
Injectable hydrogels as emerging drug-delivery platforms for tumor therapy
Yao
Cheng†
,
Haitao
Zhang†
 *,
Hua
Wei
* and
Cui-Yun
Yu
*
*,
Hua
Wei
* and
Cui-Yun
Yu
*
Hunan Province Cooperative Innovation Center for Molecular Target New Drug Study & School of Pharmaceutical Science, Hengyang Medical School, University of South China, 28 W Changsheng Road, Hengyang 421001, Hunan, China. E-mail: zhanghaitao@usc.edu.cn; weih@usc.edu.cn; yucuiyunusc@hotmail.com
First published on 30th January 2024
Abstract
Tumor therapy continues to be a prominent field within biomedical research. The development of various drug carriers has been propelled by concerns surrounding the side effects and targeting efficacy of various chemotherapeutic drugs and other therapeutic agents. These carriers strive to enhance drug concentration at tumor sites, minimize systemic side effects, and improve therapeutic outcomes. Among the reported delivery systems, injectable hydrogels have emerged as an emerging candidate for the in vivo delivery of chemotherapeutic drugs due to their minimal invasive drug delivery properties. This review systematically summarizes the composition and preparation methodologies of injectable hydrogels and further highlights the delivery mechanisms of diverse drugs using these hydrogels for tumor therapy, along with an in-depth discussion on the optimized therapeutic efficiency of drugs encapsulated within the hydrogels. The work concludes by providing a dynamic forward-looking perspective on the potential challenges and possible solutions of the in situ injectable hydrogels for non-surgical and real-time diagnostic applications.
1. Introduction
Tumors are a significant health concern and the second leading cause of death globally, affecting millions of people worldwide each year. Different types of cancer affect various body tissues, but they all share the common feature of abnormal cell proliferation due to genetic and epigenetic alterations.1 Despite the success of current therapies, such as surgery, chemotherapy, immunotherapy, radiation therapy, and targeted therapy, in treating patients, these methods are often associated with a variety of serious side effects. Additionally, patients may experience cancer recurrence due to metastasis, as well as damage to normal organs.2In recent decades, nano-drug delivery systems (NDDS) have provided a promising approach for controlled and targeted drug delivery and are a promising strategy for cancer treatment.3 Nanocarriers, including liposomes, polymeric micelles, dendritic macromolecules, carbon nanotubes, gold nanoparticles, etc., have the ability to recognize tumor cells and deliver therapeutic agents in a fixed ratio, causing little harm to healthy cells. All these nano-systems offer advantages such as tumor-targeted drug delivery, reduced systemic side effects, prolonged plasma circulation, and the ability to carry larger drug payloads while preventing recognition by efflux pumps.4,5 However, these conventional nano drug-carrying systems often suffer from several drawbacks, including poor targeting, premature release, and the need for frequent injections during the delivery process. Therefore, there is a pressing need for the development of a novel drug delivery system that can act as a “drug reservoir” and deliver directly to the tumor site for controlled on-demand drug delivery.6,7
Hydrogel is a class of polymer materials with a three-dimensional network structure, which is widely used in the fields of biomaterials, environmental engineering, and biomedicine.8–10 Nanoparticles are considered drug formulations with good biocompatibility and biodegradability. Compared to conventional nanomaterials, hydrogels are capable of designing hydrogel drug-carrying systems with stimulus-responsive properties, taking into account the complex tissue-specific physiology at the macroscopic scale in combination with dynamic biophysical/biochemical stimuli. Hydrogels can meet a variety of clinical needs, such as drug and cell delivery, tissue repair and regeneration, bio-adhesion, and immunomodulation.11 Injectable hydrogels, in particular, are becoming more and more widely used in the field of tissue engineering and drug delivery due to their tunable physical and chemical properties, controlled degradation, high water content, and the ability to achieve delivery in a minimally invasive manner.
Our group has made significant contributions to the field of injectable hydrogels in recent years, particularly in the context of osteoporosis and myocardial repair treatment. For example, Wei et al. report a double crosslinked peptide-based hydrogel that fosters cartilage regeneration through a feedback-regulated mechanism. This hydrogel integrates multiple supramolecular interactions, including host–guest interactions and extensive hydrogen bonding networks, endowing the hydrogel with superior self-healing properties and mechanical robustness for synergistically enhanced lubrication and compressive properties.12 Additionally, our group has developed a reactive oxygen species (ROS)-responsive hydrogel fabricated by mixing poly-3-amino-4-methoxybenzoicacid with TK-NH2-modified gelatin (PAMB-G-TK) and 4-arm-PEG-succinimidyl glutarate ester (4-arm-PEG-SG) for topical application, designed to directly target the mitochondria within damaged cardiomyocytes. This approach regulates, in a feedback-driven manner, the release of the encapsulated liposomes and the depletion of excess ROS, thereby improving the activity of cardiomyocytes and enhancing myocardial infarction (MI) therapy.13 These studies provide a foundation for understanding and advancing researches on the applications of injectable hydrogels in a broad biomedical field.
Based on this, injectable therapeutic hydrogels are gaining increasing attention as non-invasive multifunctional platforms for traceable nanomedicine. With the rapid development of injectable hydrogels, many reviews have elaborated on the uses of injectable therapeutic hydrogels in myocardial repair14 and tissue regeneration.15 In the context of therapeutic applications in oncology, a large number of research on injectable hydrogels have been summarized. However, most of the published papers focus on showcasing a single therapeutic aspect of injectable hydrogels for cancer therapy. There is a lack of reviews, to our knowledge, providing a comprehensive summary of injectable hydrogels used for diverse and combinatory therapeutic effects, which includes elegant integration with chemotherapy, gene therapy, radiotherapy, phototherapy, immunotherapy, and combined drug therapies within a three-dimension hydrogel network. Besides this key uniqueness of this review relative to the published data, the intrinsic properties of hydrogels are emphasized by highlighting their drug-loading capabilities and distinctive structural modulation features. Last but not least, in-depth discussion and instructive prospects on the existing challenges and possible solutions, and clinical translation potentials of injectable hydrogels are made to promote a well understanding of injectable hydrogels from a fresh perspective (Fig. 1).
 | ||
| Fig. 1 (A) Schematic diagram of a hydrogel as a drug delivery platform for generating antitumor effects. (B) The role of hydrogels in tumor therapy. | ||
2. Brief introduction to hydrogels
Injectable hydrogels should not only be stable, economical, and easy to fabricate, but also should be formulated with injectable drugs to promote hydrogel formation, enable uniform and consistent drug loading, and facilitate drug release.18–20 These hydrogels come with numerous advantages. (i) High water absorption capacity. The molecular structure of hydrogels contains a large number of hydrogen bonds, which are able to form strong interaction forces with water molecules. At the same time, small molecule water-soluble solutes penetrate easily in the hydrogel, which enables the loaded substance to remain active for a long time and ensures the continuous prolonged release of the drug in the body. (ii) Soft nature. The hydrogel can maintain a certain shape, and its meticulous structure prevents the leakage of the drug and also hinders the degradation of unstable macromolecules such as recombinant proteins and monoclonal antibodies. (iii) Commendable biocompatibility. Hydrogels are aggregates of water molecules and networks of hydrophilic polymers, and hydrogel formulations made from materials that can be absorbed and metabolized by the body have good biocompatibility and low cytotoxicity. The soft hydrophilic surface makes it easy to adhere to tissues, while its similarity to body tissues reduces its immunogenicity.17,21 Additionally, the degradability makes it easy to metabolize and facilitates the complete removal of the carrier polymer from the body after the release of the drug. (iv) Controlled release. Injectable hydrogels are ideal drug delivery systems with the potential for non-invasive drug delivery,16,22–25 and in vivo gel–sol transition enables localized drug delivery avoiding the problems of systemic drug delivery and increasing the local drug concentration.25,26 It can spatially and temporally control the release of loaded therapeutic agents and prolong their action by modulating physical or chemical properties. For oncology therapy, injectable hydrogel formulations loaded with anticancer drugs can be easily prepared without loss of anticancer drugs and can be delivered directly to and accumulate in specific tumor sites, thereby minimizing drug accumulation in other organs, enhancing therapeutic efficacy and minimizing adverse drug reactions to normal tissues.27,28 Hydrogels can also be used in situ to form bionic scaffolds for tissue regeneration while providing sustained release of encapsulated drugs.3. Hydrogel drug loading method
Injectable hydrogels are capable of encapsulating drugs via multiple mechanisms, including physical encapsulation, chemical bonding, swelling, and cross-linking etc. During the physical encapsulation process, drugs are adsorbed onto hydrogels via various supramolecular forces, such as electrostatic interactions, hydrogen bonding, and van der Waals forces between the drug molecules and the hydrogel matrix. For example, Abraham et al. developed cationic hydrogels based on fluorenylmethyloxycarbonyl-modified phenylalanine (Fmoc–Phe) for the encapsulation and controlled release of diclofenac. The charge reversal property of this hydrogel at a physiological pH promotes functional pain management.29 Sato et al. prepared poly(acrylamide) and poly(dimethylacrylamide) gels with surface significantly adsorbed submicron silica particles solely by van der Waals forces. Despite of a Hamaker constant much smaller in water than that in air, the van der Waals attraction between the particles and the gel is sufficient to facilitate adsorption.30The relatively straightforward physical drug encapsulation is inevitably associated with a rapid drug release profile. Contrary to physical encapsulation, a chemical bonding process generally offers greater stability and a better controlled drug release behavior due to dynamic drug conjugation. During this process, the drug molecules undergo a chemical reaction with the reactive sites of hydrogels, resulting in the formation of covalent or ionic bonds for stable drug entrapment within the hydrogel matrix. Popescu et al. developed an amphiphilic, pH-sensitive hydrogel conjugated with a triblock copolypeptide via an imine bond generated by a Schiff base reaction between the N-terminus of the peptide and benzaldehyde of the hydrogel. The coupling and decoupling of the benzoic acid–imine dynamic covalent bond induces a pH-responsive reversible sol–gel transition for accelerated drug release.31 Werner et al. engineered star PEG–heparin hydrogel networks that are crosslinked through Michael-type addition reactions. The incorporated MMP-cleavable peptides facilitate localized controlled VEGF delivery. Enzymatic cleavage was significantly accelerated when the gels’ accessibility to proteases increased in contrast to the slow non-enzymatic hydrolysis and degradation of ester bonds. Consequently, the combination of protease-sensitive and -insensitive cleavage sites can enhance the degradation of bio-responsive gel materials for promoted endothelial cell morphogenesis.32
Hydrogels possess an exceptional swelling capacity due to high water absorption and entrapment. Upon drug dissolution in water, the hydrogel undergoes simultaneous swelling and drug encapsulation via water absorption. This swelling-based encapsulation mechanism facilitates a sustained drug release profile but is generally limited to hydrophilic drug species. Mayte et al. developed chitosan-based covalent hydrogels through the Mannich reaction between the amino groups of chitosan and the hydroxymethyl groups of tetrakis(hydroxymethyl)phosphonium chloride as a crosslinking agent. The resulting hydrogels exhibited pH-sensitive second-order swelling kinetics and enabled a sustained release of the encapsulated camptothecin over 48 hours.33
Additionally, utilization of crosslinking agents generates an interconnected polymer network within the hydrogel for drug encapsulation. For instance, a phosphate crosslinker, bis[2-methacryloyloxy] ethyl phosphate (BMEP) was employed in the construction of Eswaramma's dual-responsive hydrogel networks (PPAD) composed of pectin and poly((2-dimethylamino) ethyl methacrylate). These hydrogel networks successfully employed for the encapsulation of the anticancer drug 5-fluorouracil (5-FU).34 Yan et al. developed a straightforward and adaptable strategy for fabricating low hysteresis hydrogels by employing porous cationic polymers (PCPs) with adjustable counteranions as dynamic physical cross-linkers. The PCPs interpenetrated and became entangled with the polymer chain segments to form the hydrogels’ topological structure. The reactive centers abundant in the PCPs effectively immobilized the polymer chains for inhibited segment slippage and minimized energy dissipation and enhanced elasticity.35
4. Response mode of injectable hydrogel
The loading of drugs on hydrogels can be divided into physical and chemical, physical binding is loading the drug after encapsulation and adsorption through hydrophobic interactions, coordination, etc., and chemical binding usually involves connecting the drug with the carrier hydrogel through bonds that can be enzymatically cleaved or hydrolyzed, and then releasing the drug after the bonds are enzymatically cleaved or hydrolyzed in vivo. Injectable hydrogels are also prepared in a very similar way to drug loading methods, which can be categorized into chemically cross-linked and physically cross-linked hydrogels, depending on the type of cross-linking used.36,37 On the one hand, physical cross-linking is transient cross-linking that relies on physical interactions, such as ionic, hydrogen, and hydrophobic bonding, as well as inter- and intra-physical interactions. Therefore, physically cross-linked hydrogels usually be used to design delivery systems with self-healing and stress-responsive features. On the other hand, chemical cross-linking, produces chemical reactions, such as covalent bonding to build a persistent three-dimensional network through light, enzymes, and clicking cross-linkers. Chemically cross-linked hydrogels are structurally stable and permanent.38,39 The following section will focus on injectable hydrogels formed by physical and chemical methods (Fig. 2). | ||
| Fig. 2 Construction strategy and response mechanism for injectable hydrogels. | ||
4.1. Physical cross-linking method
The relatively weak nature of physical cross-linking allows its reversible transformation back to solution when external conditions fluctuate. This flexibility facilitates the formation of hydrogel matrices that can adapt according to different biological conditions.40,41 Moreover, physically cross-linked hydrogels eliminate the need for cross-linkers or chemical modifications.42,43Utilizing the above advantages of hydrogen bonding, Chen successfully prepared a high-strength, injectable supramolecular hydrogel by constructing a multi-hydrogen bonding system. It is self-assembled from a single nucleoside molecular gelling agent (2-amino-2′-fluoro-2′-deoxyadenosine (2-FA)) using distilled water/phosphate-buffered saline as solvent. The remarkable energy storage modulus, reaching 1 MPa at a concentration of 5.0 wt%, positions it as the strongest supramolecular hydrogel containing an ultra-low molecular weight (MW < 300) gelling agent.47 Inspired by the slow transformation of mineral precursors during biomineralization, Li proposed a general endogenous gelation method for tannic acid-macromolecule hydrogels. The method controls the elimination or slow generation of hydrogen bonding interactions between tannins and macromolecules by regulating the pH value of the system. In order to prepare the hydrogel precursor, the pH of the tannin solution is first increased before combining the components. This process ensures that the phenolic hydroxyl groups transition into a phenol-negative ion state, which inhibits the formation of a sufficient quantity of hydrogen bonds to establish a crosslinked network when combined with the macromolecules. Subsequently, gluconolactone is added to the solution. As gluconolactone has the ability to gradually release hydrogen ions, this allows for the slow generation of hydrogen bonding interactions.48 The disintegration conditions of hydrogels formed through hydrogen bonding primarily depend on the hydrogen bonds’ interaction with surrounding water molecules, the forces exerted on the crosslinked interconnecting chains, and external loads strong enough to disrupt the crosslinks, and change the network's microstructure, leading to drug release.
The hydrophobic interaction-driven gelation mainly exhibits thermo-responsive behavior.50 Poly(N,N-diethylacrylamide) (PDEA) is a thermosresponsive polymer that can be used as a trigger for the formation of thermally reversible gels. Michael et al. synthesized ABA copolymers with the structure PDEA-b-poly(ethylene glycol) (PEG)-b-PDEA, yielding four block copolymers of PDEA and PEG with different molecular weights and high molecular weight PEG blocks to form thermally reversible gels with predominantly similar solid behavior.51 Finally, A20–B10–A20 was selected as the most promising thermoreversible gel. It can be made thermoreversible by controlling the concentration (30% w/v) and ionic strength (0.3 m NaCl) and temperature control below 37 °C, making it possible that gelation occurs upon contact with the internal temperature of the human body.
These above-mentioned nonpolar hydrophobic moieties aggregated together have strong hydrophobic interactions and can form several hydrophobic microregions (or micelles), which can accommodate small organic molecules of drugs under specific conditions and release them slowly under other specific conditions, overcoming the problem of rapid release of drugs from gels in the swelling state.40
In summary, physically crosslinked hydrogels exhibit superior biocompatibility and biodegradability due to minimal use of any chemical reagents during the hydrogel preparation process, thus are environmental friendly with significantly reduced environmental pollution. They generally have the abilities to form three-dimensional interconnected structures without the aid of any additional cross-linking agents. These advantages make physically crosslinked hydrogels undergo easy administration via injection through a fine needle and rapid gelation post-injection.
However, as the physical interactions depend substantially on the inherent properties of the used biomaterials, certain characteristics of these materials, such as gelation time, in vitro and in vivo maintenance, and mechanical properties of hydrogels are actually inherent inflexible. Consequently, precise modulation of the in vivo properties of hydrogels may constitute a significant challenge.53,54 Nevertheless, these drawbacks can be overcome by using biomaterials with different molecular weights and concentrations, as well as composite biomaterials, which generates hydrogels with diverse hardness, viscosities, rheological behaviors, swelling and disintegration properties, and biocompatibilities. Overall, injectable, in situ forming hydrogels prepared by physical interactions have been extensively used for intra-tumor injections.
4.2. Chemical crosslinking preparation
Chemical synthesis is another important and commonly used method for preparing hydrogels. Chemical cross-linking is the creation of irreversible covalent bonds through chemical reactions (e.g., free radical polymerization and click chemistry) to build a durable polymer structure. Hydrogels formed by this method have strong covalent bonds that can be preserved structurally stable and permanent, only swelling and not dissolving.A covalent bond is a permanent connection between two molecules. The formation of covalent bonds within and between chains of biomaterials can lead to permanent immobilization of biomaterials or changes in their physical properties.55 These covalent bonds can be used to prepare injectable hydrogels through chemical reactions such as thermal response, pressure, pH change, light irradiation, click reaction, or enzymatic activity.
Chemical cross-linking imparts superior stability in mechanical properties and diverse morphologies, surpassing the aforementioned deficiencies of physically cross-linked hydrogels. However, it usually involve the use and generation of certain toxic residues and complicated chemical design and synthesis with compromised scalable production and translational potentials.
Physically or chemically cross-linked hydrogels contain various reversible bonds, including metal–ligand, Diels–Alder covalent, imine, and acyl hydrazone. Activation of these bonds, triggered by a specific external temperature or pH, enables the formation of self-healing hydrogels.79 This category of hydrogels, primarily constituted by reversible chemical reactions, exhibits the unique ability to transiently fluidize under shear stress, subsequently restoring their original mechanical properties. Such self-healing hydrogels, often employed in the development of drug/cell delivery systems, can be injected in a localized, targeted, and minimally invasive manner through a narrow syringe without the necessity for invasive surgical procedures.
5. Delivery of chemotherapeutic agents
Chemotherapy, proven as a highly effective anti-cancer treatment, eliminates tumor cells primarily by obstructing DNA replication or protein synthesis. Several studies have shown that low doses and slow drug infusions are more effective than aggressive chemotherapy given in multiple high doses. Injectable hydrogels, which allow localized co-delivery of multiple drugs with different therapeutic mechanisms around tumors, have made significant advances in the treatment of cancer.80 Controlled release of chemotherapeutic agents can be achieved by modulating the properties of hydrogels or external stimuli, including pH, temperature, enzymes, reactive oxygen species, and radiation. The application of drug delivery systems not only reduces the side effects of chemotherapeutic drugs in cancer cells but also improves the efficacy of cancer treatment via controlling drug release. Chemotherapeutic agents can be categorized according to their composition and therapeutic mechanism (Table 1).| Classification of chemotherapy drugs | Mechanism of action | Representative drugs | Hydrogel matrix | Gelling mechanism | Release mechanisms | Cancer types | Tumor model (in vivo) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Alkylating agent | DNA destruction | Cisplatinum | Poly(ethylene glycol)–poly(β-aminoester urethane) (PEG–PAEU) | Chelating ligand–metal coordination cross-coupling reactions | pH and temperature responsiveness | Lung cancer | A549 | 82 |
| Anti-metabolite | Interrupts the S phase of the cell cycle | 5-Fluorouracil (5-FU) | Biodegradable temperature-sensitive hydrogel (PDLLA–PEG–PDLLA, PLEL) | Responding to physiological temperatures | Temperature response | Stomach cancer | MKN45-Luc | 84 |
| Anthracycline | Inhibition of enzymes causing DNA replication | Doxorubicin (DOX) | Poly(acrylic acid-co-4-vinyl-phenylboronic acid) nanohydrogels (P(AA-co-4-VPBA) NG) | Reflux-precipitation polymerization | pH and redox stimuli-responsive | Breast cancer | MCF-7 | 86 |
| Topoisomerase inhibitors | Inhibiting topoisomerase activity | Topotecan | Chitosan (CS) and β-glycerophosphate (β-GP) | Responding to physiological temperatures | Temperature response | Liver cancer | H22 | 88 |
| Alkaloid | Termination of M phase of cell cycle | Paclitaxel | The methoxy poly(ethylene glycol)-b-poly(ε-caprolactone-co-1,4,8-trioxa[4.6]spiro-9-undecanone) (impact) | Hydrophobic interaction | Photothermal response | Breast cancer | 4T1 | 89 |
| Corticosteroids | Inhibits tumor cell production | Dexamethasone | Dexamethasone (Dex)-loaded microspheres (Dex-Ms) mixed with click-crosslinked hyaluronic acid (Cx-HA) | Click-crosslinked | — | — | FRTL-5 | 90 |
Alkylating agents, often seen as early-discovered and diverse anti-neoplastic agents, fall under the chemotherapeutic agents’ classification. Using Cisplatin (CDDP) as a classic alkylating agent example, its robust antitumor effects stem from its irreparable interactions with nuclear DNA. Moreover, CDDP can interact with other biomolecules such as thionin and RNA to inhibit or modulate signaling pathways accountable for mediating cell death. By affecting DNA and killing cells at different stages of division (inclusive of the G0 stage), it brings about diversified side effects such as severe nephrotoxic, neurotoxic, and ototoxic side effects. Despite the potential to encapsulate CDDP into nanoscale drug delivery formulations enhancing its physicochemical properties, the stability and cancer cell-specific targeting insufficiency in formulations need novel carrier exploration for CDDP-targeted delivery.81 The cytotoxicity of the drug to other tissues is mainly reduced by restricting the drug release conditions. Lee et al. synthesized chondroitin sulfate nano gels bearing cisplatin (CS-nano gels) through a chelating ligand–metal ligand crosslinking reaction. These nano gels were incorporated into pH- and temperature-responsive bioabsorbable hydrogels based on poly(ethylene glycol)–poly(β-amino ester urethane) (PEG–PAEU) for tumor cell-specific delivery. CDDP was released gradually under physiological conditions (pH 7.4), while accelerated CDDP release was triggered upon shift to an acidic pH condition (pH 5.0), which can effectively increase the localized drug concentration in the TME for inhibiting tumor cell proliferation (Fig. 3A).82 The fluorescein 5-thiosemicarbazone-labeled CS-nano gels, released from hydrogels, selectively bind to the A549 lung cancer cell line, without any affinity for NIH 3T3 cells lacking of overexpressed CD44 receptors. Therefore, an in vitro cytotoxicity assay demonstrated the potent inhibitory effect of CS-nano gels on the growth of A549 lung cancer cells. Such an intelligent delivery technique allowed better cisplatin targeting to the tumor site, thereby improving its bioavailability.
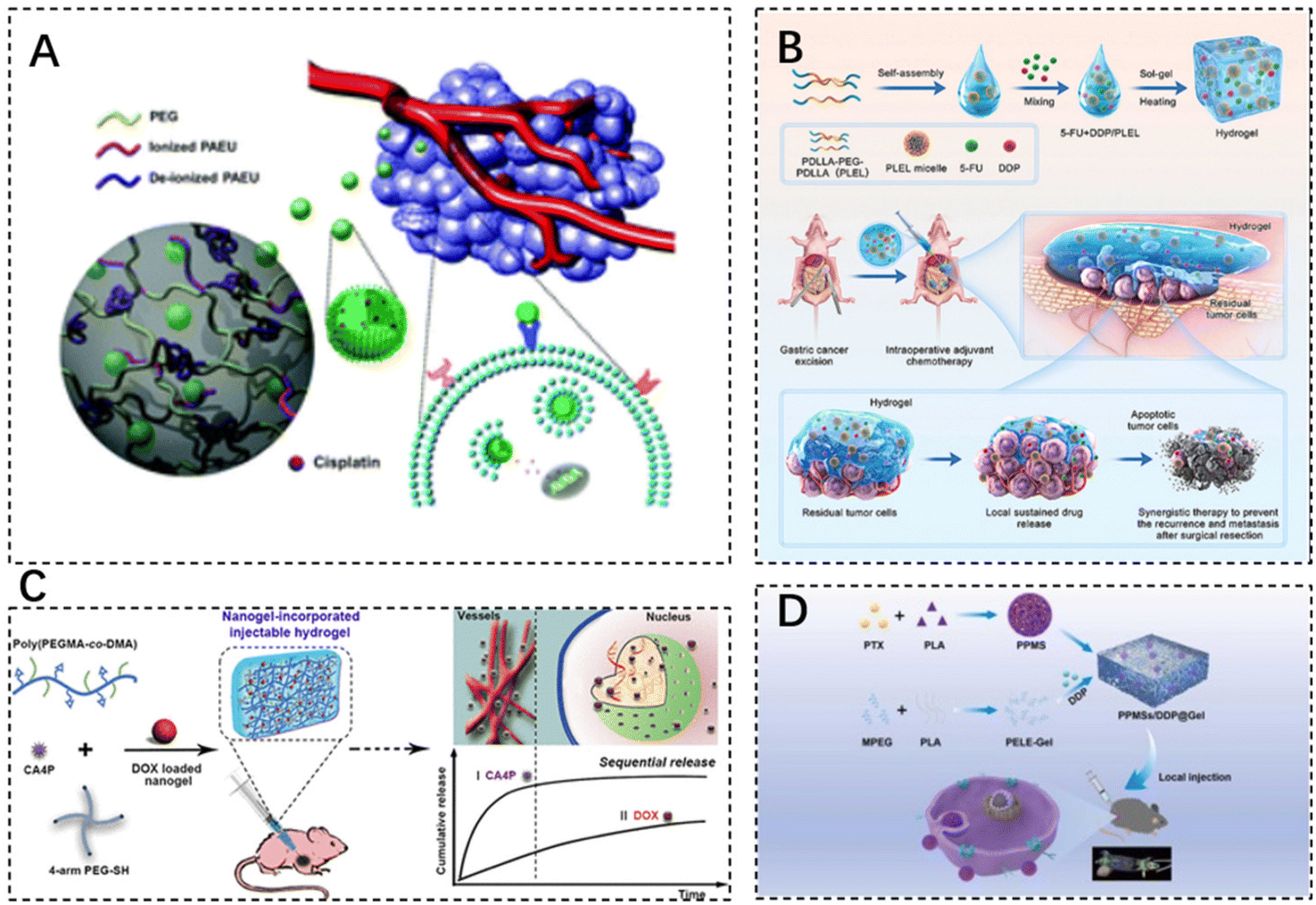 | ||
| Fig. 3 The use of injectable hydrogels in chemotherapeutic drug delivery. (A) Chondroitin sulfate nanogels (CS-nanogels) with CDDP incorporated into pH- and temperature-responsive bioabsorbable poly(ethylene glycol)–poly(β-amino ester urethane) (PEG–PAEU) hydrogels for CDDP-specific delivery to cancer cells. Reproduced with permission from ref. 82. Copyright 2017, Royal Society of Chemistry. (B) Schematic diagram of sustained co-delivery of 5-fluorouracil (5-FU) and cis-platinum (DDP) via biodegradable thermo-sensitive hydrogel. Reproduced with permission from ref. 84. Copyright 2022, KeAi. (C) Preparation process of injectable Nanogel-Incorporated Hydrogel (NHG) for Sequential Local Delivery of CA4P and DOX. Reproduced with permission from ref. 86. Copyright 2018, American Chemical Society. (D) Construction of a dual-drug co-delivery hydrogel system (PPMSs/DDP@Gel) for in situ chemotherapy by co-embedding PPMSs and cisplatin (DDP) in a thermosensitive hydrogel. Reproduced with permission from ref. 87. Copyright 2023, Elsevier Masson SAS. | ||
5-Fluorouracil (5-FU) is a type of antimetabolite that operates by interrupting the S-phase of tumor cells. It is often used as a first-line chemotherapeutic agent for rectal adenocarcinomas. However, 5-FU presents cardiotoxicity risks, including coronary vasospasm, coronary thrombosis, cardiomyopathy, and sudden cardiac death.83 Qian et al. designed a platinum (DDP) and 5-FU co-delivery system based on cis-biodegradable temperature-sensitive hydrogel (PDLLA–PEG–PDLLA, PLEL) for intraoperative adjuvant combination chemotherapy of gastric cancer. This hydrogel system presents a specific sol–gel phase transition in response to physiological temperature and exhibits sustained drug release in vitro and in vivo. As evidenced, strong synergistic cell proliferation inhibition and apoptosis promotion effects of hydrogels were observed on gastric cancer MKN45-luc cells. Dual drug-loaded hydrogel formulations showed superior antitumor effects compared to single drug-loaded analogues and the free drug combination, thus assisting in side effect minimization in a peritoneal metastasis model of gastric cancer (Fig. 3B).84
Doxorubicin (DOX), an anthracycline antibiotic example, stands out as a widely-used model cancer drug for drug delivery studies. It comprises glycosides, which contain a tetracyclic ring and a sugar portion. The drug can prevent DNA replication and RNA transcription by embedding into DNA and inhibiting DNA and RNA polymerase reactions. DOX (Adriamycin) has several cytotoxic effects, cardiotoxicity, myelosuppression, and alopecia.85 The current application of DOX is used in combination with chemotherapy to reduce toxicity and improve efficacy. Toward this end, Wang et al. designed a dual drug delivery system based on nanogel-doped injectable hydrogel (NHG) for sequential topical delivery of combretastatin A4 phosphate (CA4P) and adriamycin (DOX) toward combined antiangiogenic and anticancer therapy. Injectable hydrogels were prepared to load and rapidly release the hydrophilic drug CA4P, while pH- and redox-stimulation-responsive nanohydrogels were incorporated into the injectable hydrogels via pH-responsive borate bonds to maintain long-term DOX delivery. Such an approach offers universal drug carriers for local dual drug delivery offering sequential release behavior through simple injections (Fig. 3C).86 DOX–CA4P@NHG significantly reduced cell viability compared to single-agent-loaded hydrogels by approximately 23% and 14% after 48 and 72 hours of incubation, respectively, which confirmed that the co-administration could greatly improve the therapeutic efficiency.
Porous polypropylene oxide (PLA) microspheres (PPMSs) loaded with paclitaxel (PTX) constructed via a modified double emulsion solvent evaporation method, along with cisplatin (DDP), were co-embedded in a thermosensitive hydrogel (PPMSs/DDP@Gel) for in situ chemotherapy, which is used for the treatment of melanoma by intra-tumor injection delivery. The system allowed the slow release of both drugs and exhibited good temperature-sensitive properties (Fig. 3D).87
Injectable hydrogels have been shown to be more suitable for intra-tumor drug therapy than traditional single carriers, and they have great potential for oncology chemotherapy by acting as a companion to drugs for enhancing treatment efficiency.
6. Delivery of gene drugs
6.1. DNA hydrogels
DNA hydrogel is a hydrophilic material that exists between fluid and solid states and is composed of DNA. It can be designed to take on different shapes and sizes and is divided into two categories: single-component DNA hydrogel and multi-component DNA hydrogel.91–94 Single-component DNA hydrogels are formed by using DNA as the only component, backbone, or cross-linking agent, either chemically (chemical bonding as the cross-linking point) or physically (non-covalent interactions such as hydrogen bonding, van der Waals’ forces, or entanglement of DNA molecules between the strands, etc.). Multicomponent DNA hydrogels are usually constructed by DNA-mediated cross-linking of other materials, including biocompatible polymers such as polyacrylamide, chitosan, and polyethylene glycol,95 as well as liposomes,96 black phosphorus quantum dots,97 carbon dots,98 silica nanoparticles99 nanoparticles, metal nanoparticles,100 and other nanoparticles. Due to the high loading capacity, excellent mechanical stability, and viscoelasticity of hydrogels, effective encapsulation of biologically active molecules (e.g., nucleotides, proteins, antibodies, and drugs) can be realized while their biological activities can be preserved.101–103 DNA hydrogels, which use nucleic acid aptamers as specific detection elements, can serve as a new class of visualization platforms based on the DNA base complementary pairing principle and nucleic acid aptamer–target molecule interactions.DNA is a polymer with a large number of negative charges, and its bases have the characteristics of aromatic ring structure, while DNA can be modified with many functional groups, such as acrylate, amino, carboxyl, and thiols, etc. These unique properties make DNA can be combined with some polymers or nano-materials through covalent interactions, electrostatic force, hydrogen bonding, van der Waals force, etc., which further expands the application of DNA hydrogel.104,105 The reversible nature of the conformational changes between DNA can realize the reversible regulation of the hydrogel solution state and gel state. Through the encapsulation of color-developing substances and signal-amplifying enzymes, the DNA–hydrogel will change the state of the soul according to the response to target molecules, specifically releasing the stored signal substances and realizing the output of visual signals.
DNA structure-based nanoreactors are a promising biomaterial for antitumor therapy due to their inherent biodegradability, biocompatibility, and tunable multifunctionality. He et al. designed a smart DNA nano hydrogel that can target cancer cells, control size, respond to pH, and load glucose oxidase (GOx). GOx was successfully encapsulated in a DNA nanohydrogel by hybridizing two X-shaped DNA monomers and DNA junctions assembled together to form a DNA nano hydrogel. The DNA junctions were engineered with i-motif sequences and modified with ferrocene (Fc). In an acidic tumor microenvironment, the DNA nano hydrogel can be broken down to release GOx, which can oxidize intratumoral glucose to produce gluconic acid and H2O2. The generated H2O2 is catalyzed by Fc to induce the production of toxic hydroxyl radicals (–OH), which can effectively kill cancer cells (Fig. 4A).106 The cytotoxicity assay of DNA nanohydrogels showed elevated cell mortality following Gel–GoxFc incubation, confirming its superior cell-killing capacity. In addition, Gel–Gox–Fc exhibited a total apoptosis rate of 51%.
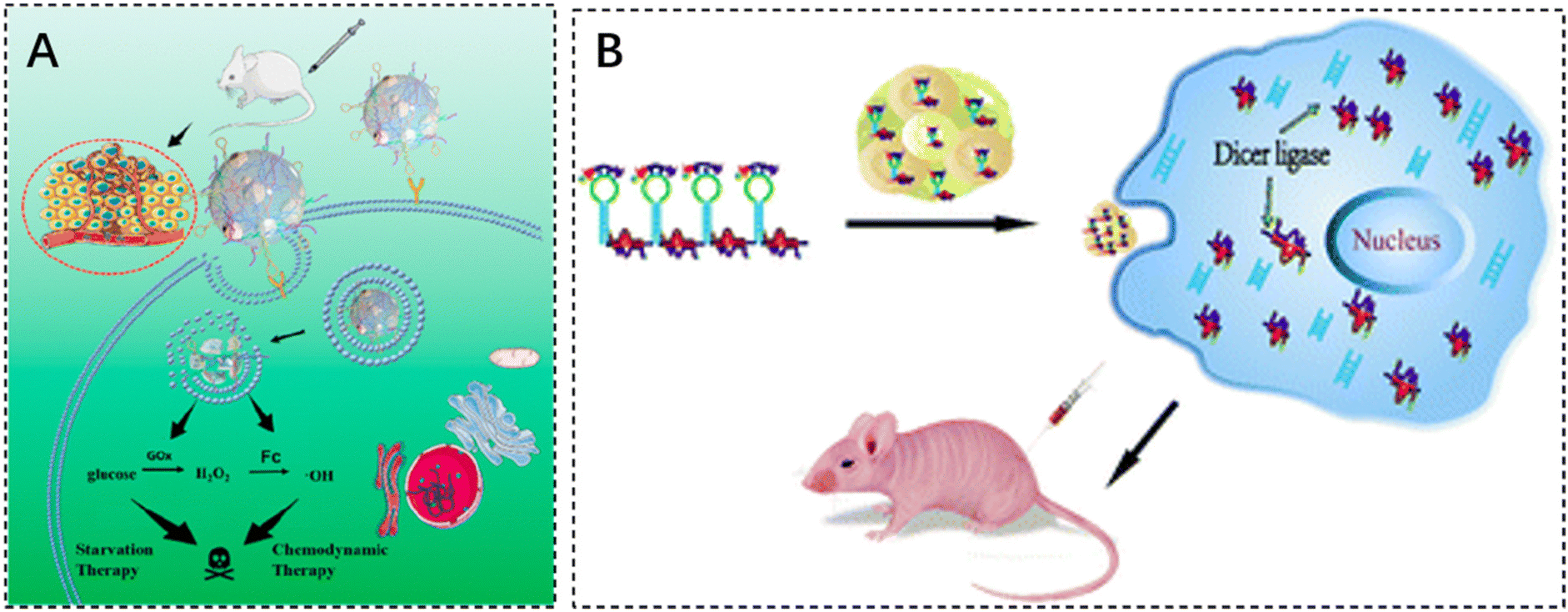 | ||
| Fig. 4 The application of injectable hydrogels in gene therapy. (A) Schematic illustration of a self-assembled DNA nano hydrogel and application of the antitumor combination therapy. Reproduced with permission from ref. 106. Copyright 2021, American Chemical Society. (B) mRNA nanoparticles bound to CXCL9-loaded polyethylene glycol (PEG) hydrogels. Reproduced with permission from ref. 107. Copyright 2022, Springer Nature. | ||
6.2. Other gene drugs in hydrogels
Gene therapy is an ideal method for the treatment of diseases related to genetic abnormalities, which can be achieved by inserting molecules such as synthetic mRNA, cyclic RNA, and self-amplifying RNA (saRNA) to achieve upregulation of gene expression, which are usually made into the form of liposomal nanoparticles, polymer nanoparticles, inorganic nanoparticles and other forms of delivery into the body, but the problems of easy degradation, poor targeting, and low efficiency of entry into the target cells continue to hinder the effective gene therapy.Maryam Rahman designed the HCM vaccine by combining mRNA nanoparticles with a polyethylene glycol (PEG) hydrogel loaded with the chemokine CXC ligand 9 (CXCL9). The HCM vaccine delivers a large mRNA payload via subcutaneous injection and recruits a diverse population of immune cells to produce significant anti-tumor efficacy (Fig. 4B).107 In addition, Zhang et al. developed a novel RNA-triple helix-hydrogel for the treatment of triple-negative breast cancer (TNBC) by doping RNA-triple helix and siRNA double-stranded bodies of CXCR4 into the same RNA nanoparticles without the addition of synthetic polycationic reagents. The RNA-triple helix consists of a tumor suppressor miRNA (miRNA-205) and an oncomiR inhibitor (miRNA-221), both of which have shown excellent performance in synergistically eliminating tumors.108 Following incubation with the RNA-triple-helix hydrogel for 36 hours, MDA-MB-231 cells exhibited a cell viability of mere 15%. Comparative analysis revealed that the RNA-triple-helix hydrogel outperformed free miRNA and RNA transcripts in silencing efficiency.
7. Other therapeutic agent delivery
7.1. For radiopharmaceutical delivery
Radiation therapy is a highly effective mainstream cancer treatment that kills cancer cells by targeting beams of high-energy radiation such as X-rays and gamma-rays to the tumor (external beam radiation therapy) or by introducing radioisotopes into the tumor through a catheter or other systemic route of administration. The main challenges of intense radiation therapy are the damage caused to adjacent normal tissues in the path of the radiation beam, the reduced response of tumors to radiation therapy, the resistance of cancer cells to radiation therapy, and the rapid elimination of radioisotope reagents. Hydrogels can be used as reservoirs of radioisotope reagents for targeted radiation therapy. Nanomaterials (e.g., radiosensitizers, such as nanoparticles containing high-Z elements) are used in the development of cancer drug carriers to focus radiation on tumors and overcome radiation resistance, thereby greatly improving the efficacy and safety of radiation therapy.109–111Liu et al. developed a radioiodinated thermosensitive supramolecular hydrogel for brachytherapy. An iodine 131 (131I)-labeled injectable thermosensitive methoxy poly(ethylene glycol)-b-poly tyrosine hydrogel (denoted as PETyr-131I) was prepared. This thermosensitive supramolecular hydrogel platform holds potential for 3D conformal radionuclide delivery and serves as a potential radiosensitizer due to its proximity to the primary tumor lesion or the postoperative cavity. The SmacN7-R9 peptide combined with PETyr-I 131 radiation demonstrated a higher radiosensitization ratio (SER) compared to the PETyr-I 131 radiation alone, suggesting the radiosensitizing effect of SmacN7-R9 peptide on HepG2 cells strongly result in synergistic therapeutic outcomes and reduced radiation-related side effects112 (Fig. 5).
 | ||
| Fig. 5 The use of injectable hydrogels in radiopharmaceutical delivery. Reproduced with permission from ref. 112. Copyright 2020, Elsevier B.V. | ||
7.2. For delivery of immunotherapeutic agents
Immune checkpoint inhibitors are effective means to prevent tumors from escaping immune surveillance. In recent years, the combination of immune checkpoint inhibitors with other tumor treatments has become a hot research topic in the field of tumor therapy. However, the adverse toxicity of many immunotherapeutic agents to T cells, natural killer (NK) cells, and dendritic cells (DC) limits their integration with immunotherapy.113 Some researchers are exploring new ways to deliver these drugs. Cui et al. designed and synthesized a peptide-based supramolecular filament (SF) hydrogel as a versatile carrier for local delivery of three immunomodulators with different mechanisms of action and different molecular weights, including αPD1 antibody, IL-15 cytokine, and STING agonist (CDA). It was demonstrated that intratumoral injection of SF solutions containing aPD1, IL-15, or CDA triggered hydrogelation, and the resulting hydrogel served as a scaffold reservoir for sustained release and response of immunotherapeutic agents in response to MMP-2, resulting in enhanced antitumor activity and reduced side effects (Fig. 6A).114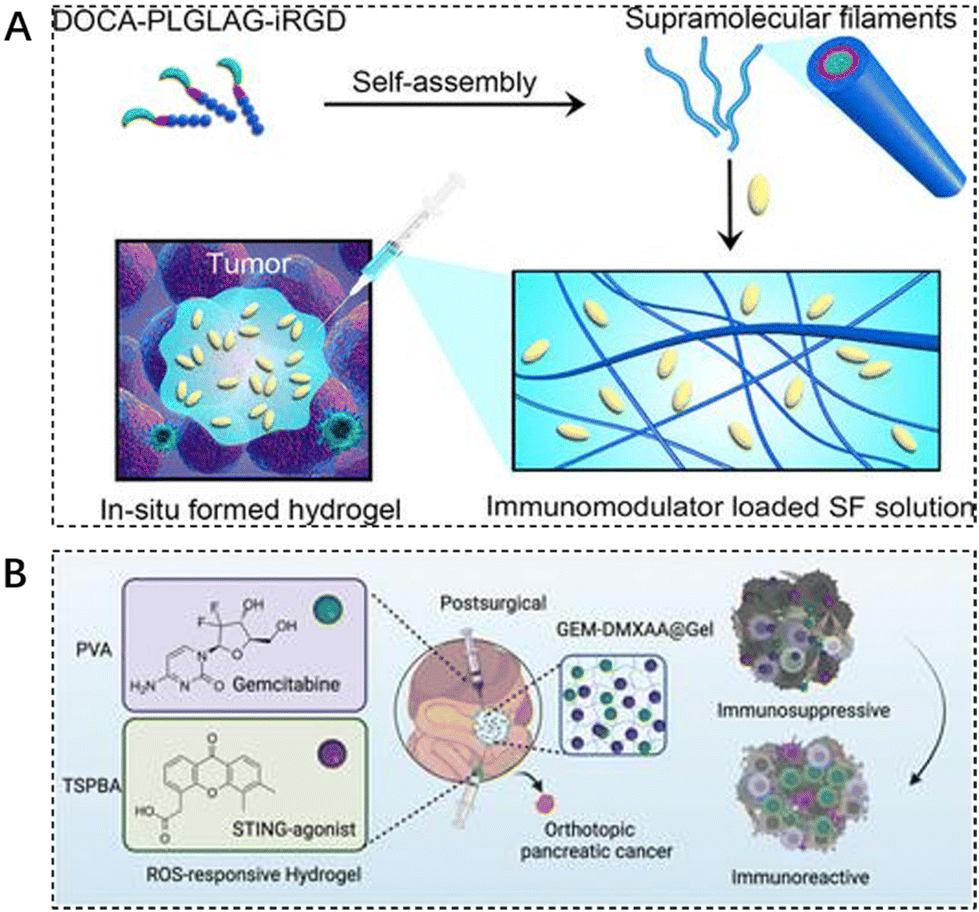 | ||
| Fig. 6 The use of injectable hydrogels in immunotherapy. (A) Schematic illustration of localized immunomodulator delivery using a DOCA–PLGLAG–iRGD supramolecular hydrogel for MMP-2-responsive drug release and tumor microenvironment regulation. Reproduced with permission from ref. 114. Copyright 2023, American Chemical Society. (B) Enhanced immunotherapy of pancreatic ductal adenocarcinoma with STING agonist and gemcitabine for in situ formation of ROS-responsive hydrogels. Reproduced with permission from ref. 115. Copyright 2023, Wiley. | ||
Immunotherapy, the most revolutionary anticancer strategy, faces major obstacles in achieving desirable outcomes in pancreatic ductal adenocarcinoma (PDAC) due to the highly immunosuppressive tumor microenvironment (TME). The traditional first-line chemotherapeutic agent gemcitabine (GEM) in the treatment of PDAC is insufficient to achieve durable efficacy when used alone. Liang et al. designed a reactive oxygen-degradable hydrogel system, denoted GEM–STING@Gel, to combine gemcitabine and the interferon gene-stimulating agonist DMXAA (5,6-dimethylxanthenone-4-acetic acid) co-delivered to the tumor site. It synergistically activates innate immunity and promotes cytotoxic T lymphocyte infiltration at the tumor site, thereby modulating immunosuppressive TME. The advantages of this integrated strategy combining chemotherapy, immunotherapy, and biomaterials-based hydrogels can be seen in the improved therapeutic efficacy and excellent biosafety (Fig. 6B).115
Hydrogels can elicit therapeutic effects by precisely delivering immune cells, such as dendritic cells (DC), directly to the tumor site. DCs stand out as the most functional antigen-presenting cells in the body. They possess the unique ability to mobilize other tumor-killing cells throughout the entire immune process, particularly in the presence of invading foreign entities, such as cancer cells. A DC vaccine could be designed to use the patients’ own DCs to activate the general immunity for attacking tumor cells. As a proof-of-concept, Yi et al. developed a CaCO3-biomineralized hydrogel DC vaccine by immobilizing the membrane proteins of 4T1 cells-DCs fusion cells (FP) into the biomineralized silk fibroin hydrogel. The FP-contained biomineralized hydrogel vaccine, SH@FP@CaCO3 exhibits improved immunogenicity by presenting a wide variety of tumor-associated antigens (TAAs) and facilitating long-term protein release for dendritic cell maturation and T cell activation. Additionally, the incorporated CaCO3 contributes to an increased pH level in the tumor microenvironment (TME), promoting the polarization of M2 to M1-type macrophages, which in turn reverses the immune-inhibitory microenvironment and alleviates the immunosuppressive effects on T cells. The outstanding immune activation effects of the biomineralized hydrogel vaccine including simultaneously enhancing immunogenicity and reversing the immunosuppressive TME represent a promising strategy for advancing cancer immunotherapy.116
Acquired cell therapy (ACT) represents a promising strategy for cancer treatment, wherein immune cells are harvested from the patients, engineered with receptors to target and destroy cancer cells, subsequently expanded in vitro, and reinfused into the patients for therapeutic intent. Hydrogels can facilitate the direct and precise delivery of immune cells to tumor sites, eliciting a therapeutic response. Grosskopf et al. developed a self-assembled, injectable biomaterials platform for CAR-T cell delivery utilizing polymer-nanoparticle (PNP) hydrogels. These hydrogels employ highly scalable chemistries and are formulated under gentle conditions that enable straightforward encapsulation of CAR-T cells and cytokines, while their shear-thinning properties and injectability afford minimally invasive locoregional delivery of the therapeutic cells to tumors. These injectable hydrogels can create a localized inflammatory microenvironment that extends the exposure of CAR-T cells and cytokines, thus enhancing localized CAR-T cell proliferation and anti-tumor efficacy.117
7.3. For delivery of phototherapeutic agents
The development of photothermal therapy (PTT) and photodynamic therapy (PDT) has also led to advances in cancer treatments that minimize damage to normal tissue. Photothermal therapy utilizes photothermal reagents that absorb external light energy and convert it into heat, which is then used to kill cancer cells. These agents are biocompatible, have a high photothermal conversion efficiency, and are able to accumulate at the target site, allowing for selective and non-invasive treatment. The volume of the surgical site, the duration of treatment, and the temperature reached during treatment should be carefully controlled to ensure optimal efficacy and to avoid damage to surrounding normal tissues.Photothermal therapy usually requires a local temperature (i.e., local hyperthermia) of less than 44 °C, while maintaining a body temperature of less than 42 °C to minimize side effects. UV and visible light have the potential to cause severe burns. Therefore, near-infrared (NIR) light with wavelengths in the range of 650–950 nm is used in most cases. Shen et al. noted that injectable thermosensitive photothermal network hydrogels (PNT-gels) were designed by guest self-assembly of photothermal conjugated polymers and β-cyclodextrin. The conjugated polymer backbone can directly convert incident light into heat, resulting in a PNT-gel with high photothermal conversion efficiency and photothermal stability. Remote controlled on-demand release of adriamycin (DOX) was achieved by photothermal-induced gel–sol transition. Since the backbone of the hydrogel absorbs NIR light and mediates the photothermal conversion itself, the PNT gel has the advantage of prolonged retention time, and NIR treatments can be repeated after a single intratumoral injection of this hydrogel. Under repeated NIR laser irradiation, synergistic photothermal chemotherapy mediated by the PNT-gel almost completely eradicated 4T1 breast cancer. The gel system integrates a multifunctional therapeutic platform with inherent photothermal properties and reversible stimulus responsiveness for on-demand delivery and a combination of photothermal chemotherapies118 (Fig. 7A).
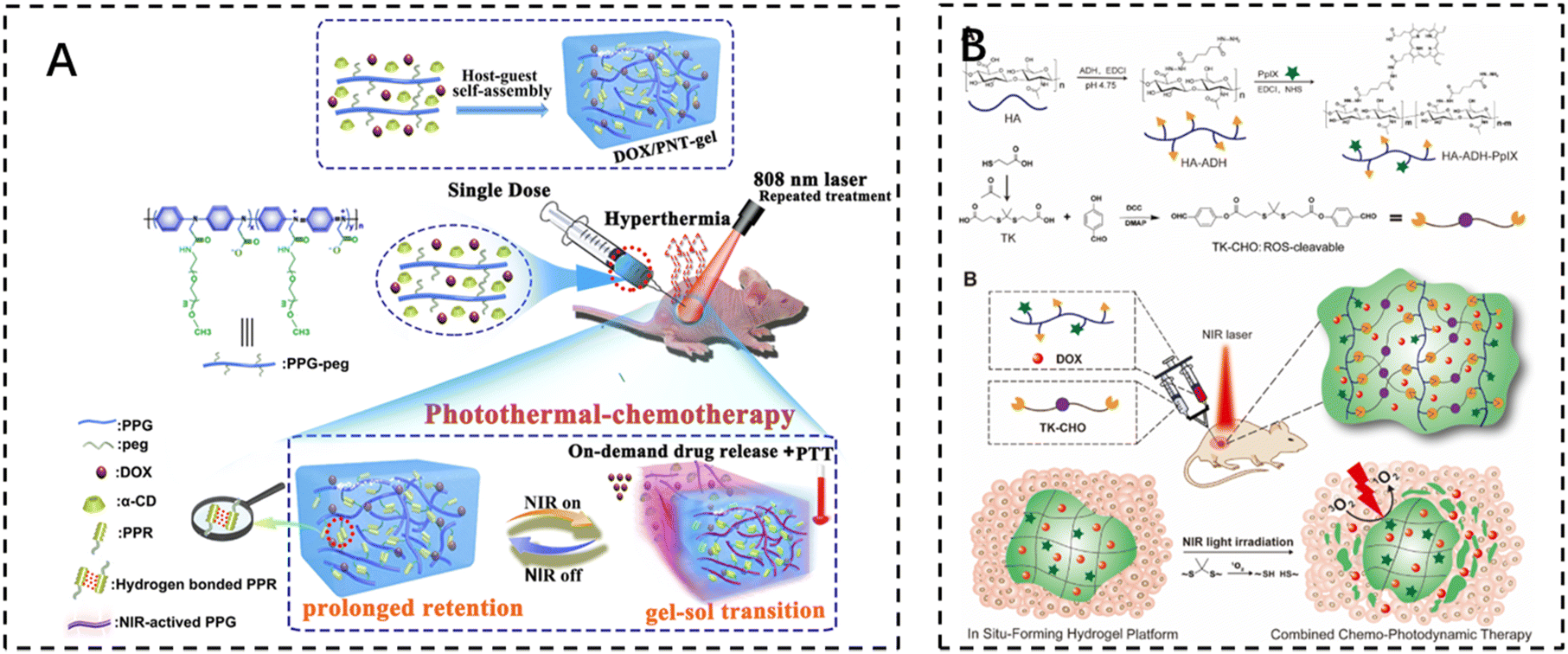 | ||
| Fig. 7 The use of injectable hydrogels in phototherapy. (A) Schematic illustration of the fabrication of the supramolecular PNT-gel and its application for repeated photothermal-chemotherapy via one-dose injection followed by multiple treatments. Reproduced with permission from ref. 118. Copyright 2019, Elsevierc. (B) Schematic illustrations depicting the synthesis of the HA–ADH–PpIX conjugate and the ROS-cleavable TK-CHO, as well as the formation of an injectable NIR light-triggered ROS degradable hydrogel for combined chemo-photodynamic therapy with light-tunable on-demand drug release. Reproduced with permission from ref. 121. Copyright 2020, Elsevierc. | ||
Photodynamic therapy (PDT) uses photosensitizers that, when exposed to light, trigger a photochemical reaction that produces cytotoxic reactive oxygen species (ROS). Cell death is then triggered through apoptosis, necrosis, or autophagy-related pathways. It can kill cancer cells directly, destroy tumor blood vessels, or induce an inflammatory response that leads to a host anti-tumor immune response.119 Photosensitizers themselves are inefficient due to their low solubility in water and self-bursting effect. It has been reported that photosensitizers encapsulated in hydrogels exhibit enhanced fluorescence and single-linear oxygen generation due to the diminished self-burst effect in the network, which reduces collisions between photosensitizer molecules.120 Zhao et al. developed an injectable and near-infrared (NIR)-triggered ROS-degradable hyaluronic acid hydrogel platform as a topical delivery vehicle for the photosensitizer protoporphyrin IX (PpIX) and the anticancer drug adriamycin (DOX) to achieve superior combined chemotherapy photodynamic therapy with on-demand modulation of drug release. A large amount of ROS produced by the hydrogel under near-infrared light irradiation not only accomplishes an efficient PDT effect but also cleaves ROS-sensitive small-molecule cross-linkers for hydrogel degradation and subsequent on-demand DOX release121 (Fig. 7B).
8. Combined drug delivery
Co-medication, the simultaneous or sequential use of two or more drugs, is often required for disease treatment due to various reasons. Reducing toxic side effects and maximizing the efficacy of the drug is the desired goal of the drug combination. Injectable hydrogels, as typical large-scale drug delivery carriers, can easily encapsulate a variety of therapeutic drugs by physical encapsulation, hydrophobic conjugation, and covalent binding, which significantly improves the in vivo anticancer effect. Various combination strategies based on injectable hydrogel platforms (e.g., chemotherapy drug combinations,122 chemotherapy and immunotherapy drug combinations123,124 chemotherapy and photothermal chemotherapy,125 chemotherapeutic drug and radioisotope combinations) have been developed and tested to enhance local anti-tumor effects (Table 2). Zhang et al. designed a novel injectable, self-crosslinked HA–SH hydrogel capable of simultaneously encapsulating multiple drugs (sorafenib, adriamycin, and metformin) simultaneously to enhance the chemotherapeutic effect. The hydrogel is relatively stable under physiological conditions and allows for rapid and direct release of the loaded drugs to the tumor site in a reductive tumor microenvironment. In vitro, antitumor activity and apoptosis assays have shown that hydrogels containing multiple drugs display significant synergistic effects on breast cancer cells. After peritumor administration, combination chemotherapy enhanced tumor cell sensitivity to drugs and promoted apoptosis of tumor cells (Fig. 8A).126 | ||
| Fig. 8 Injectable hydrogels in combination therapy. (A) A novel injectable self-crosslinking HA–SH hydrogel is able to encapsulate multiple drugs (sorafenib, adriamycin, and metformin) simultaneously to enhance chemotherapy efficacy. Reproduced with permission from ref. 126. Copyright 2017, Royal Society of Chemistry. (B) Scheme design of PNPPTX & MNPCpG@Gel for localized chemo-immunotherapy after surgical resection of glioma. Reproduced with permission from ref. 127. Copyright 2022, Elsevier B.V. (C) The preparation of ICG–MTX and ICG–MTX@PPA Gels and their application in tumor therapy. Reproduced with permission from ref. 129. Copyright 2022, Elsevier B.V. (D) The schematic diagram of design concept: formation of PECT/DOX MHg nanodrug depot as well as radionuclide reservoir for in situ chemoradiotherapy. Reproduced with permission from ref. 135. Copyright 2015, Elsevier B.V. | ||
| Type of combination therapy | Hydrogel's composition | Gelling mechanism | Drug composition | Strategies | Ref. |
|---|---|---|---|---|---|
| Multiple chemotherapy combinations | Injectable self-crosslinking HA–SH hydrogel | HA–SH polymers could self-crosslink to form the hydrogel by oxidizing free thiol groups into disulfide bonds. | Sorafenib, doxorubicin, and metformin | The combinational chemotherapy enhanced the sensitivities of tumor cells and promoted tumor cell apoptosis after peritumoral administration | 122 |
| Chemotherapy and immunotherapy combination | PLGA1750–PEG1500–PLGA1750 thermosensitive hydrogel framework (PNPPTX & MNPCpG@gel) | Response to temperature | Glioma homing peptide modified paclitaxel targeting nanoparticles (PNPPTX) and manipulated immunoadjuvant CpG targeting nanoparticles (MNPCpG) | The sustained-release PNPPTX could target the residual infiltration of glioma cells and produce tumor antigens. Meanwhile, MNPCpG targeted and activated the antigen-presenting cells, which enhanced the tumor antigen presentation ability and activated CD8+ T and NK cells to reverse immunosuppression of the glioma microenvironment | 127 |
| Photothermal and chemotherapy combination | ICG–MTX@PP A gels | Heat it at a temperature above 45 °C or under 808 nm light to form a hydrogel | Near-infrared (NIR) pho-geothermal reagents indocyanine green (ICG) and chemotherapy drugs Methotrexate (MTX) | ICG–MTX@PPA Gels can exhibit a sol–gel transition under photothermal stimulation, resulting in the accumulation at the tumor site and controlled release of nanomedicine. have the ability to NIR photothermal therapy and controlled release of the chemotherapy drug (MTX) | 129 |
| Chemotherapy and radiotherapy combination | Thermo-induced self-aggregation of poly(epsilon-caprolactone-co-1,4,8-trioxa[4.6]spiro-9-undecanone)-poly(ethylene glycol)-poly(epsilon-caprolactone-co-1,4,8-trioxa[4.6]spiro-9-undecanone) (PECT) | Response to temperature | Doxorubicin (DOX) and iodine-131 labeled hyaluronic acid ((131)I-HA) | Combination therapy significantly increased tumor growth inhibition efficiency while reducing drug-related side effects on major organs | 135 |
Chemotherapy lacks specificity in killing cancer cells and also kills normal cells, which significantly impacts the immune system of tumor patients. Excessive chemotherapy can shorten the survival time of patients. The use of chemotherapy and cellular immunotherapy can effectively make up for the shortcomings, and the immune cells not only can kill and clear the tumor cells directly or indirectly but also can reduce the toxic side effects of chemotherapy and rebuild the immune system of patients. Glioma is currently the most malignant brain tumor. Due to the presence of blood–brain barrier (BBB) and tumor cell heterogeneity, systemic chemotherapy is not effective in the treatment of surgically resected gliomas, and may even impair the body's immune system. Xin et al. designed an in situ slow-release hydrogel delivery system for the local delivery of combined chemotherapeutic agents and immune-assisted treatment of gliomas with combined chemoimmunotherapy through the resection cavity. Glioma homing peptide-modified paclitaxel-targeted nanoparticles (PNPPTX) and mannitolized immunoadjuvant CpG-targeted nanoparticles (MNPCpG) were embedded in a PLGA1750–PEG1500–PLGA1750 thermosensitive hydrogel framework (PNPPTX & MNPCpG@Gel). In vitro and in vivo results demonstrated that the targeted nanoparticle-hydrogel hybrid system could be cross-linked into a gel drug reservoir when injected into the glioma resection cavity. Then, sustained release of PNPPTX could target residual infiltrating glioma cells and produce tumor antigens. At the same time, MNPCpG targets and activates antigen-presenting cells to enhance tumor antigen presentation, activate CD8+ T and NK cells, and reverse immunosuppression in the glioma microenvironment127 (Fig. 8B).
Combined delivery of photothermal and chemotherapeutic agents is another approach. However, due to irregular tumor margins and blurred boundaries between normal and necrotic tissues, a single PTT could not achieve the desired therapeutic effect, and overheating would lead to severe inflammation of local tissues. An injectable composite hydrogel prepared from oxidized hyaluronic acid (OHA) and hydroxypropyl chitosan (HPCS) via imine bonding was used as a delivery substrate for functional substances. In the gel medium, mesoporous polydopamine (MPDA) nanoparticles were doped as an efficient photothermal agent and reservoir of DOX, which could achieve good photothermal conversion performance and slow drug release, and showed good Hepa1-6 tumor inhibition in vivo.128
Based on the former drug delivery system in which the DOX-encapsulated nanoparticles could not specifically act on tumors, the targeted nanodrugs were designed to be combined with an injectable hydrogel for photothermal chemotherapy combination therapy. The targeted nanomedicine (ICG–MTX) was prepared by combining a near-infrared (NIR) photothermal reagent (ICG) and a chemotherapeutic drug (MTX). ICG–MTX was then mixed with a hydrogel precursor and a free radical initiator to obtain an injectable hydrogel precursor solution. Under near-infrared light irradiation, the precursor solution can release alkyl radicals, which promotes the transformation of the precursor solution from a liquid to a colloidal state, allowing the nanomedicine to be effectively retained at the tumor site and continue to be released from the hydrogel. Due to the targeted nature of MTX, the released ICG–MTX can target tumor cells and improve the accuracy of combined photothermal-chemical therapy129 (Fig. 8C).
Radiation therapy, based on radioisotopes such as 131I, 67Cu, 188Re, 90Y and 177Lu, has been widely used in the clinical treatment of many types of cancer, including lung, rectal, liver, and cervical cancers. During radiation therapy, healthy tissues are also exposed to radiation.130–132 Many chemotherapeutic agents can be used as radiosensitizers to reduce radiation resistance and improve the efficacy of radiation therapy. However, their side effects still limit their clinical use.133,134 To overcome these challenges, injectable hydrogels have been used as a flexible platform for co-delivery of chemotherapeutic agents and radioisotopes. By simultaneously delivering radioisotopes and chemotherapeutic drugs, injectable thermosensitive micellar hydrogels can not only be used as micellar drug reservoirs to locally deliver concentrated chemotherapeutic nano drugs but also immobilize the radioisotopes at the thermal focus of internal irradiation. For example, a hydrogel formed from adriamycin (DOX) and 131I-labeled hyaluronic acid (131I-HA) were used as model therapeutic agents. It exhibits a sol–gel transition near body temperature, which allows for the rapid intracellular release of DOX to internalize it, while 131I can be stably immobilized at the injection site. The hydrogel allows for precise control of the dosage and ratio of the combined therapeutic agent, resulting in the desired therapeutic effect by a single administration with reduced side effects (Fig. 8D).135
9. Clinical applications of injectable hydrogels in tumor therapy
Injectable hydrogels have received extensive attention for their applications in drug delivery and tissue engineering. Various hydrogel technologies have obtained regulatory approvals for healthcare applications, including cancer treatment, cosmetic plastic surgery, and spinal fusion. Cancer therapeutic hydrogels have been utilized to encapsulate and release small molecules and biologics, enabling sustained local therapeutic drug delivery in specific tumor sites. For example, Endo's Vantas, an FDA-approved subcutaneous hormone therapy, efficiently prevents the growth of testosterone-dependent prostate cancer cells by releasing gonadotropin-releasing hormone (GnRH) or luteinizing hormone-releasing hormone (LH–RH) through a cylindrical diffusion-controlled reservoir system. The long-term convenience offered by these hydrogel reservoirs greatly enhances patient acceptance and convenience through a single injection. Nevertheless, injectable hydrogels still need further consideration regarding their polymer-dependent, mechanical and solute transport properties, degradability, compatibility, and scale-up challenges.10. Conclusions and outlook
The study of in situ hydrogel formation has recently gained attention due to its potential for local delivery of anticancer drugs via intra-tumor injection. This paper discusses the preparation of injectable hydrogels containing various anticancer drugs through chemical and physical interactions, as well as the injection therapy of these hydrogels within tumors, with a focus on the types of drugs delivered.The greatest advantage of injectable hydrogels is their potential for non-invasive administration, which offers numerous benefits to both patients and healthcare providers. Unlike traditional drug delivery methods that involve invasive procedures such as injections or surgical implantation, injectable hydrogels can be easily administered through minimally invasive techniques such as subcutaneous or intramuscular injections. This non-invasive route eliminates the need for complex surgical procedures, reduces the risk of infections, and improves patient comfort. Moreover, injectable hydrogels can be formulated to be biodegradable, eliminating the need for subsequent removal surgeries. This ease of administration and patient comfort make injectable hydrogels an attractive option for drug delivery.
In addition, injectable hydrogels have the ability to enhance the stability of drug molecules and provide controlled release. The hydrogel matrix can protect the encapsulated drug from degradation, enzymatic activity, and harsh physiological conditions, thereby increasing its shelf life. Additionally, the porous structure of hydrogels allows for the controlled release of drugs over an extended period, ensuring sustained therapeutic levels in the body. This controlled release mechanism not only improves drug efficacy but also reduces the frequency of administration, leading to enhanced patient compliance.
Finally, injectable hydrogels offer the unique advantage of targeted and localized drug delivery. By incorporating specific ligands or functional groups into the hydrogel matrix, drugs can be selectively delivered to the desired site of action. This targeted delivery minimizes systemic exposure, reduces off-target effects, and enhances therapeutic outcomes. Furthermore, the injectability of hydrogels allows for precise placement of the drug-loaded hydrogel at the desired location, enabling localized treatment of diseases such as tumors or chronic wounds. This targeted and localized drug delivery approach has the potential to revolutionize the treatment of various diseases, making it a highly desirable feature of injectable hydrogels.
However, injectable hydrogels are fraught with challenges in cancer therapy and translation to the clinic. Firstly, designing hydrogels with both desirable mechanical properties and injectability is difficult. Hydrogels need to possess sufficient mechanical strength to provide structural integrity and support tissue regeneration, while also being injectable through minimally invasive procedures. Achieving this delicate balance is crucial for effective cancer therapy, as it enables the hydrogels to be easily delivered to the tumor site and exert their therapeutic effects. Researchers are actively exploring various strategies, such as incorporating reinforcing agents or crosslinking methods, to enhance the mechanical strength of hydrogels. Suo et al. proposes to prepare hydrogels that are much more entangled than cross-linked by using unusually small amounts of water, cross-linking agents, and initiators in the synthesis process.136 Ordinary hydrogels are predominantly cross-linked and reflect a mesh-like topology, while highly entangled hydrogels have a fabric-like topology. The different topologies lead to different properties, and the dense entanglement allows the hydrogels to transfer tensile tension along the length of the polymer chain, dissipate elastic potential energy, and improve the tensile strength and toughness of the hydrogels.
Secondly, the controlled release of therapeutic agents from injectable hydrogels remains a challenge. Injectable hydrogels have the potential to encapsulate and deliver various anti-cancer drugs. However, achieving sustained and controlled release of these agents from the hydrogels is essential for optimal therapeutic outcomes. The release kinetics should be tailored to match the desired drug concentration and duration of treatment. Developing hydrogels that can release therapeutic agents in a controlled manner remains a complex task, requiring careful consideration of factors such as drug loading, hydrogel degradation, and diffusion properties.
Thirdly, the biocompatibility and biodegradability of injectable hydrogels are crucial factors for their successful translation to the clinic. Since most hydrogels are composed of chemically synthesized polymers via organic solvents or cross-linking agents, degradation of the polymer matrix may trigger an inflammatory response. Therefore, the possible immunogenicity caused by hydrogels is also an issue of concern. The short-term biocompatibility of hydrogels can be evaluated using animal models, but there is no guarantee that the long-term biocompatibility will be the same, and the long-term safety of composite hydrogels and additives is still an important issue of concern. Ensuring the safety and efficacy of injectable hydrogels in vivo is a critical step towards their clinical translation.
Finally, the clinical translation of injectable hydrogels for cancer therapy is hindered by regulatory and manufacturing challenges. The development and approval of medical devices, including injectable hydrogels, require compliance with rigorous regulatory standards and guidelines. These standards ensure the safety, efficacy, and quality of the hydrogels, but they also add complexity and time to the translation process. Additionally, the scale-up manufacturing of injectable hydrogels to meet the demands of large-scale clinical trials and commercialization poses significant challenges. Ensuring batch-to-batch consistency, reproducibility, and cost-effectiveness are key considerations for successful clinical translation.
In conclusion, although there are still many risks and challenges associated with the large-scale application of hydrogels in cancer therapy, they also offer several advantages and broader perspectives than conventional therapeutic agents (Fig. 9). Future attention will also be more focused on highly controlled and precisely tunable injectable hydrogel formulations as well as on the study of release kinetics and triggering conditions of different hydrogels.
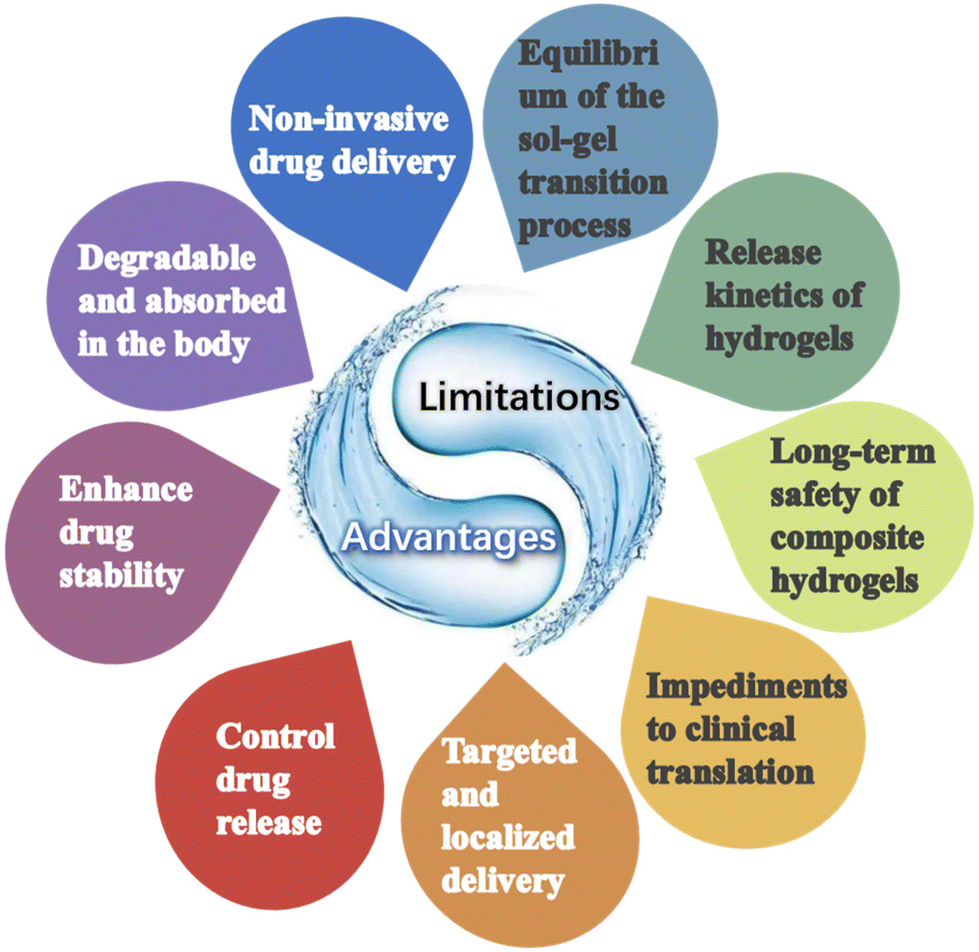 | ||
| Fig. 9 Advantages and limitations of injectable hydrogels. | ||
Author contributions
Yao Cheng & Haitao Zhang: Writing – original draft, writing – review & editing, writing – revision. Hua Wei & Cui-Yun Yu: Conceptualization, supervision, writing-review & editing writing revision.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 82373826), Key R&D Program of Hunan province (No. 2021SK2036, 2023SK2043), Hunan Science and Technology Innovation Leading Talent Project (No. 2022RC3080), Research Foundation of Education Bureau of Hunan Province (No. 21A0284), Natural Science Foundation of Hunan province (No. 2021JJ30603, 2022JJ40381), Health Research Project of Hunan Provincial Health Commission (No. 202113021875) and International Joint Laboratory for Arteriosclerotic Disease Research of Hunan Province (No. 2018WK4031).References
- R. L. Siegel, K. D. Miller, N. S. Wagle and A. Jemal, CA-Cancer J. Clin., 2023, 73(1), 17–48 Search PubMed.
- S. Y. Qin, A. Q. Zhang, S. X. Cheng, L. Rong and X. Z. Zhang, Biomaterials, 2017, 112, 234–247 CAS.
- T. A. Seidu, P. T. Kutoka, D. O. Asante, M. A. Farooq, R. N. Alolga and W. Bo, Pharmaceutics, 2022, 14(5), 1113 CAS.
- J. Wang, X. Hu and D. Xiang, Drug Delivery, 2018, 25(1), 1319–1327 CAS.
- M. Zhang, S. Gao, D. Yang, Y. Fang, X. Lin, X. Jin, Y. Liu, X. Liu, K. Su and K. Shi, Acta Pharm. Sin. B, 2021, 11(8), 2265–2285 CAS.
- L. E. Low, J. Wu, J. Lee, B. T. Tey, B. H. Goh, J. Gao, F. Li and D. Ling, J. Controlled Release, 2020, 324, 69–103 CAS.
- M. S. Yoon, Y. J. Lee, H. J. Shin, C. W. Park, S. B. Han, J. K. Jung, J. S. Kim and D. H. Shin, Pharmaceutics, 2020, 12(12), 1156 CAS.
- B. Balakrishnan and R. Banerjee, Chem. Rev., 2011, 111(8), 4453–4474 CAS.
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112(5), 2853–2888 CAS.
- W. R. Gombotz and S. Wee, Adv. Drug Delivery Rev., 1998, 31(3), 267–285 CAS.
- Y. Chao, Q. Chen and Z. Liu, Adv. Funct. Mater., 2020, 30(2), 1902785 CAS.
- Z. Zheng, J. Sun, J. Wang, S. He, Y. Huang, X. Yang, Y. Zhao, C. Yu and H. Wei, Chem. Eng. J., 2023, 473, 145228 CAS.
- Z. Zheng, C. Lei, H. Liu, M. Jiang, Z. Zhou, Y. Zhao, C. Y. Yu and H. Wei, Adv. Healthcare Mater., 2022, 11(19), e2200990 Search PubMed.
- Z. Zheng, Y. Tan, Y. Li, Y. Liu, G. Yi, C. Y. Yu and H. Wei, J. Controlled Release, 2021, 335, 216–236 CAS.
- P. Bertsch, M. Diba, D. J. Mooney and S. Leeuwenburgh, Chem. Rev., 2023, 123(2), 834–873 CAS.
- C. Liu, Y. Liao, L. Liu, L. Xie, J. Liu, Y. Zhang and Y. Li, Front. Bioeng. Biotechnol., 2023, 11, 1121887 Search PubMed.
- D. G. Leach, S. Young and J. D. Hartgerink, Acta Biomater., 2019, 88, 15–31 CAS.
- J. Li, G. Chen, X. Xu, P. Abdou, Q. Jiang, D. Shi and Z. Gu, Regener. Biomater., 2019, 6(3), 129–140 CAS.
- J. H. Lee, Biomater. Res., 2018, 22, 27 Search PubMed.
- K. Pal, V. K. Singh, A. Anis, G. Thakur and M. K. Bhattacharya, Polym.-Plast. Technol. Eng., 2013, 52(14), 1391–1422 CAS.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53(3), 321–339 CAS.
- P. Matricardi, C. Di Meo, T. Coviello, W. E. Hennink and F. Alhaique, Adv. Drug Delivery Rev., 2013, 65(9), 1172–1187 CAS.
- A. Bertz, S. Whl-Bruhn, S. Miethe, B. Tiersch, J. Koetz, M. Hust, H. Bunjes and H. Menzel, J. Biotechnol., 2013, 163(2), 243–249 CAS.
- Q. Ma, Q. Li, X. Cai, P. Zhou, Z. Wu, B. Wang, W. Ma and S. Fu, J. Drug Deliv. Sci. Tec., 2022, 76, 103817 CAS.
- F. Rizzo and N. S. Kehr, Adv. Healthcare Mater., 2021, 10(1), e2001341 Search PubMed.
- M. He, J. Sui, Y. Chen, S. Bian, Y. Cui, C. Zhou, Y. Sun, J. Liang, Y. Fan and X. Zhang, J. Mater. Chem. B, 2017, 5(25), 4852–4862 CAS.
- A. Fakhari and S. J. Anand, J. Controlled Release, 2015, 220(Pt A), 465–475 CAS.
- H. Jin, C. Wan, Z. Zou, G. Zhao, L. Zhang, Y. Geng, T. Chen, A. Huang, F. Jiang, J. P. Feng, J. F. Lovell, J. Chen, G. Wu and K. Yang, ACS Nano, 2018, 12(4), 3295–3310 CAS.
- B. L. Abraham, E. S. Toriki, N. J. Tucker and B. L. Nilsson, J. Mater. Chem. B, 2020, 8(30), 6366–6377 CAS.
- N. Sato, Y. Aoyama, J. Yamanaka, A. Toyotama and T. Okuzono, Sci. Rep., 2017, 7(1), 6099 Search PubMed.
- M. Popescu, G. Liontos, A. Avgeropoulos, E. Voulgari, K. Avgoustakis and C. Tsitsilianis, ACS Appl. Mater. Interfaces, 2016, 8(27), 17539–17548 CAS.
- K. Chwalek, K. R. Levental, M. V. Tsurkan, A. Zieris, U. Freudenberg and C. Werner, Biomaterials, 2011, 32(36), 9649–9657 CAS.
- M. Martínez-Martínez, G. Rodríguez-Berna, M. Bermejo, I. Gonzalez-Alvarez, M. Gonzalez-Alvarez and V. Merino, Eur. J. Pharm. Biopharm., 2019, 136, 174–183 Search PubMed.
- S. Eswaramma, N. S. Reddy and K. Rao, Int. J. Biol. Macromol., 2017, 103, 1162–1172 CAS.
- J. Xiong, X. Wang, L. Li, Q. Li, S. Zheng, Z. Liu, W. Li and F. Yan, Angew. Chem., Int. Ed., 2023, e202316375 Search PubMed.
- M. Norouzi, B. Nazari and D. W. Miller, Drug Discovery Today, 2016, 21(11), 1835–1849 CAS.
- S. Yu, C. He and X. Chen, Macromol. Biosci., 2018, 18(12), e1800240 Search PubMed.
- F. Rizzo and N. S. Kehr, Adv. Healthcare Mater., 2021, 10(1), 2001341 CAS.
- B. Das, D. Chattopadhyay and D. Rana, Biomater. Sci., 2020, 8(17), 4665–4691 CAS.
- R. Parhi, Adv. Pharm. Bull., 2017, 7(4), 515–530 CAS.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54(1), 3–12 CAS.
- F. Akther, P. Little, Z. Li, N. Nguyen and H. T. Ta, RSC Adv., 2020, 10(71), 43682–43703 CAS.
- G. R. Shin, H. E. Kim, J. H. Kim, S. Choi and M. S. Kim, Pharmaceutics, 2021, 13(11), 1953 CAS.
- J. Xiong, X. Wang, L. Li, Q. Li, S. Zheng, Z. Liu, W. Li and F. Yan, Angew. Chem., Int. Ed., 2023, e202316375 Search PubMed.
- S. Huang, X. Xia, R. Fan and Z. Qian, Chem. Mater., 2020, 32(5), 1937–1945 CAS.
- Z. Zheng, Z. Guo, F. Zhong, B. Wang, L. Liu, W. Ma, C. Y. Yu and H. Wei, J. Controlled Release, 2022, 347, 127–142 CAS.
- Z. Wang, Y. Zhang, Y. Yin, J. Liu, P. Li, Y. Zhao, D. Bai, H. Zhao, X. Han and Q. Chen, Adv. Mater., 2022, 34(13), 2108300 CAS.
- S. Tang, X. Ke, H. Wang, J. Xie, J. Yang, J. Luo and J. Li, ACS Appl. Mater. Interfaces, 2022, 14(39), 44890–44901 CAS.
- X. Liu, X. He, B. Yang, L. Lai, N. Chen, J. Hu and Q. Lu, Adv. Funct. Mater., 2021, 31(3), 2008187 CAS.
- L. Meng, C. Shao, C. Cui, F. Xu, J. Lei and J. Yang, ACS Appl. Mater. Interfaces, 2020, 12(1), 1628–1639 CAS.
- P. J. Haddow, M. A. Da Silva, D. B. Kaldybekov, C. A. Dreiss, E. Hoffman, V. Hutter, V. V. Khutoryanskiy, S. B. Kirton, N. Mahmoudi, W. J. McAuley and M. T. Cook, Macromol. Biosci., 2022, 22(3), 2100432 CAS.
- A. Andersen, M. Krogsgaard and H. Birkedal, Biomacromolecules, 2018, 19(5), 1402–1409 CAS.
- J. Y. Seo, B. Lee, T. W. Kang, J. H. Noh, M. J. Kim, Y. B. Ji, H. J. Ju, B. H. Min and M. S. Kim, Tissue Eng. Regener. Med., 2018, 15(5), 513–520 CAS.
- F. Akther, P. Little, Z. Li, N. T. Nguyen and H. T. Ta, RSC Adv., 2020, 10(71), 43682–43703 CAS.
- H. Y. Liu, M. Korc and C. C. Lin, Biomaterials, 2018, 160, 24–36 CAS.
- M. Okubo, D. Iohara, M. Anraku, T. Higashi, K. Uekama and F. Hirayama, Int. J. Pharm., 2020, 575, 118845 CAS.
- M. A. Haque, T. Kurokawa, G. Kamita and J. P. Gong, Macromolecules, 2011, 44(22), 8916–8924 CAS.
- X. Zhao, Soft Matter, 2014, 10(5), 672–687 CAS.
- N. Chen, H. Wang, C. Ling, W. Vermerris, B. Wang and Z. Tong, Carbohydr. Polym., 2019, 225, 115207 CAS.
- G. Choi and H. J. Cha, Biomater. Res., 2019, 23, 18 Search PubMed.
- H. Yao, J. Wang and S. Mi, Photo Processing for Biomedical Hydrogels Design and Functionality: A Review, Polymers, 2018, 10(1), 11 Search PubMed.
- E. Nicol, Biomacromolecules, 2021, 22(4), 1325–1345 CAS.
- S. Pillarisetti, S. Uthaman, K. M. Huh, Y. S. Koh, S. Lee and I. Park, Tissue Eng. Regener. Med., 2019, 16(5), 451–465 Search PubMed.
- S. Mantha, S. Pillai, P. Khayambashi, A. Upadhyay, Y. Zhang, O. Tao, H. M. Pham and S. D. Tran, Materials, 2019, 12(20), 3323 CAS.
- H. Zhu, H. Yang, Y. Ma, T. J. Lu, F. Xu, G. M. Genin and M. Lin, Adv. Funct. Mater., 2020, 30(32), 2000639 CAS.
- D. H. Yang and H. J. Chun, Adv. Exp. Med. Biol., 2020, 1249, 85–93 CAS.
- C. Lee, C. D. O'Connell, C. Onofrillo, P. F. M. Choong, C. Di Bella and S. Duchi, Stem Cells Transl. Med., 2020, 9(3), 302–315 CAS.
- W. Ji, Q. Wu, X. Han, W. Zhang, W. Wei, L. Chen, L. Li and W. Huang, Sci. China: Life Sci., 2020, 63(12), 1813–1828 Search PubMed.
- M. Arslan and M. A. Tasdelen, Polymer Nanocomposites via Click Chemistry Reactions, Polymers, 2017, 9(10), 499 Search PubMed.
- J. Gopinathan and I. Noh, Tissue Eng. Regener. Med., 2018, 15(5), 531–546 CAS.
- X. Bai, S. Lv, Z. Cao, B. Ni, X. Wang, P. Ning, D. Ma, H. Wei and M. Liu, Carbohydr. Polym., 2017, 166, 123–130 CAS.
- M. Arslan and M. A. Tasdelen, Polymer Nanocomposites via Click Chemistry Reactions, Polymers, 2017, 9(10), 499 Search PubMed.
- K. Nwe and M. W. Brechbiel, Cancer Biother. Radiopharm., 2009, 24(3), 289–302 CAS.
- A. Kasiski, M. Zieliska-Pisklak, E. Oledzka and M. Sobczak, Int. J. Nanomed., 2020, 15, 4541–4572 Search PubMed.
- K. Kulkarni, R. L. Minehan, T. Gamot, H. A. Coleman, S. Bowles, Q. Lin, D. Hopper, S. E. Northfield, R. A. Hughes, R. E. Widdop, M. Aguilar, H. C. Parkington and M. P. Del Borgo, ACS Appl. Mater. Interfaces, 2021, 13(49), 58279–58290 CAS.
- F. Morshedloo, A. B. Khoshfetrat, D. Kazemi and M. Ahmadian, J. Biomed. Mater. Res., Part B, 2020, 108(7), 2950–2960 CAS.
- S. Kim, Y. An, H. D. Kim, K. Kim, S. Lee, H. Yim, B. Kim and N. S. Hwang, Int. J. Biol. Macromol., 2018, 110, 479–487 CAS.
- K. M. Park, Y. Lee, J. Y. Son, J. W. Bae and K. D. Park, Bioconjugate Chem., 2012, 23(10), 2042–2050 CAS.
- L. Fan, X. Ge, Y. Qian, M. Wei, Z. Zhang, W. E. Yuan and Y. Ouyang, Front. Bioeng. Biotechnol., 2020, 8, 654 Search PubMed.
- M. Norouzi, B. Nazari and D. W. Miller, Drug Discovery Today, 2016, 21(11), 1835–1849 CAS.
- C. Zhang, C. Xu, X. Gao and Q. Yao, Theranostics, 2022, 12(5), 2115–2132 CAS.
- M. S. Gil, T. Thambi, V. Phan, S. H. Kim and D. S. Lee, J. Mater. Chem. B, 2017, 5(34), 7140–7152 CAS.
- C. Baldeo, C. Baldeo, K. Mody, K. Seegobin and F. Rollini, J. Am. Coll. Cardiol., 2018, 71(11, Supplement), A2324 Search PubMed.
- W. Chen, K. Shi, J. Liu, P. Yang, R. Han, M. Pan, L. Yuan, C. Fang, Y. Yu and Z. Qian, Bioact. Mater., 2023, 23, 1–15 CAS.
- K. Renu, L. P. Pureti, B. Vellingiri and A. Valsala Gopalakrishnan, Toxin Rev., 2022, 41(2), 650–674 Search PubMed.
- W. J. Yang, P. Zhou, L. Liang, Y. Cao, J. Qiao, X. Li, Z. Teng and L. Wang, ACS Appl. Mater. Interfaces, 2018, 10(22), 18560–18573 CAS.
- Y. Liu, W. Ma, P. Zhou, Q. Wen, Q. Wen, Y. Lu, L. Zhao, H. Shi, J. Dai, J. Li and S. Fu, Biomed. Pharmacother., 2023, 160, 114380 CAS.
- J. Xing, X. Qi, Y. Jiang, X. Zhu, Z. Zhang, X. Qin and Z. Wu, Pharm. Dev. Technol., 2015, 20(7), 812–819 CAS.
- M. Liu, P. Huang, W. Wang, Z. Feng, J. Zhang, L. Deng and A. Dong, J. Mater. Chem. B, 2019, 7(16), 2667–2677 CAS.
- J. Y. Heo, J. H. Noh, S. H. Park, Y. B. Ji, H. J. Ju, D. Y. Kim, B. Lee and M. S. Kim, Pharmaceutics, 2019, 11(9), 438 CAS.
- J. Li, L. Mo, C. H. Lu, T. Fu, H. H. Yang and W. Tan, Chem. Soc. Rev., 2016, 45(5), 1410–1431 CAS.
- M. Shahbazi, T. S. Bauleth-Ramos and H. L. A. Santos, Adv. Ther., 2018, 1(4), 1800042 Search PubMed.
- S. R. Deshpande, R. Hammink, R. K. Das, F. H. T. Nelissen, K. G. Blank, A. E. Rowan and H. A. Heus, Adv. Funct. Mater., 2016, 26(48), 9075–9082 CAS.
- C. Li, P. Chen, Y. Shao, X. Zhou, Y. Wu, Z. Yang, Z. Li, T. Weil and D. Liu, Small, 2015, 11(9–10), 1138–1143 CAS.
- Q. Chen, S. Wang, T. Huang, F. Xiao, Z. Wu and R. Yu, Anal. Chem., 2022, 94(14), 5530–5537 CAS.
- D. Lyu, S. Chen and W. Guo, Small, 2018, 14(15), 1704039 Search PubMed.
- L. Zhou, W. Pi, M. Hao, Y. Li, H. An, Q. Li, P. Zhang and Y. Wen, Biomater. Sci., 2021, 9(14), 4904–4921 CAS.
- S. Singh, A. Mishra, R. Kumari, K. K. Sinha, M. K. Singh and P. Das, Carbon, 2017, 114, 169–176 CAS.
- R. Bagley and S. T. Jones, Chem. Commun., 2021, 57(91), 12111–12114 Search PubMed.
- J. Song, S. Hwang, K. Im, J. Hur, J. Nam, S. Hwang, G. O. Ahn, S. Kim and N. Park, J. Mater. Chem. B, 2015, 3(8), 1537–1543 CAS.
- S. Ming, K. Lin, H. Zhang, F. Jiang, P. Liu, J. Xu, G. Nie and X. Duan, Chem. Commun., 2020, 56(39), 5275–5278 CAS.
- Y. Shao, Z. Sun, Y. Wang, B. Zhang, D. Liu and Y. Li, ACS Appl. Mater. Interfaces, 2018, 10(11), 9310–9314 CAS.
- M. He, N. Nandu, T. B. Uyar, M. Royzen and M. V. Yigit, Chem. Commun., 2020, 56(53), 7313–7316 CAS.
- A. Finke, A. Schneider, A. Spreng, M. Leist, C. M. Niemeyer and A. Marx, Adv. Healthcare Mater., 2019, 8(9), 1900080 Search PubMed.
- Z. Zhang, J. Han, Y. Pei, R. Fan and J. Du, ACS Appl. Bio Mater., 2018, 1(4), 1206–1214 CAS.
- W. Song, P. Song, Y. Sun, Z. Zhang, H. Zhou, X. Zhang and P. He, ACS Biomater. Sci. Eng., 2021, 7(11), 5165–5174 CAS.
- F. Dastmalchi, N. Thomas, G. Ebrahim, M. Frain, G. Moore, H. Mendez-Gomez, E. Sayour, M. Tomdio, V. Subramaniam, T. Angelini, D. Mitchell and M. Rahman, Neuro-Oncology, 2022, 24, 105 Search PubMed.
- L. Ding, J. Li, C. Wu, F. Yan, X. Li and S. Zhang, J. Mater. Chem. B, 2020, 8(16), 3527–3533 CAS.
- K. Haume, S. Rosa, S. Grellet, M. A. Miaek, K. T. Butterworth, A. V. Solov'Yov, K. M. Prise, J. Golding and N. J. Mason, Cancer Nanotechnol., 2016, 7(1), 8 Search PubMed.
- G. Song, L. Cheng, Y. Chao, K. Yang and Z. Liu, Adv. Mater., 2017, 29(32), 1700996 Search PubMed.
- F. Wang, J. Chen, J. Liu and H. Zeng, Biomater. Sci., 2021, 9(10), 3543–3575 CAS.
- J. Liu, Y. Zhang, Q. Li, Z. Feng, P. Huang, W. Wang and J. Liu, Acta Biomater., 2020, 114, 133–145 CAS.
- P. Agarwal and I. D. Rupenthal, Drug Discovery Today, 2013, 18(7–8), 337–349 CAS.
- F. Wang, H. Su, Z. Wang, C. F. Anderson, X. Sun, H. Wang, P. Laffont, J. Hanes and H. Cui, ACS Nano, 2023, 17(11), 10651–10664 CAS.
- M. Wang, Q. Hu, J. Huang, F. Zhang, Z. Yao, S. Shao, X. Zhao and T. Liang, Adv. Healthcare Mater., 2023, 12(20), 2203264 CAS.
- W. Huo, X. Yang, B. Wang, L. Cao, Z. Fang, Z. Li, H. Liu, X. J. Liang, J. Zhang and Y. Jin, Biomaterials, 2022, 288, 121722 CAS.
- A. K. Grosskopf, L. Labanieh, D. D. Klysz, G. A. Roth, P. Xu, O. Adebowale, E. C. Gale, C. K. Jons, J. H. Klich, J. Yan, C. L. Maikawa, S. Correa, B. S. Ou, A. I. D'Aquino, J. R. Cochran, O. Chaudhuri, C. L. Mackall and E. A. Appel, Sci. Adv., 2022, 8(14), n8264 Search PubMed.
- C. Liu, X. Guo, C. Ruan, H. Hu, B. Jiang, H. Liang and X. Shen, Acta Biomater., 2019, 96, 281–294 CAS.
- C. N. Simone, J. S. Friedberg, E. Glatstein, J. P. Stevenson, D. H. Sterman, S. M. Hahn and K. A. Cengel, J. Thorac. Dis., 2012, 4(1), 63–75 CAS.
- L. Y. Xia, X. Zhang, M. Cao, Z. Chen and F. G. Wu, Biomacromolecules, 2017, 18(10), 3073–3081 CAS.
- X. Xu, Z. Zeng, Z. Huang, Y. Sun, Y. Huang, J. Chen, J. Ye, H. Yang, C. Yang and C. Zhao, Carbohydr. Polym., 2020, 229, 115394 CAS.
- M. He, J. Sui, Y. Chen, S. Bian, Y. Cui, C. Zhou, Y. Sun, J. Liang, Y. Fan and X. Zhang, J. Mater. Chem. B, 2017, 5(25), 4852–4862 CAS.
- C. Wang, J. Wang, X. Zhang, S. Yu, D. Wen, Q. Hu, Y. Ye, H. Bomba, X. Hu, Z. Liu, G. Dotti and Z. Gu, Sci. Transl. Med., 2018, 10(429), eaan3682 Search PubMed.
- L. Yang, Y. S. Li, Y. Z. Gou, X. Wang, X. M. Zhao and L. Tao, Polym. Chem., 2017, 8(34), 3071–3076 Search PubMed.
- B. Tan, Y. Wu, Y. Wu, K. Shi, R. Han, Y. Li, Z. Qian and J. Liao, ACS Appl. Mater. Interfaces, 2021, 13(27), 31542–31553 CAS.
- M. He, J. Sui, Y. Chen, S. Bian, Y. Cui, C. Zhou, Y. Sun, J. Liang, Y. Fan and X. Zhang, J. Mater. Chem. B, 2017, 5(25), 4852–4862 CAS.
- X. Wang, L. Ye, W. He, C. Teng, S. Sun, H. Lu, S. Li, L. Lv, X. Cao, H. Yin, W. Lv and H. Xin, J. Controlled Release, 2022, 345, 786–797 CAS.
- L. Rong, Y. Liu, Y. Fan, J. Xiao, Y. Su, L. Lu, S. Peng, W. Yuan and M. Zhan, Carbohydr. Polym., 2023, 310, 120721 CAS.
- D. Qi, H. Zhu, Y. Kong and Q. Shen, Polymers, 2022, 14(24), 5547 CAS.
- A. J. Breugom, M. Swets, J. F. Bosset, L. Collette, A. Sainato, L. Cionini, R. Glynne-Jones, N. Counsell, E. Bastiaannet, C. B. van den Broek, G. J. Liefers, H. Putter and C. J. van de Velde, Lancet Oncol., 2015, 16(2), 200–207 CAS.
- A. Brunelli, A. Charloux, C. T. Bolliger, G. Rocco, J. P. Sculier, G. Varela, M. Licker, M. K. Ferguson, C. Faivre-Finn, R. M. Huber, E. M. Clini, T. Win, D. De Ruysscher and L. Goldman, Eur. Respir. J., 2009, 34(1), 17–41 CAS.
- P. L. Nguyen, P. M. Devlin, C. J. Beard, P. R. Orio, M. P. O'Leary, L. D. Wolfsberger, D. A. O'Farrell, C. M. Sweeney, B. A. Hadaschik, M. Hohenfellner and G. Hatiboglu, Brachytherapy, 2013, 12(1), 77–83 Search PubMed.
- C. M. Lee, J. I. Kwon, T. K. Lee, S. T. Lim, M. H. Sohn and H. J. Jeong, ACS Macro Lett., 2014, 3(11), 1126–1129 CAS.
- S. C. Formenti and S. Demaria, Lancet Oncol., 2009, 10(7), 718–726 Search PubMed.
- P. Huang, Y. Zhang, W. Wang, J. Zhou, Y. Sun, J. Liu, D. Kong, J. Liu and A. Dong, J. Controlled Release, 2015, 220(Pt A), 456–464 CAS.
- J. Kim, G. Zhang, M. Shi and Z. Suo, Science, 2021, 374(6564), 212–216 CAS.
Footnote |
| † These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2024 |