
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improved anti-breast cancer activity by doxorubicin-loaded super stealth liposomes†
Donatella
Paolino
ab,
Nicola
d'Avanzo
 ab,
Elena
Canato
c,
Luigi
Ciriolo
d,
Antonella
Grigoletto
c,
Maria Chiara
Cristiano
e,
Antonia
Mancuso
ab,
Christian
Celia
*fgh,
Gianfranco
Pasut
*c and
Massimo
Fresta
d
ab,
Elena
Canato
c,
Luigi
Ciriolo
d,
Antonella
Grigoletto
c,
Maria Chiara
Cristiano
e,
Antonia
Mancuso
ab,
Christian
Celia
*fgh,
Gianfranco
Pasut
*c and
Massimo
Fresta
d
aDepartment of Clinical and Experimental Medicine, University of Catanzaro “Magna Græcia”, V.le “S. Venuta”, Catanzaro, I-88100, Italy
bResearch Center “ProHealth Translational Hub”, Department of Experimental and Clinical Medicine, “Magna Graecia” University of Catanzaro, Campus Universitario “S. Venuta”—Building of BioSciences, Viale S. Venuta, I-88100 Catanzaro, Italy
cDepartment of Pharmaceutical and Pharmacological Sciences, University of Padua, Via F. Marzolo 5, I-35131 Padua, Italy. E-mail: gianfranco.pasut@unipd.it; Fax: +39-049-827-5366; Tel: +39-049-827-5694
dDepartment of Health Science, University of Catanzaro “Magna Græcia”, V.le “S. Venuta”, Catanzaro, I-88100, Italy
eDepartment of Medical and Surgical Sciences, University “Magna Græcia” of Catanzaro, Campus Universitario “S. Venuta”—Building of BioSciences, Viale S. Venuta, I-Catanzaro, Italy
fDepartment of Pharmacy, University of Chieti – Pescara “G. d'Annunzio”, Via dei Vestini 31, 66100, Chieti, Italy. E-mail: c.celia@unich.it; Fax: +3-0871-355-4911; Tel: +39-0871-355-4711
gLithuanian University of Health Sciences, Laboratory of Drug Targets Histopathology, Institute of Cardiology, A. Mickeviciaus g. 9, LT-44307 Kaunas, Lithuania
hInstitute of Nanochemistry and Nanobiology, School of Environmental and Chemical Engineering, Shanghai University, Shanghai 200444, China
First published on 18th June 2024
Abstract
PEGylation is currently used for the synthesis of stealth liposomes and to enhance the pharmacokinetic and biopharmaceutical properties of payloads. PEGylated dendron phospholipids can decrease the detachment of polyethylene glycol (PEG) from the liposomal surface owing to an increased hydrophobic anchoring effect on the phospholipid bilayer of liposomes and thus generating super stealth liposomes that are suitable for the systemic delivery of anticancer drugs. Herein, doxorubicin hydrochloride-loaded super stealth liposomes were studied for the treatment of breast cancer lung metastasis in an animal model. The results demonstrated that the super stealth liposomes had suitable physicochemical properties for in vivo administration and could significantly increase the efficacy of doxorubicin in breast cancer lung metastasis tumor-bearing mice compared to the free drug. The super stealth liposomes also increased doxorubicin accumulation inside the tumor tissue. The permanence of PEG on the surface of the super stealth liposomes favored the formation of a depot of therapeutic nanocarriers inside the tumor tissue by improving their permanence after stopping treatment. The doxorubicin-loaded super stealth liposomes increased the survival of the mouse tumor model. These promising results demonstrate that the doxorubicin-loaded super stealth liposomes could be an effective nanomedicine to treat metastatic breast cancer.
1. Introduction
Anthracycline antibiotics such as doxorubicin hydrochloride (Dox) are frequently used in chemotherapy for the treatment of orthotopic and metastatic breast cancer.1,2 Since the current chemotherapeutic drugs are associated with a range of side effects that often limit their benefits to cancer patients,3 several studies have focused on designing new nanomedicines to minimize the toxic side effects and increase the efficacy of payloads.4–7 Liposomal formulation is one of the recommended drug-delivery systems to optimize drug pharmacokinetics and distribution with an aim to reduce toxicity.8–10 For example, Dox-loaded liposomes can significantly prolong the half-life of the drug in the blood stream and reduce cardiotoxicity.11 Thus, nanomedicines based on long-circulating PEGylated liposomal-Dox, i.e., Caelyx® in Europe or Doxil® in the USA, were approved by the European Medicine Agency (EMA) and Food and Drug Administration (FDA) for clinical use. Despite their advantages, Caelyx®/Doxil® did not significantly improve the therapeutic efficacy of Dox in patients compared to the free drug.12,13Although polyethylene glycol (PEG) increases the in vivo half-life of liposomes, experimental data have shown that the PEGylation of nanocarriers is limited due to the physicochemical and biological changes of the PEG coating over the nanocarriers, achieved by the use of commercial PEGylated phospholipids, after systemic injection.14 These modifications cause PEG leakage from the external bilayer and conversion of the PEGylated liposomes into nanocarriers with a reduced PEG coating, thus affecting their stability in the blood stream as well as their long circulation properties and therapeutic efficacy.14
To overcome these drawbacks, we proposed new PEGylated dendron phospholipids where PEG is conjugated through an amino acidic spacer to multiple (two or four) phospholipids units.15 These synthetic PEGylated dendron phospholipids strengthen the interaction between the PEG and the phospholipids, allowing a stable and durable linkage to be formed between the polymers and lipids, and thus creating stable stealth nanocarriers, or “super stealth liposomes” (SSLs). SSLs have the same biopharmaceutical properties as common PEGylated liposomes, but they are more stable in the blood stream and biological fluids, with a reduced detachment of PEG after a long circulation of the nanocarriers and reduced interaction with circulating proteins and enzymes.15 SSLs can be also conjugated with targeting compounds and can deliver different payloads, such as chemotherapeutic drugs, RNAi/DNAi, therapeutic peptides, and proteins, and can be loaded with single or combined drugs for multi-drug therapy for anticancer treatment.5
The aim of this study was to synthesize and characterize SSLs-Dox and to test this with in vitro metastatic breast cancer cells and an in vivo breast cancer lung metastasis xenograft mouse model. SSLs-Dox was physicochemically characterized by dynamic light scattering (DLS) analysis and the resulting nanocarriers were found to have average sizes in the nanometer scale (below 200 nm), a narrow size distribution (polydispersity index or PDI below 0.2), and net negative surface charges (Z-potential below −38 mV), as previously reported.15 Only 0.5% of the Dox was released from the SSLs after 120 h of incubation, as previously reported,15 due to the crystallized form of the drug and the controlled release of the liposomes. Cytotoxic studies demonstrated that free- and SSLs-Dox had similar efficacies in vitro on MDA-MB-231 cells and the cell viability percentage after 72 h of incubation was calculated to be 10%, while free-Dox better accumulated on MDA-MB-231 cells in vitro than SSLs-Dox. The in vivo tests showed that SSLs-Dox can circulate for a long time in the blood stream and these nanocarriers were better biodistributed than the free-Dox. Conversely, the accumulation of SSLs-Dox was found to be decreased in the heart, liver, and kidneys, while it was increased in the lungs compared to the free-Dox because of the long circulation of the SSLs. The SSLs-Dox significantly decreased tumor growth in a nude mice xenograft model of breast cancer lung metastasis and improved the overall survival of the animals at the tested set incubation times. This effect depended on apoptosis of the breast cancer lung metastasis cells occurring after treatment with the SSLs-Dox. Overall, the results demonstrate that SSLs-Dox can provide an effective treatment strategy for metastatic breast cancer.
2. Materials and methods
2.1. Materials
1,2-Dipalmitoyl-sn-glycero-3-phosphocholine monohydrate (DPPC), and N-(carbonylmethoxypolyethyleneglycol-2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE-mPEG 2000) were purchased from Avanti Polar (Avanti Polar Lipids, Inc., Alabaster, AL, USA). Doxorubicin hydrochloride (Dox) was purchased from LC Laboratories (MA, USA). CellTiter 96® aqueous non-radioactive cell proliferation, [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium], inner salt (MTS) assay was purchased from Promega (Madison, WI, USA). Also purchased were the QIA33 FragEL™ DNA fragmentation detection kit (Millipore, Billerica, MA, USA) and Ki67 antibody (ab66155, Abcam®, Cambridge, MA, USA). High-glucose Dulbecco's modified Eagle's minimal essential medium (DMEM), Roswell Park Memorial Institute medium (RPMI-1640), heat-inactivated fetal bovine serum (FBS), penicillin–streptomycin solution, glutamine, pcDNA6.2-GFP-DEST vector, 293FT cell, lipofectamine 2000, and blasticidin were purchased from Invitrogen™ (ThermoFisher Scientific, Waltham, MA, USA). The MDA-MB-231 breast cancer cell line was obtained from the American type culture collection (ATCC, LGC Standards, Teddington, UK). D-Luciferin, potassium salt and Synergy H4 hybrid readers were purchased from Gold Biotechnology® (GOLDBIO®, St Louis, MO, USA). Sodium dodecyl sulfate (SDS), daunorubicin (Dau), radio-immunoprecipitation assay (RIPA) buffer, and phosphate saline tablets (for the preparation of phosphate buffer solution pH 7.4) were obtained from Sigma-Aldrich (Milan, Italy). The Eclipse 80i microscope was obtained from Nikon Corporation (Nikon Instruments Inc, Melville, NY, USA), while the IVIS imaging system 200 series was purchased from Caliper Life Sciences (PerkinElmer, Winter St Waltham, MA, USA). Female nude mice were obtained from Charles River Laboratories (Boston, MA, USA). All other chemical reagents and solvents from Sigma-Aldrich and Thermo Fisher Scientific were analytical grade and used without further purification.2.2. Synthesis of mPEG-βGlu(βGlu)2(DSPE)4
First, mPEG-βGlu(βGlu)2(DSPE)4 was synthesized and physicochemically characterized as previously reported.15 Briefly, βGlu residuals were conjugated to mPEG-p-nitrophenyl carbonate (5 kDa) in 0.1 M borate medium pH 8. The intermediate derivative from the reaction was extracted from the reaction buffer by dichloromethane and then precipitated in diethyl ether. The resulting dried intermediate was activated using dicyclohexylcarbodiimide (DCC) and N-hydroxysuccinimide (NHS) and reacted with βGlu. The intermediate product was extracted as above reported and then, after another step of activation, it was conjugated to four macromolecules of DSPE.15 The mPEG-βGlu(βGlu)2(DSPE)4 was physicochemically characterized by NMR spectroscopy (1H-NMR and DOSY 1H-NMR) and the Snyder assay as previously reported.152.3. Synthesis of doxorubicin hydrochloride-loaded super stealth liposomes (SSLs-Dox)
SSLs were formulated using phospholipids and mPEG-dendron-derivatives at different molar ratios as previously reported.15,16 Briefly, 40 mg of lipids mixture containing DPPC, Chol, and the mPEG dendron derivative, i.e., mPEG-βGlu(βGlu)2(DSPE)4, for SSLs, in a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:0.5 molar ratio, respectively, were dissolved in a round-bottom glass tube containing 2 ml of a chloroform/methanol (3:1 v/v) solution. The organic solvent was evaporated using a Büchi R210 Rotavapor® (Büchi, Milan, Italy) under vacuum until the formation of a thin lipid film on the surface of the round-bottom flask. The residual organic solvents were removed overnight at 30 ± 1 °C using a Büchi T51 (Büchi, Milan, Italy) glass oven drier connected to a high-vacuum pump. A pH-gradient method remote loading procedure was carried out to increase the Dox concentration inside the SSLs. The lipid film was hydrated using ammonium sulfate solution (250 mM; pH 4) to obtain a final lipid concentration of 20 mg ml−1. Three alternative cycles of warming (55 ± 0.5 °C in a warmed water-bath apparatus) and vortex-mixing (700 rpm using an MS1 Minishaker, IKA-WERKE GMBH and Co., Staufen, Germany) were applied to make multilamellar SSLs with a pH acidic aqueous core. The SSLs were further extruded through a stainless-steel extrusion device (Lipex Biomembranes, Northern Lipids Inc., Vancouver, BC, Canada) warmed at 55.0 ± 0.5 °C. A nitrogen flux of 430, 600, and 900 kPa and stacked polycarbonate membrane filters (Costar, Corning, Inc., New York) of 400, 200, and 100 nm, were used during the preparation procedure. Ten passages through the different polycarbonate membrane filters were carried out to decrease the average size and increase the size distribution of the SSLs. The resulting unilamellar SSLs were precipitated using a Beckman Optima™ ultracentrifuge (1 h, 4 °C), re-suspended in a Dox solution (2 mg ml−1, pH 7.4), and continuously stirred in a dark atmosphere for 2 h at 55 ± 0.5 °C. The un-entrapped drug was removed through dialysis (polycarbonate membrane, cutoff 10 kDa, Spectrum Laboratories, Inc, Rancho Dominguez, CA, USA) before use. The therapeutic SSLs were finally collected and used in the further experiments.
:3:0.5 molar ratio, respectively, were dissolved in a round-bottom glass tube containing 2 ml of a chloroform/methanol (3:1 v/v) solution. The organic solvent was evaporated using a Büchi R210 Rotavapor® (Büchi, Milan, Italy) under vacuum until the formation of a thin lipid film on the surface of the round-bottom flask. The residual organic solvents were removed overnight at 30 ± 1 °C using a Büchi T51 (Büchi, Milan, Italy) glass oven drier connected to a high-vacuum pump. A pH-gradient method remote loading procedure was carried out to increase the Dox concentration inside the SSLs. The lipid film was hydrated using ammonium sulfate solution (250 mM; pH 4) to obtain a final lipid concentration of 20 mg ml−1. Three alternative cycles of warming (55 ± 0.5 °C in a warmed water-bath apparatus) and vortex-mixing (700 rpm using an MS1 Minishaker, IKA-WERKE GMBH and Co., Staufen, Germany) were applied to make multilamellar SSLs with a pH acidic aqueous core. The SSLs were further extruded through a stainless-steel extrusion device (Lipex Biomembranes, Northern Lipids Inc., Vancouver, BC, Canada) warmed at 55.0 ± 0.5 °C. A nitrogen flux of 430, 600, and 900 kPa and stacked polycarbonate membrane filters (Costar, Corning, Inc., New York) of 400, 200, and 100 nm, were used during the preparation procedure. Ten passages through the different polycarbonate membrane filters were carried out to decrease the average size and increase the size distribution of the SSLs. The resulting unilamellar SSLs were precipitated using a Beckman Optima™ ultracentrifuge (1 h, 4 °C), re-suspended in a Dox solution (2 mg ml−1, pH 7.4), and continuously stirred in a dark atmosphere for 2 h at 55 ± 0.5 °C. The un-entrapped drug was removed through dialysis (polycarbonate membrane, cutoff 10 kDa, Spectrum Laboratories, Inc, Rancho Dominguez, CA, USA) before use. The therapeutic SSLs were finally collected and used in the further experiments.
2.4. Physicochemical characterization of SSLs-Dox
Physicochemical characterization of the SSLs was carried out as previously reported.17,18 The average size, size distribution (polydispersity index or PDI), and zeta potential (Z-potential) tests were carried out using Zetasizer NanoZS (Malvern Instruments Ltd, Worchestershire, UK) apparatus equipped with a laser diode/Ne (4.5 mW), operating with a light source at 670 nm and a backscattering photon angle detector of 173°. A real refractive index, an imaginary index, and a medium refractive index of 1.59, 0.0, and 1.330, respectively, were used during the analysis. Samples were measured by applying a medium viscosity of 1.0 mPa s and a medium dielectric constant of 80.4. A suitable dilution of SSLs was carried out before the analysis to avoid multi-scattering phenomena. The SSLs were diluted (1:100 v/v) using isotonic double-distilled pyrogen-free water, which was previously filtered through 0.22 μm polypropylene pore-sized membranes (Whatman Inc., Clifton, NJ, USA). The diluted samples were analyzed using a disposable cuvette (Malvern Instruments Ltd, Worchestershire, UK), and the analysis was carried out at 25 °C. Laser doppler anemometry was used to evaluate the Z-potential of the SSLs. The results were expressed as a function of the electrophoretic mobility of colloidal suspensions by using Smoluchowsky's constant F (Ka) of 1.5. The following parameters were set up before the analysis: He/Ne laser (633 nm) with a nominal power of 5.0 mW. A suitable dilution (1:100 v/v) of the samples was carried out before the experiments. The morphology of the empty-SSLs and SSLs-Dox was evaluated by transmission electron microscopy (TEM).
The long-term stability of SSLs-Dox in PBS at 4 °C and 25 °C was studied for up to 70 days of incubation (Fig. S5†) and compared to the PEGylated liposomes (SLs-Dox).
The stability of the SSLs-Dox in serum was also tested and compared to that of SLs-Dox (Fig. S6†). In detail, 400 μl of nanocarriers was incubated with 2 ml of a PBS:serum mixture (50:50 volume ratio) at 37 °C and the average size was measured at specific time points for up to 72 h of incubation. Nanocarriers incubated in PBS buffer at 37 °C were used as the negative control.
2.5. Cell lines and culture
MDA-MB-231 cells, a breast cancer cell line derived from humans, were obtained from the American type culture collection (ATCC) and seeded in DMEM, supplemented by 10% (v/v) heat-inactivated fetal bovine serum (FBS), 1% (v/v) glutamine, and 0.1% (v/v) penicillin/streptomycin.The bioluminescent MDA-MB-231 cell line was engineered with an overexpression of luciferase (Luc) and green fluorescent protein (GFP). Briefly, the firefly luciferase gene (luc2, Promega) was first cloned into the pcDNA6.2-GFP-DEST vector to obtain the fused luciferase/GFP cassette. After purification, the cassette was further cloned using the lentivirus backbone to form an engineered plasmid, which was then packaged into virus particles. The virus particles were used to transfect breast cancer cells and the resulting GFP-positive MDA-MB-231 cells (Luc/GFP) were selected by FACS sorting and maintained in complete DMEM medium supplemented with 6 μg ml−1 of blasticidin.
2.6. In vitro cytotoxicity
MDA-MB-231 cells were seeded in to 96-well culture plates (5000 cells per well) and incubated overnight in 100 μl DMEM medium supplemented with 10% (v/v) FBS. The day before, the medium was replaced with fresh medium and different concentrations of empty-SSLs, free-Dox, or SSLs-Dox, were added and incubated at different time points, i.e., 24, 48, and 72 h. At the end of the set incubation times, the medium was withdrawn, and cells were washed by PBS buffer twice. Also, 100 μl of fresh medium was finally added to each well before the analysis. The cell viability was carried out using the MTS assay (CellTiter 96® aqueous non-radioactive cell proliferation assay, Promega). Briefly, 20 μl of MTS/PMS solution (20/1 v/v) was added to each well and incubated for 4 h at 37 °C in a humidified 5% CO2 atmosphere. The resulting absorbance from each well was recorded at 490 nm using Synergy H4 hybrid readers (BioTek, Instruments, Inc., Highland Park, Winooski, VT, USA).2.7. In vitro intracellular uptake of free- and SSLs-Dox
First, 1.5 × 105 of MDA-MB-231 cells per ml were seeded into 24-well plates and incubated at 37 °C. The next day, the medium was replaced with fresh medium supplemented with 0.5 μM equivalent dosage of free-Dox and SSLs-Dox. The cells were incubated for 0.5, 1, 2, 4, 6, 24, and 48 h. At the different time points, the cells were washed with cold PBS buffer three times and collected in 100 μl lysis buffer (RIPA buffer and protease inhibitors). The Dox concentrations in the cell lysates were measured using a fluorescence spectrophotometer (Synergy H4 hybrid readers, BioTek, Instruments, Inc., Highland Park, Winooski, VT, USA) at an excitation wavelength of 490 nm and an emission wavelength of 590 nm. Standardization curves were prepared using cellular lysates containing a series of known concentrations of free-Dox and SSLs-Dox. The cellular uptake of Dox was expressed as nmols per milligram of protein. The protein concentrations of the cell lysates were tested using the micro BCA protein assay kit according to the manufacturer's instructions (ThermoFisher, Waltham, MA USA).2.8. Animal studies
All the animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of the University of Catanzaro (Italy) and University of Padua (Italy); and approved by the National Directorate of Veterinary Services permit no. 235 of June 30th, 2011, and permit no. 938/2016-PR of October 10th, 2016, for the University of Catanzaro and University of Padua, respectively.For the pharmacokinetic studies, female Sprague–Dawley rats (140–190 g) were randomly divided into groups of 3 rats each. A dose of 2.5 mg kg−1 Dox of either the free drug or SSLs-Dox was administered via the tail vein to the rats previously anesthetized with isoflurane gas (mixed with O2 in enclosed cages). At scheduled time points, blood samples (∼200 μl) were collected from the tail into heparin-treated tubes and thereafter immediately centrifuged for 15 min at 1500g to separate the plasma. Dox was extracted by treating 50 μl of plasma with 10 μl of 1 M Triton X-100 and 580 μl of 81 mM HCl in isopropanol to allow precipitation of the plasma proteins. After overnight incubation at 4 °C, the samples were centrifuged for 3 min at 3000 rpm and the supernatants were analyzed using an FP-6500 Jasco spectrofluorometer (λex = 470 nm; λem = 584 nm). The Dox concentration in each plasma sample was extrapolated through a calibration curve of the standard solutions of either Dox. The pharmacokinetic analysis was performed using PKSolver 2.0 software by applying a bicompartmental model.
For the in vivo activity studies, female nude mice (6 weeks old, 20–30 g) were purchased from the Charles River Laboratories (Calco (Lecco), Italy). The animals were housed in a specific pathogen-free facility under a 12-hour light–dark cycle and fed with a pathogen-free diet and water ad libitum.
The engineered MDA-MB-231 cells (Luc/GFP), which express both firefly luciferase and green fluorescent proteins, were used to constitute the mouse model of breast cancer lung metastasis. Here, 3 × 105 MDA-MB-23 (Luc/GFP) cells per ml were suspended in PBS buffer and injected into the lateral tail vein in a volume of 100 μl. The breast cancer lung metastasis tumor size was tracked via an IVIS imaging system, 200 series (Caliper Lifesciences, USA). Prior to imaging, the mice were first anesthetized with isoflurane, and then treated by an intraperitoneal injection of D-luciferin (150 mg per kg body weight in PBS). To collect consistent and the strongest bioluminescence, all the images were captured by IVIS imaging system 10 min after luciferin injection. A semiquantitative region of interest (ROI) analysis was performed with Live Image v4.2 software (Caliper Lifesciences, PerkinElmer Inc., Waltham, MA, USA), and the bioluminescence intensity was used to measure the growth of lung metastasis.
Sixty female nude mice engrafted with breast cancer lung metastasis were used for the in vivo antitumor effect experiments. The mice were divided into four groups (n = 15 per group): PBS, free-Dox, empty-SSLs, and SSLs-Dox. One week after MDA-MB-231 (Luc/GFP) cell inoculation, 100 μl PBS, empty-SSLs, free-Dox, and SSLs-Dox was administrated into the mice via intravenous injection, with an equivalent dosage of 3 mg kg−1 of Dox. The treatments were administrated weekly for three times. The breast cancer lung metastasis tumor size was tracked every week.
The tumor volumes were measured using a caliper as previously reported19 and were calculated with the following equation (eqn (1)):
| V = 0.5 × ab2 | (1) |
The body weight, feeding, and motor activity of the mice were used to monitor the general health of the tumor-bearing mice during the therapeutic treatment.
After 7 weeks, 10 mice in each group were carefully recorded for performing a Kaplan–Meier plot and the analysis was carried out at up to 210 days.20
Five mice per group were dissected for pathology and immunohistochemistry analyses, according to the Standard Operating Procedures of the veterinary animal house facility at the University of Catanzaro “Magna Graecia”.
2.9. Hematoxylin and eosin (H&E) staining
Formalin-fixed paraffin embedded sections were cut at 4 microns, baked at 60 °C overnight in an oven, dewaxed in xylene and hydrated through increasing the concentration of the reagent alcohol to distilled water, stained for 3 minutes in 3% (v/v) acetic acid/Harris hematoxylin, washed in running tap water for 1 min, rinsed in 95% (v/v) reagent alcohol prior to dipping 10 times in alcoholic Eosin Y stain, dehydrated in increasing concentration reagent alcohol, cleared in xylene and placed under a permanent coverslip. Images of the tissue sections after H&E staining were captured by an Eclipse 80i microscope (Nikon Metrology Inc. Brighton, MI, USA).2.10. Immunohistochemistry (IHC)
Ki-67 antibody was used with a dilution of 1:400 (v/v). The immunohistostaining was carried out according to the standard protocol according to the manufacturer's instruction. Briefly, paraffin sections were first dewaxed and hydrated using distilled water. Then, high temperature antigen retrieval was performed in a steamer using Antigen Unmasking solution from Vector Laboratories according to the company's recommendations. After washing in PBS buffer (pH 7.4), the resulting sections were stained according to the standard IHC protocol using the Vector Laboratories HRP-conjugated horse anti-rabbit polymer detection system. Then, the sections were incubated overnight at 4 °C and developed in DAB for 5 min and counterstained in Mayer's hematoxylin. Finally, all the sections were dehydrated and cleared in xylene and permanently placed under a coverslip. All sections were analyzed under an Eclipse 80i microscope (Nikon Metrology Inc. Brighton, MI, USA).
2.11. Analysis of tissue distribution
Experiments were performed on MDA-MB-231 (Luc/GFP) breast cancer lung metastasis nude mice, which were dosed intravenously (i.v.) with 3 mg kg−1 free-Dox and SSLs-Dox. The mice were sacrificed after 1 h, 24 h, 4 days, and 7 days, respectively. Three mice per group were used for each time point, and the lung, liver, heart, kidney, spleen, and blood were harvested for the biodistribution study. The total amount of Dox in the different organs was analyzed by HPLC.The HPLC analyses were performed with a Waters Liquid Chromatography instrument equipped with a model 1525 binary solvent pump and a 2996 photodiode array detector (Waters Spa, Milford, MA, USA). The mobile phase was on-line degassed directly using the degasser integrated inside the Waters 1525 binary solvent pump (Waters Spa, Milford, MA, USA). A Gemini reverse phase C18 packing column (4.6 mm × 250 mm; 5 μm particle size; Phenomenex, Torrance, CA, USA) and a disposable Security Guard column (4.0 × 3.0 mm, 5 μm particle size; Phenomenex, Torrance, CA, USA), warmed at 25 ± 1 °C with a Waters Temperature Control Module II (Waters Spa, Milford, MA, USA), were used for the detection of Dox. The mobile phase was an isocratic mixture of water and acetonitrile (40:60 v/v) containing 0.05% (v/v) trifluoroacetic acid; the flow rate was adjusted to 1.0 ml min−1 and the fluorescence detection of Dox was carried out at an excitation wavelength of 490 nm and an emission wavelength of 590 nm using a fluorescent detector (Waters 2475 Multi Fluorescence detector). Empower v.2 Software (Waters Spa, Milford, MA, USA) was used for the HPLC data monitoring and acquisition.
Empty-SSLs were used as the blank during the analysis and the retention time of Dox was 4.29 min as previously reported.21 An external calibration curve in the linear range of concentration from 0.25–25 μg ml−1 was used to quantify the Dox. The following equation (eqn (2)) was applied for the quantification of the samples:
| AUC = 8 × 106x + 3 × 106; R2 = 0.9997 | (2) |
The quantification of Dox in the mice serum and tissues was carried out using Dau as an internal standard as previously reported.22,23 Here, 300 μl of serum or tissues (i.e., heart, liver, spleen, lung, kidney, 300 mg homogenized in 1 ml of PBS buffer) and 10 μl of Dau (aqueous stock solution at 50 μg ml−1) were mixed with a 4-fold volume of the extraction solution, which was a mixture of chloroform and methanol (3:1, v/v). The mixture was vortexed for 2 min and centrifuged at 13000 rpm, 4 °C, for 10 min to separate the aqueous and organic phases. After centrifugation, the organic phases were collected and evaporated to dryness at 25 °C under a nitrogen flow. The dry residues from the serum and tissues were dissolved in 100 μl of methanol and 10 μl of the resulting solution was injected for HPLC analysis.
To preserve Dox from degradation, all the standard solutions for the calibration and the resulting samples were frozen and stored at −20 °C until the HPLC analysis.
2.12. Statistics
All data represent the average of different measurements ± standard error (S.E.). The statistical significance for different experiments was analyzed using Student's t-test. A P-value ≤0.05 was considered statistically significant, and P-value ≤0.01 was considered statistically very significant.3. Results and discussion
3.1. Physicochemical characterization of SSLs-Dox
The mPEG-βGlu(βGlu)2(DSPE)4 derivative made stable SSLs thanks to the increased phospholipid/PEG ratio, which allowed a stable interaction between PEG chains and the bilayer of liposomes, as previously reported.15 In fact, the dendron structure of mPEG derivatives stabilizes the PEGylated liposomes and decreases the detachment of PEGs in the blood circulation after systemic injection.15 The mPEG-βGlu(βGlu)2(DSPE)4 was characterized by 1H-NMR spectroscopy (Fig. S1(a)†). The DSPE degree of binding was 98%, based on the ratio between the experimental value of –CH3 DSPE's protons (23.61) with respect to the theoretical value (24.00). The DOSY spectrum confirmed that the DSPE molecules were covalently linked to the polymer backbone, since all the protons in the spectrum had the same diffusion coefficient (D = 1.25 × 10−6 cm2 s−1) (Fig. S1(b)†). Also, the Snyder assay confirmed the absence of free DSPE.The supramolecular structure of SSLs enabled stable and long-circulating liposomes to be obtained, with average sizes below 200 nm, a narrow size distribution (PDI 0.2), and a net negative surface charge (below −38 mV) (Fig. S2†). The physicochemical parameters were independent from Dox, and the resulting SSLs can pass easily through endothelial fenestrations (300–500 nm) in the neo-formed tumor vasculature of solid carcinomas.24–26 The remote-loading and pH-gradient method allowed the precipitation of Dox, as a sulfate salt, into the SSLs. In fact, the internal acidic pH (4.5) of the liposomes leads to a gel-like structure, caused by ammonium sulfate salts,27 which crystallizes Dox into the aqueous core of the SSLs and forms a coffee bean structure for payloads, like Doxil®/Caelyx® nanomedicines.28 Here, 88% of Dox was loaded into the SSLs by the pH-gradient remote-loading procedure (Fig. S2†) according to Barenholz's computational modeling, which allows predicting physicochemical parameters for potential hydrophilic drug candidates expected to provide a high remote loading efficiency if they are formulated as liposomes.29 In fact, Dox hydrochloride had a logD at pH 7, in the range from 2.5 to 2.0, and a pKa ≥ 3; while the drug and lipid (D/L) ratio, before and after the entrapment of chemotherapeutic drugs into liposomes, was <0.3 (ratio 1), as previously reported for Doxil®/Caelyx® nanomedicines.30 The liposome morphology and narrow size distribution were confirmed by TEM (Fig. S3†).
SSLs-Dox showed a slow drug release and less than 0.5% of the Dox leaked from the SSLs after 120 h of incubation (Fig. S4†), as previously reported.15 The SSLs-Dox showed excellent stability after incubation in solution (PBS) at either 4 °C or 25 °C, for up to 70 days of incubation in terms of liposomal size changes and polydispersity; the resulting data were similar to classic Dox-loaded stealth liposomes (SLs) (Fig. S5†).
The SSLs incubated in physiological buffer (PBS:serum mixture) were stable up to 72 h. The stability of the SSLs-Dox increased after 6 and 24 h incubation compared to SLs-Dox, and the sizes of the SSLs were decreased significantly compared to those of SLs-Dox. These results may depend on the reduced leakage of PEG from the liposomal bilayer and the permanence of the synthetic PEGylated dendron phospholipids on the surface of the SSLs, which improved their stealth properties15 (Fig. S6†).
3.2. In vitro cytotoxicity of free-Dox and SSLs-Dox
We tested the in vitro cytotoxicity of free-Dox and SSLs-Dox using the MTS assay in MDA-MB-231 cells (Fig. 1(a and b)). There was no statistically significant difference between the SSLs-Dox and free-Dox after 24 h of incubation (except for the two highest drug concentrations); while free-Dox showed an increase in cytotoxicity compared to SSLs-Dox at 48 and 72 h of incubation (Fig. 1(c and d) and Table S1†). The empty-SSLs did not show any toxicity to MDA-MB-231 cells in the range of the tested concentrations (Fig. S7†). These results agreed with data previously reported for Doxil®/Caelyx® nanomedicines.28 The reduced in vitro cytotoxicity for SSLs-Dox compared to free-Dox may also depend on the release kinetics of the drug from the liposomes, as published in a previous paper.15 As expected, the crystallized Dox could not be easily released from the SSLs and precipitated into the aqueous core of liposomes without diffusing through the liposomal bilayer. Therefore, smaller amounts of Dox were slowly released into the cancer cells while free-Dox was suitably available to perform its cytotoxic activity.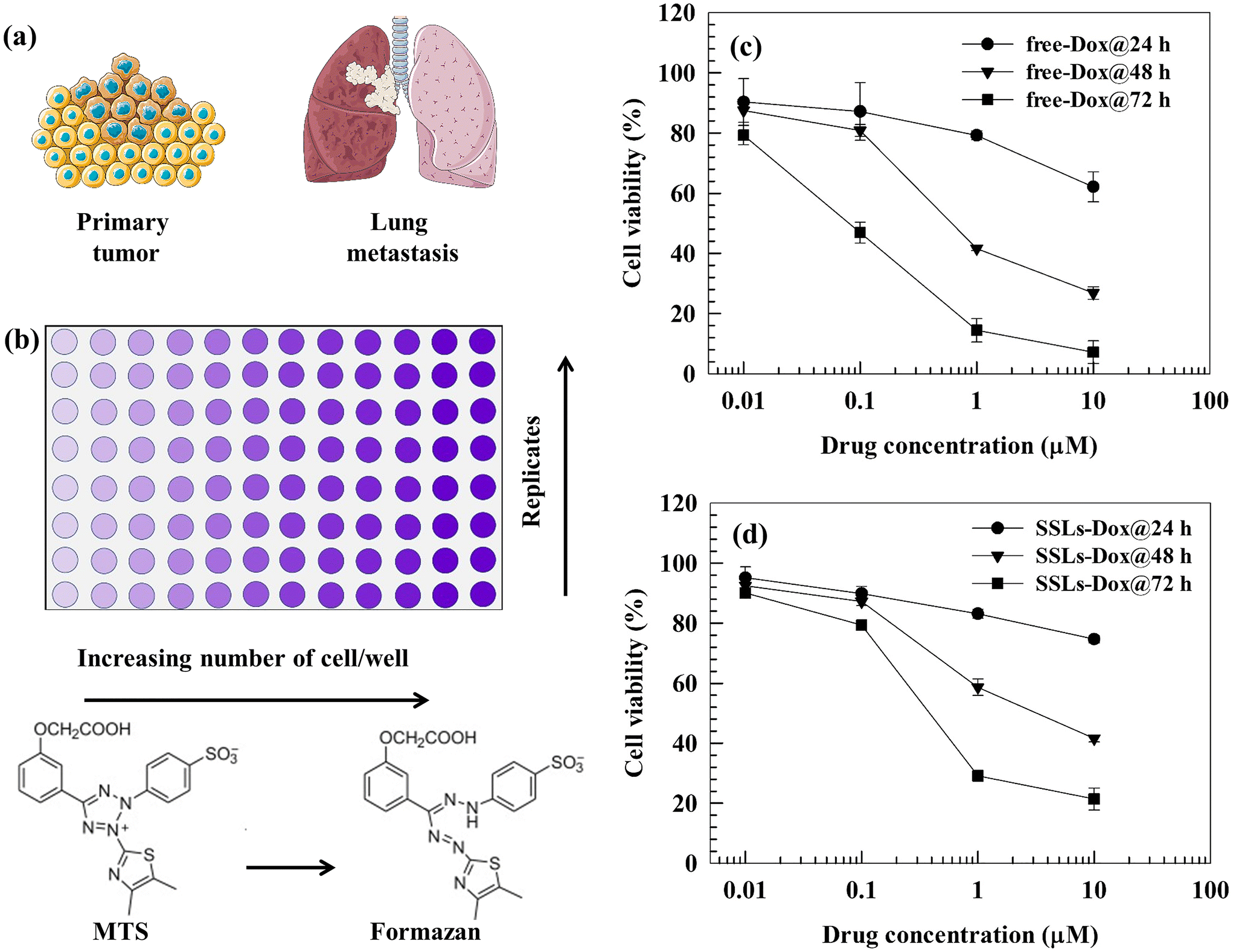 | ||
| Fig. 1 (a) and (b) Schematic representations of the primary/metastatic breast cancer and MTS tests, respectively. (c) and (d) Cytotoxic effects of free-Dox and SSLs-Dox on the MDA-MB-231 cell line. MDA-MB-231 cells (5000 cells per well) were seeded into a 96-well plate. Free-Dox or SSLs-Dox, were added at different concentrations (0.01, 0.1, 1, and 10 μM), and the cells were then incubated at 37 °C for 24, 48, and 72 h. The MTS test was used to evaluate the cellular viability. Data (n = 6) are presented as the percentage (mean ± S.E.) of viable cells versus the control (corresponding to 100%) at the Dox concentration herein reported. The error bars, if not shown, are within the symbols. Statistical analysis (free-Dox versus SSLs-Dox) and the cytotoxic effect of empty liposomes are reported in the ESI (Table S1 and Fig. S7,† respectively). | ||
The stable PEGylation, and consequently the permanence of the intact PEG coating on the surface of SSLs,15 may favor a reservoir system for Dox. The PEG layer may hinder, like a physical barrier, the release of the payload into the bulk solution and cellular cytoplasm.24,31 In fact, only Dox, accumulated into the nucleus of cancer cells, can inhibit DNA directly, causing a significant anticancer effect on solid tumors, and blocking the cell cycle by initiating DNA damage.32 The intra-nuclear accumulation of Dox can also be affected by an acidic pH, particularly the acidic pH of lysosomes, which allows the release of Dox from nanocarriers and increases the drug accumulation inside the nucleus of cancer cells.33 The data seemed to suggest that the in vitro anticancer activity of SSLs-Dox on MDA-MB-231 cells may depend on the amount of the chemotherapeutic drug that is therapeutically available after the intracellular uptake of liposomes, and its accumulation in cells.31 Previous data agreed with our results and demonstrated that Dox-loaded PEGylated liposomes do not improve the cell growth inhibition in human colorectal adenocarcino HT-29 and retinoblastoma Y79 cell lines.34 In fact, the inhibition concentration (IC) 50 values for Dox-loaded PEGylated liposomes were similar to those of free-Dox in the tested cell lines after 24 and 48 h of incubation.34 Conversely, free-Dox showed higher in vitro cytotoxicity on murine B16 melanoma cell lines than the PEGylated polyamidoamine dendrimer-Dox conjugates at 48 h of incubation. The IC50 values for different Dox-polymer conjugates were in the range from 12–70 μM, respectively, compared to 0.4 μM for free-Dox.31 Based on this in vitro data, it is possible to suppose that the intracellular uptake and release of Dox may affect the drug cytotoxicity; while PEGylated liposomes aid in decreasing the presence of free-Dox in the blood stream and, therefore, overcoming the Dox-associated side effects, i.e., cardiotoxicity and myeloid-suppression.35–37
3.3. In vitro cellular uptake of free-Dox and SSLs-Dox
As previously discussed, the main advantages of using PEGylated liposomes to deliver Dox are the decrease in free-Dox concentration into the blood stream and its preferential accumulation inside tumor tissues. These advantages improve the therapeutic efficacy of Dox and decrease its side effects. The intracellular uptake and tumor accumulation of Dox decrease its cardiotoxicity and prevent side effects, which can otherwise reduce patient compliance and compromise the efficacy of therapy.28,38–40 We previously discussed the relationship between in vitro cytotoxicity and the intracellular uptake of Dox and Dox nanomedicines. Based on the in vitro results, we studied the intracellular uptake of free-Dox and SSLs-Dox (Fig. 2(a)). No significant differences were obtained by testing free-Dox and SSLs-Dox on MDA-MB-231 cells (Fig. 2(b)). The resulting data showed that the free-Dox and SSLs-Dox had a similar uptake up to 6 h of incubation. At this incubation time, only 4 nmol and 3.8 nmol per gram of proteins were taken up into MDA-MB-231 cells for the free-Dox and SSLs-Dox, respectively (Fig. 2(b)). The uptake for the free-Dox (12 nmol per gram of proteins) was higher than for SSLs-Dox (8 nmol per gram of proteins) at 24 h of incubation, while it showed the same concentration at 48 h (Fig. 2(b)). Although, the PEGylated liposomes decrease the non-specific interaction with circulating cellular proteins and avoid the opsonin attachment to liposomal surface,41,42 PEGylation can decrease the liposomal uptake in the targeting cells and modify the intracellular accumulation of free-Dox.43 This effect may depend on the drug release from liposomes, which plays an important role in the anticancer effect of chemotherapeutics and becomes slow for nanomedicines that have been formulated using a pH-gradient remote-loading method.15,27 In fact, the Dox is crystallized as a coffee bean structure and forms a gel-like structure that precipitates inside the aqueous core of liposomes. These crystals reduce the Dox release and result in a low amount of drug being freely available inside the cancer cells. This effect may have provide an apparent decrease in the availability of free-Dox inside the MDA-MB-231 cells for SSLs-Dox compared to free-Dox due to the formulation in the liposomes, which confines Dox inside their core. Conversely, the SSLs may increase in vivo the Dox accumulation inside the tumor tissues and decrease its cardiotoxicity by extravasating the chemotherapeutic drug through the tumor vessels, which are more fenestrated than normal vessels.44,45 Additionally, the free-Dox may accumulate quickly in the nucleus of cancer cells, while SSLs-Dox present a slow accumulation in the nucleus in short incubation times, as previously reported for self-assembled chimeric Dox nanoparticles, where the Dox intracellular uptake was reported to depend on nanoparticles, intracellular drug release, and trafficking.46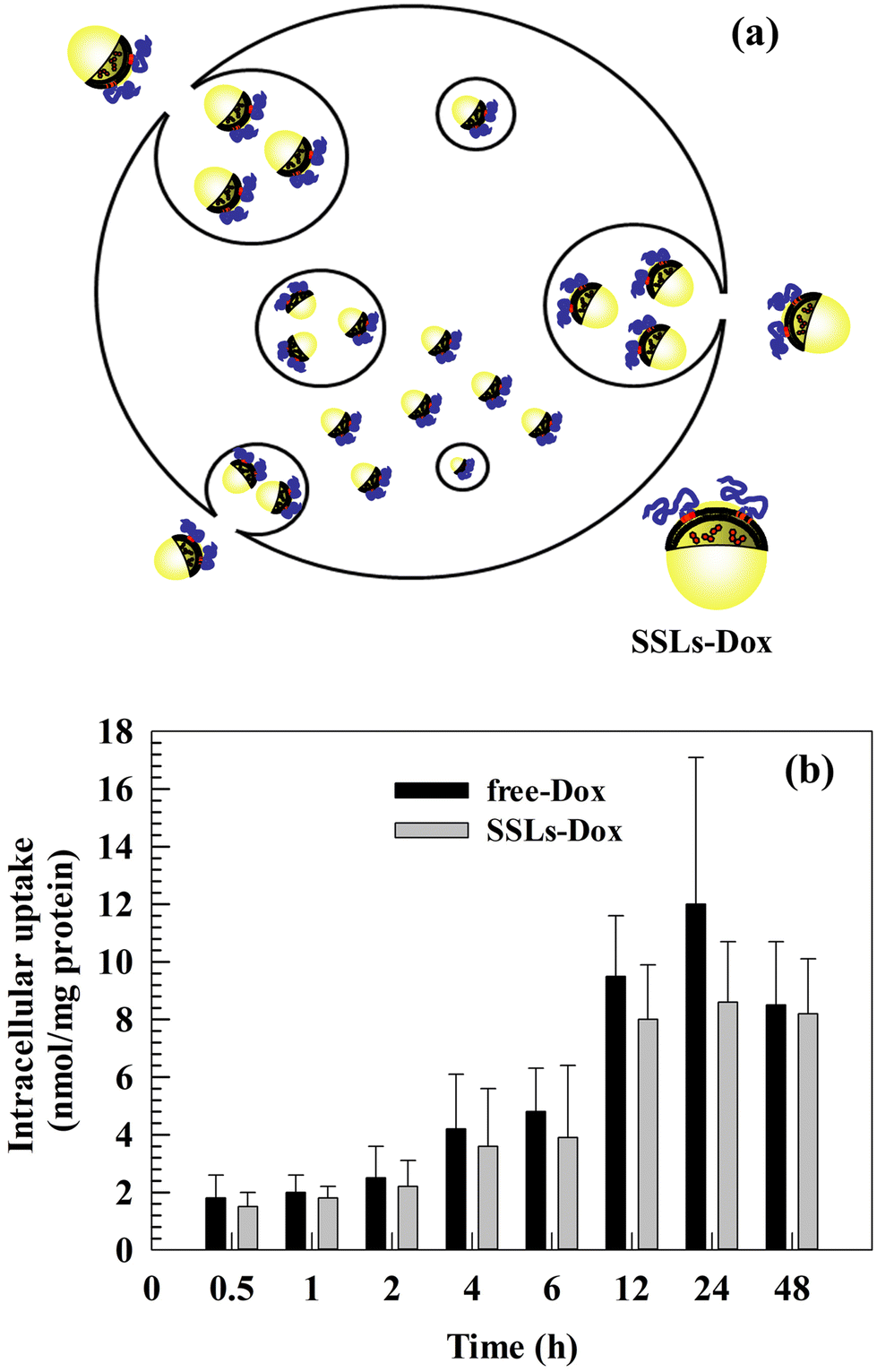 | ||
| Fig. 2 Intracellular uptake of free-Dox and SSLs-Dox in MDA-MB-23 cells. Illustration of the intracellular uptake of SSLs into cancer cells (a). Cells were incubated with free-Dox and SSLs-Dox up to 48 h (b). Results are the average of three different experiments ±S.E. Each point is collected from 6 wells. The error bars, if not shown, are within the symbols. | ||
3.4. Pharmacokinetic and biodistribution studies
The size and shape of liposomes can affect their long circulation after systemic injection and their biodistribution in animal models as well as in patients.47 Basically, PEGylation significantly prolongs the circulation of nanoparticles after systemic injection and avoids their uptake from macrophages of the reticuloendothelial system (RES), which are located preferentially in the liver, spleen, bone marrow, and lymph nodes. The pharmacokinetic study here confirmed, as expected, the long circulation time of Dox formulated with SSLs, as shown in Fig. S8 and reported in Table S2,† also shown in comparison to SLs. The T½ of SSLs-Dox was increased about 8-fold and 3-fold with respect to Dox and SLs-Dox, respectively, and the area under the curve (AUC) reached almost an increase of 300-fold and 11-fold by the same comparison reported above (Table S2†).Also, an in vivo biodistribution study of free-Dox and SSLs-Dox was carried out using the MDA-MD-231 lung metastasis tumor-bearing mouse model (Fig. 3). Plasma and tissues were collected at different time points after the i.v. injection of free-Dox and SSLs-Dox. The results demonstrated that free-Dox accumulated quickly in the liver at 1 h post injection and the concentration of the drug decreased quickly at 24 h. The free-Dox was cleared 4 days after injection and no drug was still in the liver after 7 days. Conversely, a low concentration of SSLs-Dox was still present in the liver at 1 and 24 h and decreased slowly for up to 7 days after injection (Fig. 3(a)). The free-Dox concentration was higher in the spleen and kidneys than that of SSLs-Dox at 1 h and decreased quickly for up to 7 days. The spleen and kidneys uptake of SSLs-Dox showed a lower concentration at 1 and 24 h after injection than the free drug (Fig. 3(b and c)). The partial accumulation of SSLs-Dox in the liver and spleen after 1 h of incubation could also have been affected by the enhanced permeability and retention (EPR) effect. In fact, the large gaps of the endothelial cells, from a few nanometers up to 400 nm in size, allow accumulating nanoparticles inside the tumor tissue. However, the large fenestration of the endothelial cells is not only present in the tumor tissues but represents the main component of the endothelium for the liver and spleen, which contain fenestrations with physiological characteristics, like the tumor tissue.48 The large fenestration of liver sinusoid49,50 and spleen51 showed that a large amount of nanoparticles could be filtered through these organs, and the liver and spleen are the two major organs competing with tumor tissues to accumulate nanoparticles.48 The differences in the biodistribution patterns between free-Dox and SSLs-Dox also depend on the overall physicochemical properties of liposomes. In particular, the addition of synthetic PEGylated dendron phospholipids modifies the hydrophobic/hydrophilic balance of surface nanoparticles, thus reducing the opsonin interaction and their liver/spleen uptake.52 The PEGylation of liposomes, and the presence of intact PEG chains on liposomal surface for a long time, limits the access of liposomes through the capillaries of the liver and spleen, which show a discontinuous endothelium with approximately 100 nm fenestrae and 6%–8% porosity.31,53,54 The free-Dox was accumulated in the heart 1 h after injection, while the SSLs decreased the accumulation of the chemotherapeutic drug in the heart and prevented its dose-related side effects, particularly cardiotoxicity (Fig. 3(d)).28 This result suggested that the cardiotoxicity of Dox versus heart tissue was decreased by loading the chemotherapeutic drug into the SSLs. Conversely, SSLs-Dox accumulated more in the lung tissue than free-Dox at 1 and 24 h of incubation, and the SSLs-Dox could still be detected in the lung tissue 7 days after injection (Fig. 3(e)). The SSLs-Dox thus increased the long circulation of the payload after its systemic injection. The SSLs-Dox concentration in the blood was higher than that of free-Dox, which decreased rapidly 1 h after injection and had disappeared completely by 24 h (Fig. 3(f)). The resulting data seem to suggest that the free-Dox is metabolized from systemic enzymes and cleared through the kidneys.28 In fact, the SSLs protect Dox from metabolic inactivation, while the PEGylation of liposomes increases their long circulation in the blood stream and prevents their rapid clearance from the blood circulation.15,41
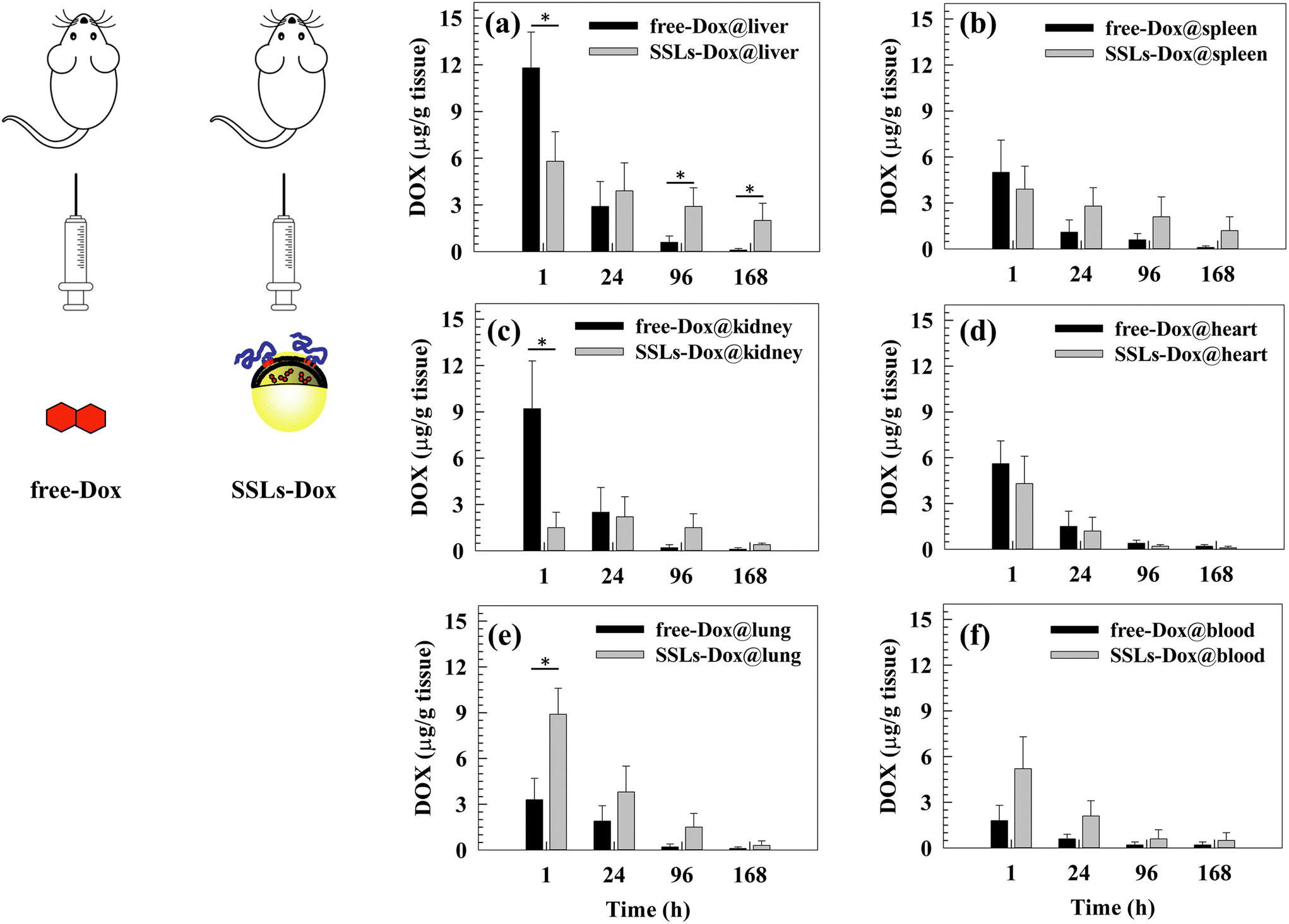 | ||
| Fig. 3 Doxorubicin hydrochloride biodistribution in the liver (a), spleen (b), kidney (c), heart (d), lung (e), and blood (f) of MDA-MB-231 tumor-bearing mice treated by free-Dox and SSLs-Dox. Mice were sacrificed at different time points, i.e., 1 h, 24 h, 4 days, and 7 days. The Dox was extracted from the tissues or blood and measured by HPLC. *p < 0.05, **p < 0.01 represent statistically significant differences of free-Dox versus SSLs-Dox. | ||
The blood concentration of SSLs-Dox was maintained for up to 7 days after systemic injection, while the free-Dox had disappeared by 24 h after injection (Fig. 3(f)).
3.5. In vivo antitumor experiment
The growth of metastatic tumors was monitored using bioluminescent MDA-MB-231 cells (Fig. S9†), which were engineered by transfecting breast cancer cells with the luciferase gene. The mouse model of human breast cancer lung metastasis was established in 6-week-old nude female mice by tail vein inoculation of 3 × 105 MDA-MB-231/luciferase cells per ml. The tumor size became similar among all the groups within 1 week after the tumor cells inoculation and the treatment started at this time, according to the therapeutic schedule reported in the Materials and methods section, by injecting 3 mg kg−1 equivalent dose of the drug weekly (Fig. 4(a)). After 2 cycles of treatment, the tumor size had significantly increased in the PBS and empty-SSLs groups, but not in the free-Dox and SSLs-Dox groups (Fig. 4(b)).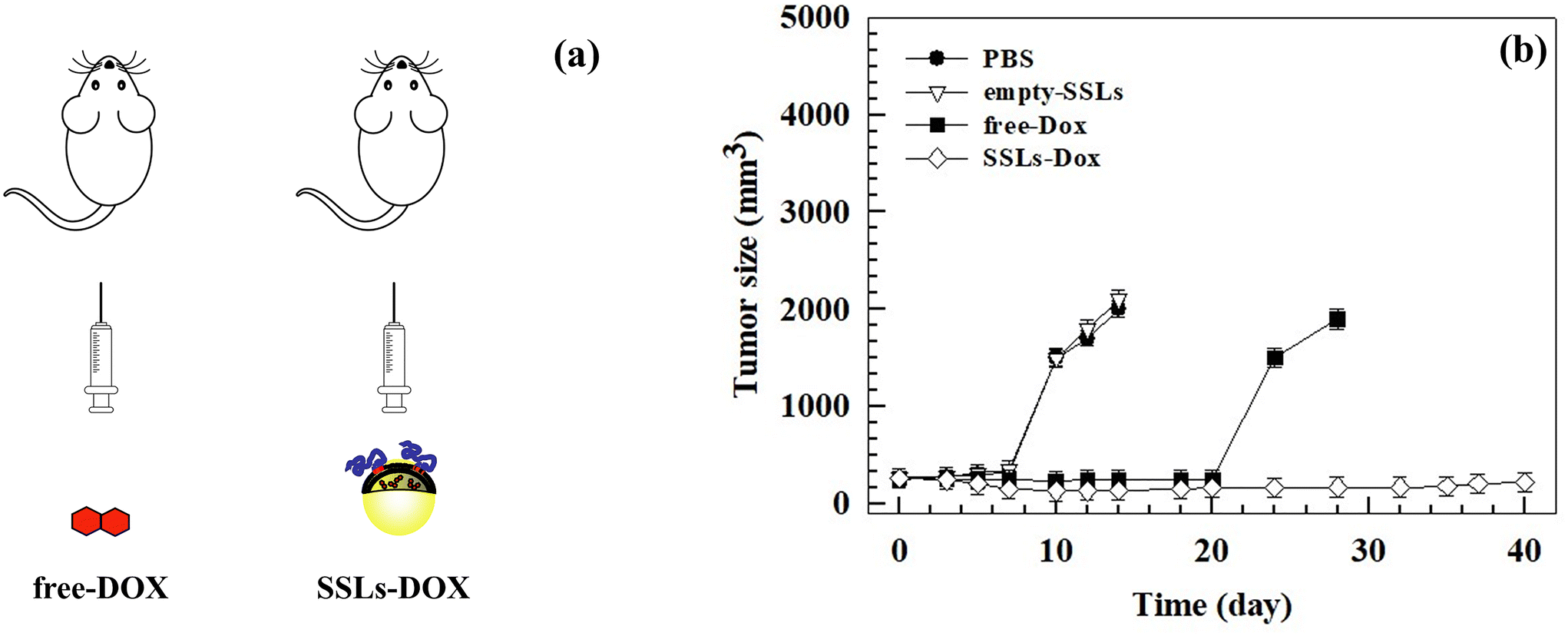 | ||
| Fig. 4 In vivo antitumor activity of free-Dox versus SSLs-Dox by using female mice engrafted with MDA-MB-231 cells. Female nude mice were injected intravenously with 3 × 105 MDA-MB-231/luciferase cells per ml. After tumor implantation and development, the mice were injected intravenously using PBS, empty-SSLs, free-Dox, and SSLs-Dox. The injected dose of SSLs-Dox was 3 mg kg−1 equivalent to free-Dox for each administration. (a) Schematic representation of the therapeutic schedule. (b) Tumor volumes of different groups were measured with a caliper and calculated according to eqn (1) reported in the Materials and methods section. The statistical analysis is reported in the ESI (Table S3†). At day 14 and day 28, the mice in the PBS/empty-SSLs and free-Dox groups were sacrificed because the tumor volume exceeded 2000 mm3. | ||
The tumor sizes were monitored at different times and tumor sizes of ∼250 mm3 were measured before starting the treatment (Fig. 4(b)). The mice were sacrificed when the tumor volumes exceeded 2000 mm3.
The increase in anticancer efficacy of SSLs-Dox compared to the free-Dox and controls, i.e., PBS and empty-SSLs, depended on the SSLs, which could protect Dox after systemic injection and increase its accumulation inside the tumor tissue. In fact, conventional and modified liposomes protect chemotherapeutic drugs, particularly Dox, after systemic injection and increase its half-life over 2 h, as shown in the literature55 and here with SSLs-Dox (Fig. S8 and Table S2†). PEGylated liposomes can easily escape intact through the leaky tumor vasculature and accumulate inside the tumor tissues. This is the special case for Doxil®, a PEGylated liposome formulation approved for clinical cancer treatment, which can pass the fenestrated tumor vessels by an EPR effect.56,57 Furthermore, the deficient lymphatic drainage of tumor tissues, as well the fenestration of the vascular tumor endothelium, can modify the fluid transport dynamics of nanoparticles mediated by the EPR effect and thus increase their retention inside the tumor tissue.48 PEGylated liposomes, having a hydrodynamic radius well exceeding that of macromolecules of 40 kDa, can escape through tumor vessels and accumulate inside the tumor tissue.58 The retention of Doxil®, as well as other PEGylated liposomes, inside the tumor tissues also affects the increase in Dox anticancer activity in vivo after systemic injection. In fact, the drug is released from Doxil®, intracellularly metabolized, and then taken up by the tumor cells.28 Dox can be released in the intracellular interstitial fluid of tumor cells and accumulate inside the tumor, where it is converted into active metabolites, which allow the drug anticancer activity.28 Additionally, phospholipases, overexpressed in the tumor tissues, hydrolyze the phospholipids of the liposomal bilayer, and destabilize the supramolecular architecture of Doxil®, which totally or partially collapses under the effect of an ammonium sulfate gradient and causes the release of Dox.59,60 A similar effect can occur by using in vivo SSLs-Dox, which have a lipid composition like Doxil® and are made up using an ammonium sulfate pH-gradient remote-loading procedure. The differences in the in vivo anticancer activity between free-Dox and SSLs-Dox can also depend on the surface modification of liposomes. The presence of PEGylated dendron phospholipids allows stabilizing PEG in the liposomal bilayer as previously reported.15
Despite the metastatic breast cancer tumor growths in the MDA-MD-231 lung metastasis tumor-bearing mouse model, the treatment with SSLs-Dox decreased the tumor size, thus maintaining tumor growth inhibition for up to 20 days after the end of the treatment (Fig. 4(b)). Conversely, the free-Dox showed lower tumor growth inhibition than SSLs-Dox (Fig. 4(b)). Indeed, in the mice treated with free-Dox, the tumor mass re-started growing after the end of treatment, thus showing a tumor size greater than 1500 mm3 after 25 days and then reached the threshold of 2000 mm3 at day 28 (Fig. 4(b)). The PBS and empty-SSLs groups had a continuous and constant growth of the tumors, which quickly reached 2000 mm3 after 14 days (Fig. 4(b)). These results demonstrated that SSLs allowed the distribution and accumulation of Dox inside the tumor mass and enabled a significant decrease in tumor size, which was consistent with liposomal accumulation in the metastatic loci and tumor microenvironment. The anatomic location and metastatic lesions, as well as the properties of the tumor microenvironment, can further improve the anticancer activity of SSLs-Dox because of the well vascularized area of lung, the high perfusion of this tissue, and the extensive lymphatic vessel density, which can increase the diffusion of lung metastasis but also the accumulation of SSLs-Dox. These results agreed with data previously reported for MM-302 liposomes that were used to treat breast cancer and inhibit lung, liver, and brain metastases.61
This difference may depend on the rapid clearance and metabolism of free-Dox from the blood stream after i.v. injection.62 The bioluminescent intensity results (Fig. S9†) agreed with data showing a significant inhibition of tumor growth in the mouse model and further demonstrated that SSLs-Dox could arrest tumor growth. No significant differences in the body weight of the mice were found in any of the groups during the experiments despite the potent anticancer efficiency of SSLs-Dox (Fig. 5(b)). The overall results demonstrated that all the treatments were well tolerated and did not cause any toxicity in the tested groups (Fig. 5(b)).
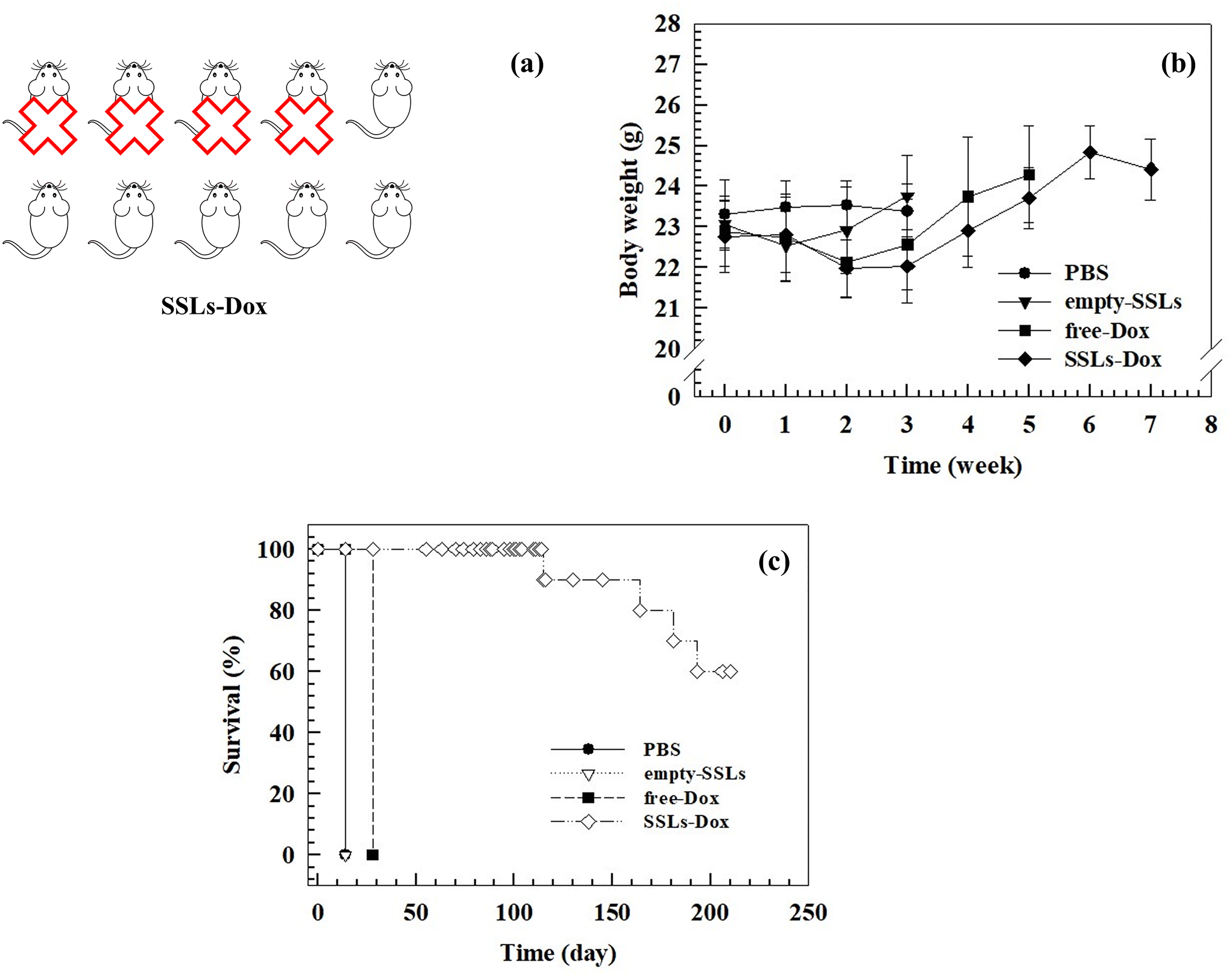 | ||
| Fig. 5 (a) Schematic description of the dead mice after treatment with PBS, empty-SSLs, free-Dox, and SSLs-Dox. The illustration is representative of the dead mouse trend reported in the survival curve and represents the number of animals surviving at the end of treatment for each group. (b) Changes in body weight were monitored weekly. No significant differences between these four groups were found, but a drop in body weight was obtained for the groups after the first injection. (c) Survival rate of tumor-bearing mice was studied for up to 210 days (n = 10 per group). The survival rate tests of the PBS, empty-SSLs, and free-Dox groups were discontinued due to the overgrowth of the tumor size (volume >2000 mm3). | ||
The presence of a stable PEG on the bilayer of the liposomes may increase the retention of nanocarriers in the bloodstream and promote tumor accumulation, thus improving the in vivo antitumor activity of liposomes compared to the free drug (Fig. 4(b)). The tumor bioluminescence results agreed with these data, thus demonstrating a significant reduction of the signal in the SSLs-Dox group compared to the free-Dox group (Fig. S9†). To further study the SSLs-Dox treatment efficiency, the mice were monitored for up to 210 days, revealing a survival rate of 60% (Fig. 5).
To carry out morphological studies, lung tissue samples were collected from 3 mice from each group. Many macroscopic tumor nodules were observed on the lung tissues from the PBS and empty-SSLs groups, and adhesions were also found between the lung and thoracic cavity of the mice from the above two groups. The status of the lungs of the mice in the free-Dox and SSL-Dox groups were much better than those for the two control groups (PBS and empty-SSLs). The lung tissue samples showed tumor nodules in the free-Dox group, while nodules were decreased and reduced in number in the SSLs-Dox group (Fig. S10(a)†). The lung tissue in the SSLs-Dox group was like normal tissue and no macroscopic tumor nodules or pathological adhesions to the thoracic cavity were observed (Fig. S10(a)†). The lung tissues were further examined by H&E staining (Fig. S10(b)†). Similar results were carried out after H&E staining analysis, and tumor nodules were found to be present in lung tissue treated with PBS and empty-SSLs. Conversely, few tumor nodules were found in the free-Dox group, while the nodules were strongly decreased in the SSL-Dox group. These results were further supported by the Ki-67-staining-positive cells; in fact, there were fewer numbers of Ki-67-staining-positive cells in the SSLs-Dox treated mice compared to the PBS, empty-SSLs, and free-Dox groups (Fig. S10(c)†).
4. Conclusions
We tested the anticancer efficacy of SSLs-Dox and their potential ability to inhibit the proliferation of breast cancer cells and decrease the induction of lung metastasis. We demonstrated that SSLs could enable a long circulation time of Dox after systemic injection, guarantee the distribution of chemotherapeutic drug in the bloodstream, and thus favor the efficacy of anticancer treatment. SSLs also decreased the drug's side effects and increased the accumulation of Dox in the lungs, where metastasis occurred. SSLs allowed for the long circulation of Dox in the blood stream, and the liver and spleen uptake was decreased compared to the free-Dox, which showed accumulation in both tissues 1 h after injection. The intravenous injection of SSLs-Dox decreased tumor growth and the development of the tumor sizes in the mice as well as the tumor nodules, compared to the free-Dox. We hope that this promising data could support the potential use of SSLs-Dox in clinical trials and the future development of an efficacious breast cancer lung metastasis therapy.Author contributions
Nicola d'Avanzo (N. d'A.): data curation, investigation, formal analysis, and writing – original draft. Donatella Paolino (D. P.): data curation, formal analysis, conceptualization, and investigation. Gianfranco Pasut (G. P.): conceptualization, writing – review and funding acquisition. Antonella Grigoletto (A. G.), Elena Canato (E. C.): investigation. Luigi Ciriolo (L. C.): investigation. Maria Chiara Cristiano (M. C. C.): writing – review & editing. Antonia Mancuso (A. M.): data curation. Christian Celia (C. C.): writing – original draft, investigation, data curation, conceptualization and funding acquisition. Massimo Fresta (M. F.): writing – review, conceptualization, supervision and data curation.Data availability
The data supporting this article have been included as part of the ESI.†The online freely available software version of grammarly has been used to check and correct the spelling and grammar errors, wording, and punctuation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by Ministero dell'Università e della Ricerca (MUR) [FAR 2018 (D56C18000780005), FAR 2019 (D54I19002790005)] to C. C., and AIRC (IG2017, Cod. 20224) to G. P. Authors thank Prof. Sara De Martin for the support on pharmacokinetic analysis.References
- A. G. Waks and E. P. Winer, J. Am. Med. Assoc., 2019, 321, 288–300, DOI:10.1001/jama.2018.19323.
- H. A. Wahba and H. A. El-Hadaad, Cancer Biol. Med., 2015, 12, 106, DOI:10.7497/j.issn.2095-3941.2015.0030.
- M. Sterba, O. Popelova, A. Vavrova, E. Jirkovsky, P. Kovarikova, V. Gersl and T. Simunek, Antioxid. Redox Signal., 2013, 18, 899–929, DOI:10.1089/ars.2012.4795.
- H. Li, H. Jin, W. Wan, C. Wu and L. Wei, Nanomedicine, 2018, 13, 1639–1656, DOI:10.2217/nnm-2018-0007.
- S. Z. Vahed, R. Salehi, S. Davaran and S. Sharifi, Mater. Sci. Eng., C, 2017, 71, 1327–1341, DOI:10.1016/j.msec.2016.11.073.
- A. Chrastina, K. A. Massey and J. E. Schnitzer, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3, 421–437, DOI:10.1002/wnan.143.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2019, 4, e10143, DOI:10.1002/btm2.10143.
- A. J. Thorley and T. D. Tetley, Pharmacol. Ther., 2013, 140(2), 176–185, DOI:10.1016/j.pharmthera.2013.06.008.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822, DOI:10.1126/science.1095833.
- C. Celia, M. C. Cristiano, F. Froiio, M. Di Francesco, N. d'Avanzo, L. Di Marzio and M. Fresta, Adv. Ther., 2021, 4, 2000121, DOI:10.1002/adtp.202000121.
- A. Gabizon, H. Shmeeda and Y. Barenholz, Clin. Pharmacokinet., 2003, 42, 419–436, DOI:10.2165/00003088-200342050-00002.
- I. Judson, J. A. Radford, M. Harris, J. Y. Blay, Q. van Hoesel, A. le Cesne, A. T. van Oosterom, M. J. Clemons, C. Kamby, C. Hermans, J. Whittaker, E. Donato di Paola, J. Verweij and S. Nielsen, Eur. J. Cancer, 2001, 37, 870–877, DOI:10.1016/s0959-8049(01)00050-8.
- M. E. O'Brien, N. Wigler, M. Inbar, R. Rosso, E. Grischke, A. Santoro, R. Catane, D. G. Kieback, P. Tomczak, S. P. Ackland, F. Orlandi, L. Mellars, L. Alland and C. Tendler, Ann. Oncol., 2004, 15, 440–449, DOI:10.1093/annonc/mdh097.
- M. J. Parr, S. M. Ansell, L. S. Choi and P. R. Cullis, Biochim. Biophys. Acta, Biomembr., 1994, 1195, 21–30, DOI:10.1093/annonc/mdh097.
- G. Pasut, D. Paolino, C. Celia, A. Mero, A. S. Joseph, J. Wolfram, D. Cosco, O. Schiavon, H. Shen and M. Fresta, J. Controlled Release, 2015, 199, 106–113, DOI:10.1016/j.jconrel.2014.12.008.
- D. Catanzaro, S. Nicolosi, V. Cocetta, M. Salvalaio, A. Pagetta, E. Ragazzi, M. Montopoli and G. Pasut, Oncotarget, 2018, 9, 16847, DOI:10.18632/oncotarget.24708.
- D. K. Kirui, C. Celia, R. Molinaro, S. S. Bansal, D. Cosco, M. Fresta, H. Shen and M. Ferrari, Adv. Healthcare Mater., 2015, 4, 1092–1103, DOI:10.1002/adhm.201400738.
- C. Marianecci, L. Di Marzio, E. Del Favero, L. Cantù, P. Brocca, V. Rondelli, F. Rinaldi, L. Dini, A. Serra and P. Decuzzi, Langmuir, 2016, 32, 1241–1249, DOI:10.1021/acs.langmuir.5b04111.
- D. Paolino, D. Cosco, M. Gaspari, M. Celano, J. Wolfram, P. Voce, E. Puxeddu, S. Filetti, C. Celia and M. Ferrari, Biomaterials, 2014, 35, 7101–7109, DOI:10.1016/j.biomaterials.2014.04.088.
- P. Sedgwick, Br. Med. J., 2014, 349, g5608, DOI:10.1136/bmj.g5608.
- M. Di Francesco, C. Celia, M. C. Cristiano, N. d'Avanzo, B. Ruozi, C. Mircioiu, D. Cosco, L. Di Marzio and M. Fresta, ACS Omega, 2021, 6, 2973–2989, DOI:10.1021/acsomega.0c05350.
- K. Alhareth, C. Vauthier, C. Gueutin, G. Ponchel and F. Moussa, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 887–888, 128–132, DOI:10.1016/j.jchromb.2012.01.025.
- S. Wohlfart, A. S. Khalansky, S. Gelperina, D. Begley and J. Kreuter, J. Controlled Release, 2011, 154, 103–107, DOI:10.1016/j.jconrel.2011.05.010.
- D. C. Drummond, O. Meyer, K. Hong, D. B. Kirpotin and D. Papahadjopoulos, Pharmacol. Rev., 1999, 51, 691–744 CAS.
- A. V. Kalra, J. Kim, S. G. Klinz, N. Paz, J. Cain, D. C. Drummond, U. B. Nielsen and J. B. Fitzgerald, Cancer Res., 2014, 74, 7003–7013, DOI:10.1158/0008-5472.CAN-14-0572.
- M. H. Kang, J. Wang, M. R. Makena, J.-S. Lee, N. Paz, C. P. Hall, M. M. Song, R. I. Calderon, R. E. Cruz and A. Hindle, Clin. Cancer Res., 2015, 21, 1139–1150, DOI:10.1158/1078-0432.CCR-14-1882.
- A. Fritze, F. Hens, A. Kimpfler, R. Schubert and R. Peschka-Süss, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1633–1640, DOI:10.1016/j.bbamem.2006.05.028.
- Y. C. Barenholz, J. Controlled Release, 2012, 160, 117–134, DOI:10.1016/j.jconrel.2012.03.020.
- A. Cern, Y. Barenholz, A. Tropsha and A. Goldblum, J. Controlled Release, 2014, 173, 125–131, DOI:10.1016/j.jconrel.2012.03.020.
- A. Cern, A. Golbraikh, A. Sedykh, A. Tropsha, Y. Barenholz and A. Goldblum, J. Controlled Release, 2012, 160, 147–157, DOI:10.1016/j.jconrel.2011.11.029.
- S. Zhu, M. Hong, G. Tang, L. Qian, J. Lin, Y. Jiang and Y. Pei, Biomaterials, 2010, 31, 1360–1371, DOI:10.1016/j.biomaterials.2009.10.044.
- D. Gewirtz, Biochem. Pharmacol., 1999, 57, 727–741, DOI:10.1016/s0006-2952(98)00307-4.
- R. Molinaro, J. Wolfram, C. Federico, F. Cilurzo, L. Di Marzio, C. A. Ventura, M. Carafa, C. Celia and M. Fresta, Expert Opin. Drug Delivery, 2013, 10, 1653–1668, DOI:10.1517/17425247.2013.840286.
- L. Serpe, M. Guido, R. Canaparo, E. Muntoni, R. Cavalli, P. Panzanelli, C. D. Pepa, A. Bargoni, A. Mauro and M. R. Gasco, J. Nanosci. Nanotechnol., 2006, 6, 3062–3069, DOI:10.1166/jnn.2006.423.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160, DOI:10.1038/nrd1632.
- A. A. Gabizon, Cancer Res., 1992, 52, 891–896 CAS.
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229, DOI:10.1124/pr.56.2.6.
- A. J. Smith, Front. Cardiovasc. Med., 2021, 8, 624028, DOI:10.3389/fcvm.2021.624028.
- M. Bosman, K. Favere, C. H. Neutel, G. Jacobs, G. R. De Meyer, W. Martinet, E. M. Van Craenenbroeck and P.-J. D. Guns, Toxicol. Lett., 2021, 346, 23–33, DOI:10.1016/j.toxlet.2021.04.015.
- S. Sritharan and N. Sivalingam, Life Sci., 2021, 278, 119527, DOI:10.1016/j.lfs.2021.119527.
- G. Pasut and F. M. Veronese, J. Controlled Release, 2012, 161, 461–472, DOI:10.1016/j.jconrel.2011.10.037.
- A. Gabizon, H. Shmeeda and T. Grenader, Eur. J. Pharm. Sci., 2012, 45, 388–398, DOI:10.1016/j.ejps.2011.09.006.
- S. Biswas, N. S. Dodwadkar, P. P. Deshpande, S. Parab and V. P. Torchilin, Eur. J. Pharm. Biopharm., 2013, 84, 517–525, DOI:10.1016/j.ejpb.2012.12.021.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951, DOI:10.1038/nbt.3330.
- R. Islam, H. Maeda and J. Fang, Expert Opin. Drug Delivery, 2022, 19, 199–212, DOI:10.1080/17425247.2021.1874916.
- J. A. MacKay, M. Chen, J. R. McDaniel, W. Liu, A. J. Simnick and A. Chilkoti, Nat. Mater., 2009, 8, 993–999, DOI:10.1038/nmat2569.
- S. M. Moghimi, A. Hunter and T. Andresen, Annu. Rev. Pharmacol. Toxicol., 2012, 52, 481–503, DOI:10.1146/annurev-pharmtox-010611-134623.
- S. Taurin, H. Nehoff and K. Greish, J. Controlled Release, 2012, 164, 265–275, DOI:10.1016/j.jconrel.2012.07.013.
- J. Li, C. Chen and T. Xia, Adv. Mater., 2022, 34, 2106456, DOI:10.1002/adma.202106456.
- T. Horn, J. H. Henriksen and P. Christoffersen, Liver, 1986, 6, 98–110, DOI:10.1111/j.1600-0676.1986.tb00275.x.
- H. Sarin, J. Angiog. Res., 2010, 2, 1–19, DOI:10.1186/2040-2384-2-14.
- I. Brigger, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2012, 64, 24–36, DOI:10.1016/s0169-409x(02)00044-3.
- R. Kaur, V. W. Bramwell, D. J. Kirby and Y. Perrie, J. Controlled Release, 2012, 164, 331–337, DOI:10.1016/j.jconrel.2012.07.012.
- M. Urbanczyk, A. Zbinden and K. Schenke-Layland, Adv. Drug Delivery Rev., 2022, 114323, DOI:10.1016/j.addr.2022.114323.
- H. D. Han, A. Lee, T. Hwang, C. K. Song, H. Seong, J. Hyun and B. C. Shin, J. Controlled Release, 2007, 120, 161–168, DOI:10.1016/j.jconrel.2007.03.020.
- R. K. Jain, R. T. Tong and L. L. Munn, Cancer Res., 2007, 67, 2729–2735, DOI:10.1158/0008-5472.CAN-06-4102.
- H. Maeda, K. Tsukigawa and J. Fang, Microcirculation, 2016, 23, 173–182, DOI:10.1111/micc.12228.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151, DOI:10.1016/j.addr.2010.04.009.
- D. Shi, C. Feng, J. Xie, X. Zhang, H. Dai and L. Yan, J. Mater. Chem. B, 2022, 10(37), 7349–7360, 10.1039/d2tb00608a.
- E. Oude Blenke, E. Mastrobattista and R. M. Schiffelers, Expert Opin. Drug Delivery, 2013, 10, 1399–1410, DOI:10.1517/17425247.2013.805742.
- N. Dumont, S. Merrigan, J. Turpin, C. Lavoie, V. Papavasiliou, E. Geretti, C. W. Espelin, L. Luus, W. S. Kamoun, O. Ghasemi, G. G. Sahagian, W. J. Muller, B. S. Hendriks, T. J. Wickham and D. C. Drummond, Nanomedicine, 2019, 17, 71–81, DOI:10.1016/j.nano.2018.12.010.
- Y. H. Choi, Y. K. Lee and M. G. Lee, Xenobiotica, 2013, 43, 901–907, DOI:10.3109/00498254.2013.783250.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4bm00478g |
| This journal is © The Royal Society of Chemistry 2024 |