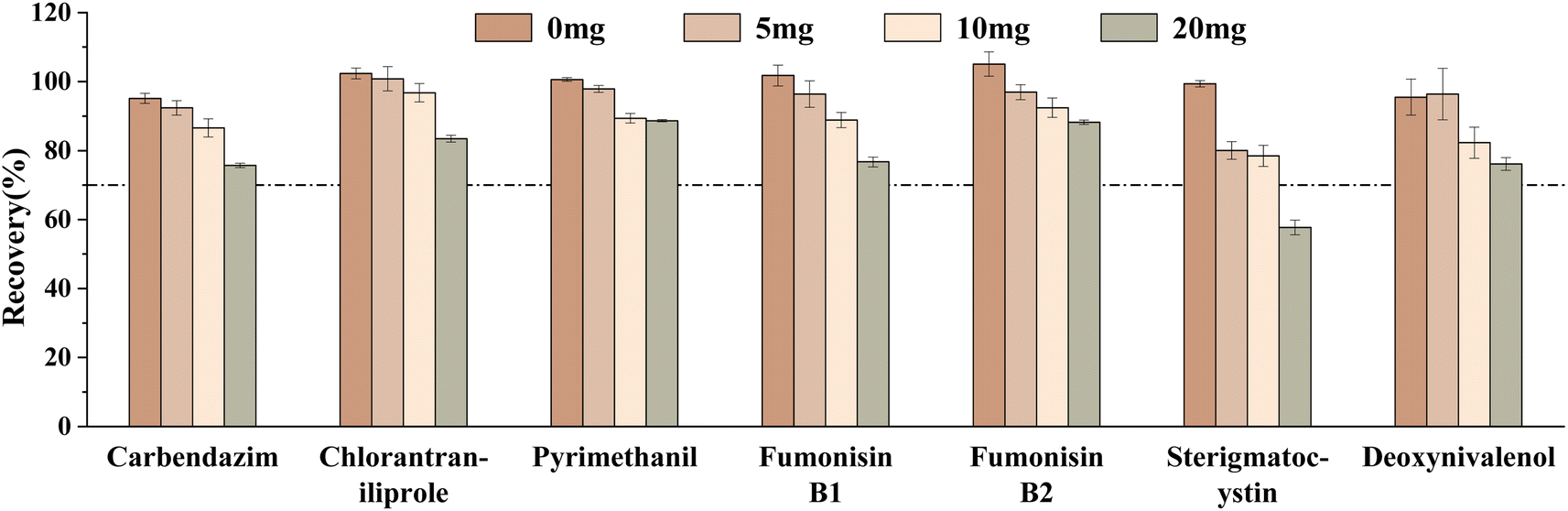
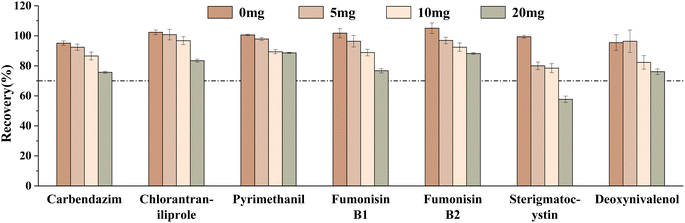
Finally, different amounts of GCB (0 mg, 5 mg, 10 mg, and 20 mg) on the target compounds were investigated. The results showed that the recoveries of carbendazim, chlorantraniliprole, pyrimethanil, and sterigmatocystin gradually decreased with the increase of the GCB amount (Fig. 8). This was due to the planar-structured compounds which were easily adsorbed by GCB. It was found that the number of target compounds with recovery ranging from 70 to 120% was the greatest without GCB. Therefore, 100 mg PSA and 150 mg C18 were finally selected for adsorption.
 |
| | Fig. 8 Effect of different contents of GCB on the recovery of pesticides and mycotoxins (n = 3). | |
3.3. Validation of the approach
3.3.1. Matrix effect.
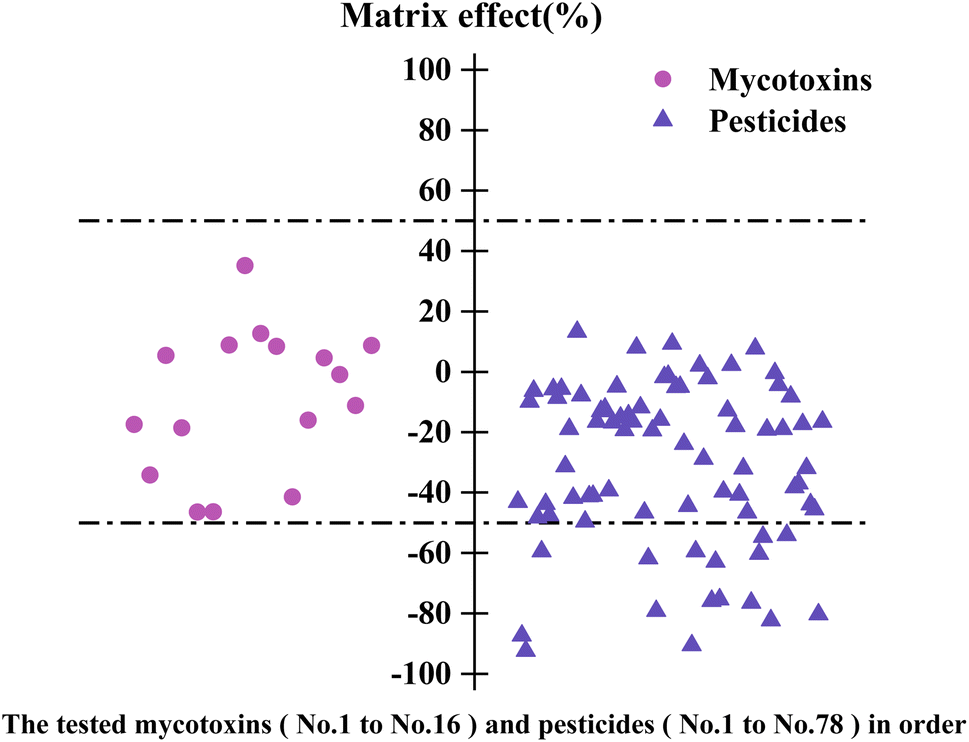
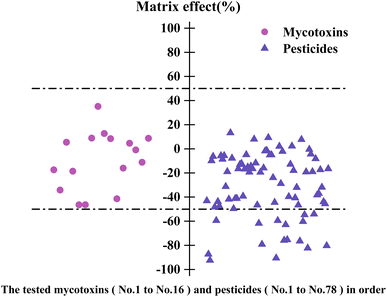
During sample pre-treatment, the co-extract in the matrix may affect the ionization of the target compounds, which results in matrix interference and affects the detection and quantification of the target, thus affecting the precision and accuracy of the method.36 The matrix effect (ME) of tsampa was assessed according to the formula: ME (%) = [(KA/KB) − 1] × 100, where KA: slope of the matrix-matched standard curve; KB: slope of the solvent standard curve. If the ME is positive, it indicates ion signal enhancement, whereas if the ME is negative, it indicates ion signal suppression. According to the ME value, the ME could be classified as weak (|ME| ≤ 20%), moderate (20% < |ME| ≤ 50%), and strong (|ME| > 50%).37 In this study, the matrix-matched standard curve (0.4, 1, 2, 4, 10, 20, 40, 100, 200 μg kg−1) and solvent standard curve (0.2, 0.5, 1, 2, 5, 10, 20, 50, 100 μg L−1) were established to calculated ME. The results showed that 86.2% of the target compounds exhibited matrix inhibition in the tsampa (Fig. 9), which may be attributed to the components in the matrix that reduced the efficiency of charged droplets or reduced the number of ions formed in the LC-MS/MS.38 Among the 94 target compounds, 53.2% of them exhibited weak ME and 29.8% exhibited moderate ME, indicating that the method was highly resistant to interference with tsampa.
 |
| | Fig. 9 ME of 78 pesticides and 16 mycotoxins in tsampa. | |
3.3.2. Linearity, limits of detection (LODs) and limits of quantification (LOQs).
The blank matrix extracts of tsampa were prepared according to the pretreatment described in Section 1.3 which were formulated into a series of matrix-matched standard solutions at a series of concentrations. The matrix-matched standard curves were plotted with the compound concentration as the horizontal axis and the peak area as the vertical axis. The limit of detection (LOD) was determined as the concentration of the compound that produced a 3-fold signal-to-noise ratio, while the limit of quantification (LOQ) was determined as the concentration of the compound that produced a 10-fold signal-to-noise ratio. In Table 2, the results show that all the target compounds exhibited good linear correlation with determination coefficient (R2) greater than 0.9990. The LODs and LOQs were 0.10–3.00 μg kg−1 and 0.40–10.00 μg kg−1, respectively, which were lower than the MRL of pesticide residues and mycotoxin in the Regulation on Contaminants in Foods (EU) No. 2023/915 (EN 2023/915).39
Table 2 Linear range, R2, LOD, LOQ, recovery and RSD of pesticides and mycotoxins in tsampa
| No. |
Compound |
Linear range (μg L−1) |
R
2
|
LOD (μg kg−1) |
LOQ (μg kg−1) |
1 × LOQ |
2 × LOQ |
10 × LOQ |
| Recovery (%) |
RSD (%) |
Recovery (%) |
RSD (%) |
Recovery (%) |
RSD (%) |
|
Pesticides (78)
|
| 1 |
Acetamiprid |
0.2–100 |
0.9994 |
0.12 |
0.40 |
111.2 |
0.8 |
108.5 |
0.6 |
111.2 |
0.5 |
| 2 |
Aldicarb |
2–100 |
0.9990 |
1.10 |
4.00 |
103.2 |
2.4 |
105.9 |
1.7 |
97.6 |
5.2 |
| 3 |
Aldicarb sulfone |
2–100 |
0.9993 |
1.20 |
4.00 |
94.1 |
2.6 |
83.3 |
4.2 |
114.8 |
1.4 |
| 4 |
Aldicarb sulfoxide |
0.2–100 |
0.9996 |
0.10 |
0.40 |
88.4 |
1.6 |
106.1 |
0.8 |
101.8 |
2.3 |
| 5 |
Azoxystrobin |
0.2–100 |
0.9991 |
0.10 |
0.40 |
107.2 |
1.4 |
111.5 |
0.6 |
107.6 |
0.9 |
| 6 |
Bitertanol |
0.5–100 |
0.9991 |
0.25 |
1.00 |
88.3 |
6.1 |
96.4 |
5.3 |
100.6 |
0.7 |
| 7 |
Butralin |
0.2–100 |
0.9995 |
0.10 |
0.40 |
103.7 |
0.5 |
109.0 |
1.9 |
99.7 |
5.8 |
| 8 |
Cadusafos |
0.2–100 |
0.9994 |
0.12 |
0.40 |
95.9 |
5.1 |
107.5 |
1.1 |
95.8 |
5.4 |
| 9 |
Carbendazim |
0.2–100 |
0.9997 |
0.12 |
0.40 |
107.2 |
0.6 |
102.7 |
3.5 |
94.8 |
3.1 |
| 10 |
Carbofuran |
0.2–100 |
0.9990 |
0.10 |
0.40 |
103.8 |
0.9 |
114.6 |
1.1 |
111.5 |
1.4 |
| 11 |
Carbofuran-3-hydroxy |
0.5–100 |
0.9997 |
0.12 |
0.40 |
106.3 |
1.3 |
90.3 |
6.2 |
110.8 |
0.7 |
| 12 |
Chlorantraniliprole |
1–100 |
0.9993 |
0.60 |
2.00 |
85.9 |
3.2 |
105.7 |
3.8 |
88.4 |
1.6 |
| 13 |
Chlorbenzuron |
0.2–100 |
0.9994 |
0.10 |
0.40 |
109.1 |
0.6 |
101.4 |
2.2 |
101.0 |
1.8 |
| 14 |
Chlordimeform |
2–100 |
0.9993 |
1.15 |
4.00 |
84.8 |
5.0 |
104.6 |
4.1 |
106.5 |
2.9 |
| 15 |
Chlorfenvinphos |
0.5–100 |
0.9993 |
0.30 |
1.00 |
95.4 |
3.8 |
86.6 |
11.7 |
105.5 |
0.5 |
| 16 |
Chlorsulfuron |
0.2–100 |
0.9993 |
0.12 |
0.40 |
100.7 |
4.0 |
103.4 |
2.3 |
108.5 |
1.5 |
| 17 |
Chlortoluron |
0.2–100 |
0.9997 |
0.12 |
0.40 |
108.8 |
1.2 |
106.5 |
1.1 |
108.7 |
1.3 |
| 18 |
Clothianidin |
1–100 |
0.9998 |
0.60 |
2.00 |
86.7 |
11.4 |
82.0 |
2.4 |
87.7 |
2.5 |
| 19 |
Cymoxanil |
0.2–100 |
0.9996 |
0.10 |
0.40 |
109.5 |
0.8 |
103.2 |
3.2 |
111.7 |
1.8 |
| 20 |
Difenoconazole |
1–100 |
0.9992 |
0.60 |
2.00 |
91.4 |
5.6 |
102.5 |
1.3 |
90.5 |
5.7 |
| 21 |
Diflubenzuron |
0.2–100 |
0.9995 |
0.10 |
0.40 |
113.3 |
4.1 |
103.1 |
4.7 |
107.8 |
1.5 |
| 22 |
Ethametsulfuron |
0.5–100 |
0.9998 |
0.30 |
1.00 |
100.4 |
6.6 |
87.0 |
4.1 |
97.2 |
2.2 |
| 23 |
Ethoprophos |
0.2–100 |
0.9993 |
0.10 |
0.40 |
79.9 |
4.6 |
102.3 |
5.1 |
106.2 |
0.7 |
| 24 |
Etoxazole |
0.2–100 |
0.9998 |
0.10 |
0.40 |
91.1 |
2.8 |
86.4 |
7.9 |
85.0 |
4.5 |
| 25 |
Fenamiphos |
0.2–100 |
0.9993 |
0.10 |
0.40 |
112.6 |
1.0 |
99.5 |
3.0 |
100.1 |
1.7 |
| 26 |
Fenamiphos sulfone |
0.2–100 |
0.9993 |
0.12 |
0.40 |
99.3 |
3.5 |
111.1 |
0.7 |
107.2 |
1.1 |
| 27 |
Fenamiphos sulfoxide |
0.2–100 |
0.9996 |
0.10 |
0.40 |
99.0 |
3.1 |
105.5 |
2.1 |
103.6 |
1.8 |
| 28 |
Fenbuconazole |
0.5–50 |
0.9993 |
0.30 |
1.00 |
100.3 |
5.2 |
98.7 |
3.5 |
99.8 |
2.2 |
| 29 |
Fenoxanil |
1–100 |
0.9991 |
0.60 |
2.00 |
100.5 |
5.4 |
94.9 |
0.5 |
91.9 |
2.9 |
| 30 |
Fonofos |
0.5–50 |
0.9999 |
0.30 |
1.00 |
93.9 |
10.8 |
89.5 |
2.2 |
88.2 |
3.0 |
| 31 |
Heptenophos |
0.2–100 |
0.9994 |
0.10 |
0.40 |
111.6 |
0.8 |
102.3 |
1.8 |
104.1 |
1.0 |
| 32 |
Hexaconazole |
0.2–100 |
0.9993 |
0.10 |
0.40 |
84.5 |
5.8 |
102.9 |
4.0 |
106.8 |
2.2 |
| 33 |
Hexaflumuron |
5–100 |
0.9992 |
3.00 |
10.00 |
104.8 |
0.6 |
80.4 |
5.8 |
80.1 |
9.8 |
| 34 |
Imidacloprid |
2–100 |
0.9998 |
1.25 |
4.00 |
101.0 |
4.9 |
92.5 |
1.8 |
106.6 |
0.8 |
| 35 |
Indanofan |
0.2–100 |
0.9991 |
0.12 |
0.40 |
73.7 |
2.6 |
83.1 |
4.0 |
73.0 |
3.2 |
| 36 |
Isofenphos-methyl |
1–100 |
0.9992 |
0.50 |
2.00 |
87.3 |
4.1 |
93.0 |
8.0 |
75.2 |
7.9 |
| 37 |
Isoprothiolane |
0.2–100 |
0.9995 |
0.10 |
0.40 |
85.6 |
1.4 |
103.1 |
4.0 |
105.9 |
1.6 |
| 38 |
Malathion |
0.2–100 |
0.9994 |
0.12 |
0.40 |
99.0 |
2.9 |
109.7 |
1.4 |
103.1 |
2.0 |
| 39 |
Metalaxyl |
0.2–100 |
0.9993 |
0.10 |
0.40 |
99.9 |
2.3 |
103.7 |
1.3 |
111.7 |
0.8 |
| 40 |
Methamidophos |
0.5–100 |
0.9993 |
0.30 |
1.00 |
79.0 |
1.9 |
74.6 |
2.8 |
79.9 |
2.6 |
| 41 |
Methidathion |
0.2–100 |
0.9995 |
0.10 |
0.40 |
99.4 |
5.1 |
108.2 |
1.7 |
105.5 |
1.5 |
| 42 |
Methiocarb |
0.2–100 |
0.9995 |
0.12 |
0.40 |
105.2 |
1.8 |
108.6 |
1.0 |
108.9 |
1.0 |
| 43 |
Methiocarb sulfone |
2–100 |
0.9993 |
1.00 |
4.00 |
90.9 |
0.8 |
93.2 |
2.3 |
95.5 |
3.1 |
| 44 |
Methiocarb sulfoxide |
0.2–100 |
0.9996 |
0.10 |
0.40 |
104.0 |
0.5 |
108.6 |
1.5 |
100.3 |
2.3 |
| 45 |
Methomyl |
1–100 |
0.9997 |
0.50 |
2.00 |
73.2 |
0.2 |
94.9 |
4.7 |
83.2 |
2.0 |
| 46 |
Metrafenone |
0.2–100 |
0.9993 |
0.10 |
0.40 |
104.8 |
2.2 |
100.9 |
2.1 |
101.9 |
2.6 |
| 47 |
Metsulfuron-methyl |
0.2–100 |
0.9991 |
0.10 |
0.40 |
100.8 |
6.9 |
110.5 |
0.6 |
109.4 |
0.2 |
| 48 |
Mevinphos |
0.5–100 |
0.9993 |
0.25 |
1.00 |
100.6 |
4.9 |
98.9 |
2.0 |
102.5 |
4.1 |
| 49 |
Myclobutanil |
0.5–100 |
0.9994 |
0.30 |
1.00 |
99.9 |
2.3 |
94.4 |
1.1 |
105.2 |
1.1 |
| 50 |
Monocrotophos |
0.5–100 |
0.9993 |
0.30 |
1.00 |
94.8 |
6.5 |
85.9 |
4.9 |
97.4 |
1.0 |
| 51 |
Nitenpyram |
1–50 |
0.9995 |
0.50 |
2.00 |
84.3 |
5.2 |
99.9 |
1.1 |
74.3 |
0.7 |
| 52 |
Omethoate |
1–100 |
0.9995 |
0.55 |
2.00 |
77.3 |
2.3 |
104.5 |
4.4 |
83.5 |
8.4 |
| 53 |
Phorate |
1–100 |
0.9997 |
0.50 |
2.00 |
112.1 |
1.2 |
106.8 |
2.5 |
79.0 |
7.2 |
| 54 |
Phorate sulfone |
0.2–100 |
0.9993 |
0.12 |
0.40 |
85.8 |
3.1 |
98.6 |
4.9 |
111.6 |
2.4 |
| 55 |
Phorate sulfoxide |
0.2–100 |
0.9991 |
0.10 |
0.40 |
98.0 |
2.0 |
102.2 |
1.1 |
110.8 |
0.9 |
| 56 |
Pirimicarb |
0.2–100 |
0.9998 |
0.12 |
0.40 |
94.5 |
2.2 |
108.3 |
1.7 |
106.8 |
0.9 |
| 57 |
Profenofos |
0.5–100 |
0.9993 |
0.30 |
1.00 |
93.4 |
4.6 |
89.5 |
5.8 |
95.2 |
3.9 |
| 58 |
Propiconazole |
0.5–100 |
0.9990 |
0.30 |
1.00 |
110.5 |
11.2 |
87.3 |
5.1 |
95.7 |
2.1 |
| 59 |
Pyraclostrobin |
0.2–100 |
0.9997 |
0.10 |
0.40 |
97.6 |
7.3 |
104.2 |
3.4 |
107.0 |
1.7 |
| 60 |
Pyridaben |
0.2–100 |
0.9993 |
0.10 |
0.40 |
74.0 |
7.5 |
91.6 |
2.9 |
82.6 |
6.5 |
| 61 |
Pyrimethanil |
0.2–100 |
0.9997 |
0.10 |
0.40 |
92.6 |
6.4 |
100.3 |
4.5 |
102.5 |
3.6 |
| 62 |
Quinoxyfen |
0.5–100 |
0.9995 |
0.25 |
1.00 |
100.0 |
4.4 |
73.7 |
4.3 |
97.4 |
4.9 |
| 63 |
Spirodiclofen |
1–50 |
0.9993 |
0.55 |
2.00 |
84.7 |
5.2 |
86.2 |
1.5 |
77.8 |
3.1 |
| 64 |
Tebuconazole |
0.5–100 |
0.9994 |
0.25 |
1.00 |
98.8 |
3.3 |
102.4 |
5.2 |
105.5 |
2.3 |
| 65 |
Terbufos |
2–100 |
0.9995 |
1.25 |
4.00 |
81.6 |
1.9 |
92.2 |
2.3 |
99.2 |
3.5 |
| 66 |
Terbufos-sulfone |
0.5–100 |
0.9994 |
0.30 |
1.00 |
95.4 |
2.2 |
90.6 |
0.3 |
91.9 |
5.7 |
| 67 |
Terbufos-sulfoxide |
0.2–100 |
0.9994 |
0.10 |
0.40 |
99.1 |
3.7 |
111.2 |
1.7 |
113.4 |
1.0 |
| 68 |
Tetrachlorvinphos |
1–100 |
0.9994 |
0.60 |
2.00 |
95.3 |
1.6 |
94.8 |
1.4 |
92.7 |
3.1 |
| 69 |
Thiamethoxam |
0.5–100 |
0.9993 |
0.30 |
1.00 |
102.1 |
3.7 |
98.8 |
2.7 |
94.8 |
4.3 |
| 70 |
Triadimefon |
0.5–100 |
0.9998 |
0.25 |
1.00 |
95.5 |
0.9 |
106.4 |
2.3 |
104.7 |
2.6 |
| 71 |
Triadimenol |
0.5–100 |
0.9991 |
0.25 |
1.00 |
104.6 |
0.9 |
97.0 |
2.0 |
100.4 |
3.2 |
| 72 |
Triallate |
0.5–100 |
0.9992 |
0.30 |
1.00 |
75.2 |
3.2 |
82.9 |
11.6 |
80.8 |
5.6 |
| 73 |
Triazophos |
0.2–100 |
0.9992 |
0.10 |
0.40 |
105.6 |
4.2 |
114.9 |
0.7 |
104.6 |
1.4 |
| 74 |
Tricyclazole |
0.5–100 |
0.9995 |
0.25 |
1.00 |
109.5 |
0.5 |
94.9 |
3.2 |
95.3 |
4.4 |
| 75 |
Phoxim |
0.2–100 |
0.9995 |
0.30 |
0.40 |
105.4 |
2.8 |
104.0 |
7.1 |
101.2 |
3.4 |
| 76 |
Diazinon |
0.2–100 |
0.9994 |
0.12 |
0.40 |
88.5 |
3.7 |
79.4 |
1.5 |
93.1 |
1.9 |
| 77 |
Deltamethrin |
5–100 |
0.9995 |
2.60 |
10.00 |
81.5 |
4.2 |
78.8 |
0.7 |
96.2 |
4.0 |
| 78 |
Sedaxane |
0.2–100 |
0.9993 |
0.10 |
0.40 |
102.7 |
2.7 |
105.3 |
1.4 |
106.6 |
2.2 |
|
|
Mycotoxins (16)
|
| 1 |
15-O-Acetyl-4-deoxynivalenol |
5–100 |
0.9990 |
2.80 |
10.00 |
95.9 |
2.3 |
79.3 |
3.2 |
96.6 |
2.3 |
| 2 |
3-Acetyldeoxynivalenol |
5–100 |
0.9994 |
2.60 |
10.00 |
86.9 |
7.1 |
99.4 |
2.2 |
100.2 |
2.3 |
| 3 |
Aflatoxin B1 |
0.5–100 |
0.9993 |
0.25 |
1.00 |
102.8 |
4.0 |
85.8 |
5.7 |
99.2 |
1.0 |
| 4 |
Aflatoxin B2 |
1–100 |
0.9994 |
0.50 |
2.00 |
107.0 |
0.6 |
101.4 |
3.6 |
89.1 |
3.0 |
| 5 |
Aflatoxin G1 |
0.2–100 |
0.9993 |
0.10 |
0.40 |
103.0 |
2.1 |
92.8 |
7.6 |
102.3 |
1.1 |
| 6 |
Aflatoxin G2 |
2–100 |
0.9998 |
1.10 |
4.00 |
98.3 |
5.1 |
103.6 |
4.9 |
105.8 |
1.1 |
| 7 |
Deoxynivalenol |
1–100 |
0.9993 |
0.50 |
2.00 |
95.9 |
5.8 |
89.1 |
4.5 |
85.9 |
2.4 |
| 8 |
Fumonisin B1 |
5–100 |
0.9991 |
2.70 |
10.00 |
101.3 |
2.7 |
74.8 |
1.0 |
90.1 |
4.3 |
| 9 |
Fumonisin B2 |
2–100 |
0.9993 |
1.10 |
4.00 |
81.6 |
6.4 |
91.3 |
2.6 |
87.5 |
5.4 |
| 10 |
HT-2 toxin |
1–100 |
0.9994 |
0.60 |
2.00 |
88.0 |
10.0 |
91.4 |
1.3 |
91.2 |
2.3 |
| 11 |
Neosolaniol |
1–100 |
0.9994 |
0.60 |
2.00 |
98.6 |
3.0 |
95.4 |
2.1 |
90.3 |
4.3 |
| 12 |
Sterigmatocystin |
0.5–100 |
0.9998 |
0.25 |
1.00 |
109.9 |
2.1 |
102.4 |
2.2 |
94.5 |
5.9 |
| 13 |
T-2 toxin |
1–100 |
0.9994 |
0.60 |
2.00 |
100.1 |
3.4 |
103.5 |
2.0 |
91.9 |
1.3 |
| 14 |
Zearalenone |
0.5–100 |
0.9991 |
0.24 |
1.00 |
95.1 |
3.9 |
101.6 |
3.2 |
87.2 |
1.7 |
| 15 |
Ochratoxin A |
0.5–100 |
0.9997 |
0.30 |
1.00 |
104.3 |
1.7 |
84.4 |
1.4 |
87.5 |
1.6 |
| 16 |
Stachybotrylactam |
1–100 |
0.9992 |
0.60 |
2.00 |
104.1 |
1.3 |
101.5 |
1.2 |
80.7 |
3.2 |
3.3.3. Recovery and precision.
In this study, the accuracy and precision of the method were assessed by analyzing blank samples spiked at 3 levels (1 × LOQ, 2 × LOQ, 10 × LOQ) according to SANTE/11312/2021.40 The results showed that the recovery of the target compounds in the tsampa spiked at 1, 2, and 10 × LOQ were 73.2–113.3%, 73.7–115.2%, and 73.0–114.8%, respectively, with the RSDs in the range of 0.2–11.7% (Table 2), the results indicate that the method has superior accuracy and precision, and could satisfy the needs of daily co-detection of pesticide residues and mycotoxins.
3.4. Real tsampa sample analysis
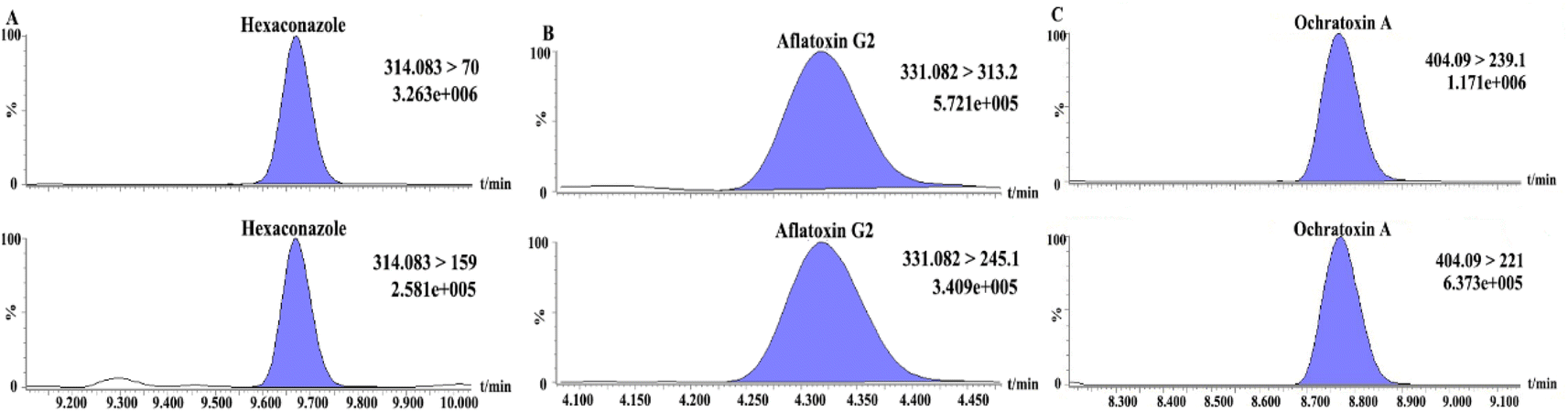
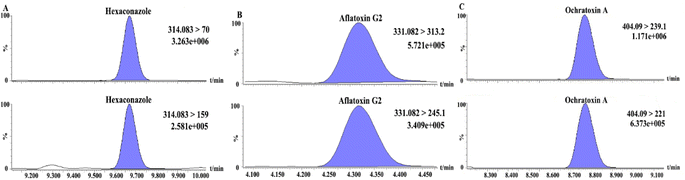
Thirty batches of tsampa samples were tested using the method developed in this study to assess the applicability of the proposed method. The results in Table 3 show that the tsampa samples contained five mycotoxins (aflatoxin G2, deoxynivalenol, HT-2 toxin, ochratoxin A, and T-2 toxin) and four pesticide residues (cadusafos, carbendazim, triadimefon, and hexaconazole), The detection rates of aflatoxin G2, cadusafos and hexaconazole were 26.32%, 36.84% and 94.74%, respectively, while the others were relatively low. The analysis revealed that 5 samples exceeded the European Union (EU) maximum residue limit (MRL) for aflatoxin G2, 1 sample exceeded the MRL for hexaconazole, and 1 sample exceeded the MRL for ochratoxin A. Additionally, 32% of the samples did not meet the qualification criteria set by EN. Therefore, attention needs to be paid to the safety of mycotoxins in tsampa and the daily monitoring of pesticides exceeding the MRL. The MRM chromatograms of pesticides and mycotoxins exceeding the MRL (hexaconazole, aflatoxin G2, and ochratoxin A) are shown in Fig. 10. The test results of all samples are shown in Table S1.†
Table 3 Results of pesticides and mycotoxins in real samples of tsampaa
| No. |
Compound |
Detected quantity |
Concentration range (μg kg−1) |
EN MRL (μg kg−1) |
Exceeding (EN) MRL quantity |
|
N.D.: not detected.
|
| 1 |
Aflatoxin G2 |
5 |
4.1–7.43 |
4 |
5 |
| 2 |
Cadusafos |
4 |
0.47–1.06 |
10 |
N.D. |
| 3 |
Carbendazim |
1 |
3.38 |
2000 |
N.D. |
| 4 |
Deoxynivalenol |
1 |
13.71 |
750 |
N.D. |
| 5 |
HT-2 toxin |
2 |
6.14–17.63 |
— |
N.D. |
| 6 |
Hexaconazole |
20 |
1.18–11.77 |
10 |
2 |
| 7 |
Ochratoxin A |
3 |
1.66–18.12 |
3 |
1 |
| 8 |
T-2 toxin |
2 |
6.42–12.67 |
— |
N.D. |
| 9 |
Triadimefon |
1 |
1.45 |
10 |
N.D. |
 |
| | Fig. 10 The MRM chromatogram of hexaconazole (A), aflatoxin G2 (B), ochratoxin A (C) in real samples. | |
3.5. Comparison with other reported methods
The method developed in this study was exhaustively compared with other reported liquid chromatographic methods for the determination of pesticides or mycotoxins in barley species. The results, as shown in Table 4, showed that the LOQs of the present method for pesticide residues were significantly lower than those of ref. 41–43. Meanwhile, for the detection of mycotoxins, the LOQs of the present method were all less than or equal to those of ref. 44. Compared with the other methods, the recovery of the present study showed a superior performance. What's more, compared with other methods, the method in this study can detect more targets and achieve the simultaneous detection of pesticides and mycotoxins, showing its unique advantages.
Table 4 Comparison with reported assays for the determination of pesticide residues and mycotoxins in barley
| Method |
Instrument |
Matrix |
Targets (quantities) |
Pesticide LOQ (μg kg−1) |
Mycotoxin LOQ (μg kg−1) |
Recovery (%) |
Ref. |
| QuEChERS |
UPLC-MS/MS |
Barley |
Pesticides (57) |
3.00–36.00 |
— |
29.0–85.0 |
41
|
| QuEChERS |
UPLC-(Q-ToF)-MS |
Barley |
Pesticides (54) |
12.00–17.00 |
— |
61.0–81.0 |
42
|
| QuEChERS |
HPLC-MS/MS |
Barley |
Mycotoxins (23) |
— |
0.10–6.34 |
70.1–93.3 |
43
|
| QuEChERS |
UPLC-MS/MS |
Barley |
Mycotoxins (15) |
— |
0.50–132.00 |
42.1–100.1 |
44
|
| QuEChERS |
UPLC-MS/MS |
Tsampa |
Pesticides (78) |
0.10–3.00 |
0.10–2.80 |
73.0–115.2 |
This study |
| Mycotoxins (16) |
4. Conclusions
In this study, an efficient and rapid method for the simultaneous determination of 78 pesticides and 16 mycotoxins in tsampa was established, using UPLC-MS/MS technology coupled with improved QuEChERS clean-up. The established method was robust and has been validated by methodology with the LOQs lower than the MRLs of the EU. This method was applied in real samples in which 32% of samples exceeded the MRL of EN. It proved to be excellent, efficient, greatly simplified, and highly applicable, which could significantly reduce the workload and time for the daily monitoring of the pesticides and mycotoxins in tsampa. It also provides important data support for the monitoring of tsampa.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article [and/or its ESI†].
Author contributions
Each author contributed to the article's design and conceptualization. All authors read the final draft before giving their approval.
Conflicts of interest
The authors confirm that they have no competing interests.
Acknowledgements
This study was supported by the Fundamental Research Funds for the Public Research Institutes of Chinese Academy of Inspection and Quarantine (2023JK001).
References
- L. Li, C. Baima, J. Jiang, Z. Liu, J. Wang, X. D. Chen and P. Wu, Food Eng., 2022, 372, 111054, DOI:10.1016/j.jfoodeng.2022.111054.
- T. Guo, C. Horvath, L. Chen, J. Chen and B. Zheng, Trends Food Sci. Technol., 2020, 103, 109–117, DOI:10.1016/j.tifs.2020.07.011.
- H. Gao, D. Zhao, L. Wang, H. Zhi, N. Bai, H. Dong, H. Chen and W. Li, Arabian J. Chem., 2024, 17(2), 105408, DOI:10.1016/j.arabjc.2023.105408.
- M. A. Gonzalez-Curbelo, S. Dionis-Delgado, M. Asensio-Ramos and J. Hernandez-Borges, J. Sep. Sci., 2012, 35, 299–307, DOI:10.1002/jssc.201100811.
- M. Roca, A. Miralles-Marco, J. Ferre, R. Perez and V. Yusa, Environ. Res., 2014, 131, 77–85, DOI:10.1016/j.envres.2014.02.009.
- J. L. da Silva, S. Lombardi, L. Castaldo, E. Morelli, J. Garda-Buffon, L. Izzo and A. Ritieni, Toxins, 2023, 15, 562, DOI:10.3390/toxins15090562.
- F. Y. Eker, K. Muratoglu, M. Ozturk, B. Cetin and S. K. Buyukunal, Foods, 2023, 12, 2744, DOI:10.3390/foods12142744.
- T. W. Zhang, D. L. Wu, W. D. Li, Z. H. Hao, X. L. Wu, Y. J. Xing, J. R. Shi, Y. Li and F. Dong, Mycotoxin Res., 2023, 39, 193–200, DOI:10.1007/s12550-023-00487-1.
- V. Navale, K. R. Vamkudoth, S. Ajmera and V. Dhuri, Toxicol Rep, 2021, 8(3), 1008–1030, DOI:10.1016/j.toxrep.2021.04.013.
- D. Klingelhöfer, Y. Zhu, M. Braun, M. H. K. Bendels, D. Brüggmann and D. A. Groneberg, Food Control, 2018, 280–290, DOI:10.1016/j.foodcont.2018.02.017.
- A. R. Soares Mateus, S. Barros, A. Pena and A. Sanches Silva, Toxins, 2021, 13(10), 682, DOI:10.3390/toxins13100682.
- K. M. N. Burgess, J. B. Renaud, T. McDowell and M. W. Sumarah, ACS Chem. Biol., 2016, 11(9), 2618–2625, DOI:10.1021/acschembio.6b00438.
- A. Das Trisha, J. M. Hafsa, A. Hasan, A. Habib, H. R. Tuba, G. H. Degen and N. Ali, Mycotoxin Res., 2023, 40, 135–146, DOI:10.1007/s12550-023-00510-5.
- F. Agahi, C. Juan, G. Font and A. Juan-García, Food Chem. Toxicol., 2020, 146, 111818, DOI:10.1016/j.fct.2020.111818.
- M. W. Sumarah, J. Agric. Food Chem., 2022, 70(31), 9619–9624, DOI:10.1021/acs.jafc.2c03690.
- C. Yue, C. Q. Zhao, S. H. Mao, Z. H. Wang, B. Shi, X. F. Xu and J. J. Liang, Chin. J. Chromatogr., 2023, 41(9), 807–813, DOI:10.3724/SP.J.1123.2023.03017.
- B. Nakhjavan, J. Bland and M. Khosravifard, Molecules, 2021, 26(21), 6627, DOI:10.3390/molecules26216627.
- Q. F. Wang, R. J. Chen, W. Shatner, Y. Cao and Y. Bai, Ultrason. Sonochem., 2018, 51(5), 369–377, DOI:10.1016/j.ultsonch.2018.08.010.
- S. A. Salim, R. Sukor, M. N. Ismail and J. Selamat, Toxins, 2021, 13(4), 280, DOI:10.3390/toxins13040280.
- L. R. Amaral, C. F. Zanchin, L. Floriano, K. Ues, O. D. Prestes and R. Zanella, Food Anal. Methods, 2023, 16, 771–780, DOI:10.1007/s12161-023-02471-y.
- B. X. Yang, S. Wang, W. Ma, G. L. Li, M. L. Tu, Z. Y. Ma, Q. H. Zhang, H. M. Li and X. J. Li, Foods, 2023, 12(4), 699, DOI:10.3390/foods12040699.
- J. Martiník, R. Bosko, Z. Svoboda, S. Beláková, K. Benesová and M. Pernica, Mycotoxin Res., 2023, 39(3), 285–302, DOI:10.1007/s12550-023-00492-4.
- A. Ghazaleh, M.-M. Masoumeh and M. Mansoureh, Food Control, 2023, 152, 109820, DOI:10.1016/j.foodcont.2023.109820.
- Q. B. Fu, Z. Z. Xia, X. Sun, H. L. Jiang, L. L. Wang, S. Y. Ai and R. S. Zhao, TrAC, Trends Anal. Chem., 2023, 162, 117054, DOI:10.1016/j.trac.2023.117054.
- T.-T. Tran-Lam, M. Q. Bui, H. Q. Nguyen, Y. H. Dao and G. T. Le, Foods, 2021, 10(10), 2455, DOI:10.3390/foods10102455.
- Q. X. Zhou, F. H. Li, L. L. Chen and D. F. Jiang, J. Food Sci., 2016, 81(11), T2886–T2890, DOI:10.1111/1750-3841.13524.
- S. Ma, L. g. Pan, T. You and K. Wang, J. Agric. Food Chem., 2021, 69(16), 4874–4882, DOI:10.1021/acs.jafc.1c00141.
- S. L. Chau, A. Zhao, W. Jia and L. Wang, Molecules, 2023, 28(19), 6883, DOI:10.3390/molecules28196883.
- J. He, B. Zhang, H. Zhang, L.-L. Hao, T.-Z. Ma, J. Wang and S.-Y. Han, J. Food Sci., 2019, 84(9), 2688–2697, DOI:10.1111/1750-3841.14695.
- N. B. Cech and C. G. Enke, Mass Spectrom. Rev., 2001, 20(6), 362–387, DOI:10.1002/mas.10008.
- L.-Y. Tong, B.-Z. Xu, X.-M. Nie, X.-J. Wang, J.-H. Ma, W. Guo, G.-R. Li, Y.-K. Gong and X.-L. Xu, Chin. J. Chromatogr., 2023, 41(11), 986–994, DOI:10.3724/sp.j.1123.2023.07010.
-
BS EN 15662:2018, Foods of plant origin. Multimethod for the determination of pesticide residues using GC- and LC-based analysis following acetonitrile extraction/partitioning and clean-up by dispersive SPE. Modular QuEChERS-method, 2018, available from: https://img.antpedia.com/standard/files/pdfs_ora/20230612/bs/BSEN/BSEN15662-2018.pdf, accessed 05 06 2018.
- A. Narváez, L. Izzo, L. Castaldo, S. Lombardi, Y. Rodríguez-Carrasco and A. Ritieni, Toxins, 2023, 15(2), 148, DOI:10.3390/toxins15020148.
- N. P. Kalogiouri, E.-N. Papadakis, M. G. Maggalou, G. S. Karaoglanidis, V. F. Samanidou and U. Menkissoglu-Spiroudi, Appl. Sci., 2021, 11(22), 10931, DOI:10.3390/app112210931.
- S. J. Lehotay, J. AOAC Int., 2007, 90(2), 485–520, DOI:10.1093/jaoac/90.2.485.
- R. Romero-González, A. Garrido Frenich, J. L. Martínez Vidal, O. D. Prestes and S. L. Grio, J. Chromatogr. A, 2011, 1218(11), 1477–1485, DOI:10.1016/j.chroma.2011.01.034.
- C. Ferrer, A. Lozano, A. Agüera, A. J. Girón and A. R. Fernández-Alba, J. Chromatogr. A, 2011, 1218(42), 7634–7639, DOI:10.1016/j.chroma.2011.07.033.
- F. Gosetti, E. Mazzucco, D. Zampieri and M. C. Gennaro, J. Chromatogr. A, 2010, 1217(25), 3929–3937, DOI:10.1016/j.chroma.2009.11.060.
-
Contaminants in Foods Regulation (EC) No 2023/915, 2023, available from: https://eur-lex.europa.eu/eli/reg/2023/915/oj, accessed 08 10 2023.
-
Document No. SANTE 11312/2021, Analytical quality control and method validation procedures for pesticide residues analysis in food and feed, 2024, available from: https://food.ec.europa.eu/system/files/2023-11/pesticides_mrl_guidelines_wrkdoc_2021-11312.pdf, accessed Jan 01, 2024.
- M. Dušek, S. Běláková, K. C. Piacentini and V. Jandovská, J. Agric. Food Chem., 2021, 69(31), 8649–8659, DOI:10.1021/acs.jafc.1c01120.
- R. Rodríguez-Ramos, Á. Santana-Mayor, B. Socas-Rodríguez, A. V. Herrera-Herrera and M. Rodríguez-Delgado, Food Chem., 2023, 400, 134089, DOI:10.1016/j.foodchem.2022.134089.
- H. Kim, E. J. Baek, B. G. Shin, H. J. Kim and J. E. Kim, Toxins, 2022, 14(7), 457, DOI:10.3390/toxins14070457.
- C. K. Mahdjoubi, N. Arroyo-Manzanares, N. Hamini-Kadar, A. M. García-Campaña, K. Mebrouk and L. Gámiz-Gracia, Toxins, 2020, 12(3), 194, DOI:10.3390/toxins12030194.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Hongyan
Zhang
ab,
Zhehui
Zhu
c,
Yujie
Xie
b,
Xingqiang
Wu
b,
Zhihong
Shi
*a,
Chunlin
Fan
b and
Hui
Chen
*b
ab,
Hongyan
Zhang
ab,
Zhehui
Zhu
c,
Yujie
Xie
b,
Xingqiang
Wu
b,
Zhihong
Shi
*a,
Chunlin
Fan
b and
Hui
Chen
*b