
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineered triphenylphosphonium-based, mitochondrial-targeted liposomal drug delivery system facilitates cancer cell killing actions of chemotherapeutics†
Subramaniyam
Sivagnanam‡
 a,
Kiran
Das‡
b,
Ieshita
Pan
c,
Adele
Stewart
e,
Atanu
Barik
d,
Biswanath
Maity
*b and
Priyadip
Das
*a
a,
Kiran
Das‡
b,
Ieshita
Pan
c,
Adele
Stewart
e,
Atanu
Barik
d,
Biswanath
Maity
*b and
Priyadip
Das
*a
aDepartment of Chemistry, SRM Institute of Science and Technology, SRM Nagar, Potheri, Kattankulathur, Tamil Nadu-603203, India. E-mail: priyadipcsmcri@gmail.com; priyadip@srmist.edu.in
bCentre of Biomedical Research, Sanjay Gandhi Post Graduate Institute of Medical Sciences (SGPGI) campus, Raebareli Road, Lucknow, Uttar Pradesh 226014, India. E-mail: bmaity28@gmail.com; bmaity@cbmr.res.in
cDepartment of Biotechnology, Saveetha School of Engineering, Saveetha Institute of Medical and Technical Sciences, Saveetha University, Chennai 602105, Tamil Nadu, India
dRadiation & Photochemistry Division, Bhabha Atomic Research Centre, Trombay, Mumbai 400085, Maharashtra, India
eDepartment of Biomedical Science, Charles E. Schmidt College of Medicine, Florida Atlantic University, Jupiter, FL 33458, USA
First published on 21st December 2023
Abstract
In addition to their classical role in ATP generation, mitochondria also contribute to Ca2+ buffering, free radical production, and initiation of programmed cell death. Mitochondrial dysfunction has been linked to several leading causes of morbidity and mortality worldwide including neurodegenerative, metabolic, and cardiovascular diseases as well as several cancer subtypes. Thus, there is growing interest in developing drug-delivery vehicles capable of shuttling therapeutics directly to the mitochondria. Here, we functionalized the conventional 10,12-pentacosadiynoic acid/1,2-dimyristoyl-sn-glycero-3-phosphocholine (PCDA/DMPC)-based liposome with a mitochondria-targeting triphenylphosphonium (TPP) cationic group. A fluorescent dansyl dye (DAN) group was also included for tracking mitochondrial drug uptake. The resultant PCDA-TPP and PCDA-DAN conjugates were incorporated into a 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC)-based lipid bilayer, and these modified liposomes (Lip-DT) were studied for their cellular toxicity, mitochondrial targeting ability, and efficacy in delivering the drug Doxorubicin (Dox) to human colorectal carcinoma (HCT116) and human breast (MCF7) cancer cells in vitro. This Lip-DT-Dox exhibited the ability to shuttle the encapsulated drug to the mitochondria of cancer cells and triggered oxidative stress, mitochondrial dysfunction, and apoptosis. The ability of Lip-DT-Dox to trigger cellular toxicity in both HCT116 and MCF7 cancer cells was comparable to the known cell-killing actions of the unencapsulated drug (Dox). The findings in this study reveal a promising approach where conventional liposome-based drug delivery systems can be rendered mitochondria-specific by incorporating well-known mitochondriotropic moieties onto the surface of the liposome.
Introduction
Cancer is one of the most complex and challenging diseases and represents the second leading cause of the global morbidity and mortality.1,2 Cancer chemotherapeutics are frequently employed for cancer therapy to decrease tumor burden and prevent the metastasis of malignant cells.3,4 Despite of significant advances in cancer treatment, there is still demand for new improved, and effective therapies lacking adverse side effects. Poor solubility, poor in vivo stability, inferior cell penetration and low specificity towards targeted cancer cells, and undesirable side effects often restrict the efficacy of conventional chemotherapy treatment. In this context, one of the promising approaches is the employment of effective drug delivery vehicles, such as nanoparticles5,6 liposomes,7 and micelles,8 which can improve the pharmacokinetics and biodistribution of anticancer drugs, leading to better therapeutic outcomes. Such drug delivery vehicles can also be engineered for specific cancer cell types or tumor microenvironments, permitting more precise and effective drug delivery and decreasing unwanted side effects.9–11Design and development of an effective drug delivery system, capable of encapsulating hydrophilic and hydrophobic drugs has proven challenging due to their variable affinities towards the carrier molecule. To address this issue, amphiphilic molecules containing several functional groups, each with differing affinity towards hydrophilic and hydrophobic drugs, have been developed. However, the application of these drug delivery systems has been limited by several factors, such as lower encapsulation efficiency, drug leakage, carrier stability issues, and burst release of loaded drug molecules. Nano-drug delivery systems (NDDS) have proven superior and allowed for efficient chemotherapeutic drug encapsulation, improved tumor microenvironment response, and target-specific drug release.
NDDS approved for clinical use include micelles (Genexol-PM®), nanoparticles (Abraxene®) and liposomes (Doxil®, Lipusu®).12 Among these NDDS, liposomes are favoured owing to their biocompatibility and biodegradability.13,14 However, conventional liposomes have certain drawbacks, such as structural instability, oxidation, premature drug release, and hydrolysis, which limits their applicability for drug delivery.12,15 Liposome polymerization can overcome some of these issues, and, in particular, polymerized polydiacetylene (PDA) based liposomes are biocompatible, display low cytotoxicity, and exhibit improved stability over unpolymerized liposomes.16 Most importantly, diacetylene monomers have a carboxylic acid as a functional head group, which opens up the possibility of functionalization with various receptors, antibodies, ligands to make this system target-specific.17
We have known for over a century that cancer cells are heavily reliant on mitochondria to fuel rapid cell growth and division with the shift in metabolic strategy exhibited by many malignant cells nicknamed the “Warburg effect”.18 Notably, in addition to their role in oxidative phosphorylation, mitochondria are critical mediators of programmed cell death and translocation of cytochrome c from the mitochondrial inner membrane into the cytosol activates key executioners of apoptosis (i.e., caspases).19–22 Thus, while disruption of mitochondrial energy production may slow tumour growth, recruitment of the intrinsic mitochondrial apoptosis pathway also represents a strategy to kill malignant cells.23,24 Current strategies to develop mitochondrial-targeted drug delivery systems (MTDDS) depend on two key mitochondrial characteristics: (i) the high membrane potential across the inner mitochondria membrane (ΔΨm) and (ii) the protein import machinery present within the organelle.25 The high ΔΨm characteristic of mitochondria strongly attracts lipophilic cations through electrostatic interactions allowing for their accumulation in the mitochondrial matrix. Notably, cancer cells exhibit an elevated mitochondrial membrane potential compared to non-malignant cells raising the possibility that mitochondrial drug delivery might be particularly facile in cancer cells.26,27
Delocalized lipophilic cations (DLCs) with positive charge have large hydrophobic surface areas, which make them permeable through the negatively charged mitochondrial membranes.23 The lipophilic nature of DLCs permit their easy penetration through the membrane bilayers, and their permanent cationic charge eventually leads to their accumulation inside the mitochondrial matrix.28 Most DLCs are tailored with two common structural features necessary for mitochondrial accumulation: (i) amphiphilicity provided by a charged hydrophilic centre with a hydrophobic core and (ii) delocalized positive-charge density.28 DLCs examples include Rhodamine123 (Rh123), dequaliniumchloride (DQA) and methyltriphenylphosphonium (TPP). TPP has a single positive charge, which is stabilized by the resonance over three phenyl rings.28–30 S. Biswas et al. reported a mitochondria-targeting drug delivery system (TPP–PEG–PE), in which polyethylene glycol-phosphatidylethanolamine (PEG–PE) was conjugated with TPP at the distal end of the PEG block. This TPP–PEG–PE conjugate was incorporated into a liposomal lipid bilayer to produce TPP–PEG–PE modified liposomes (TPP-PE-L). TPP-PEG-L showed lower cytotoxicity compared with the liposome modified with stearyl triphenylphosphonium (STTP) (STPP-L or PEGylated STPP-L) and was proven to be an effective tool for targeting mitochondria in HeLa cancer cells.7 It also enhanced the cytotoxicity and anti-tumor efficacy of Paclitaxel (PTX) in both in vitro and in vivo models. On the other hand, PTX encapsulated bare liposome without any chemical modification failed to exhibit site-specific drug delivery.
On major limitation to prior studies is the inference of enhanced cellular uptake and release of the desired drug molecule in cancer cells due to improved cell adhesion of the delivery vehicle. However, it was not possible to monitor drug delivery and release in real time precluding determination of when, where, and how the encapsulated drug is delivered. We hypothesized that coupling of an organic fluorophore to a site-specific drug delivery system would allow us to monitor the cellular uptake and release of a drug at the subcellular level.31–34
In this report, we describe the development of a stable, nontoxic, mitochondrial specific and efficient drug-delivery system for chemotherapeutic drugs that allows for imaging of drug release in real time. We functionalized 10,12-pentacosadiynoic acid (PCDA) with a mitochondria targeting triphenylphosphonium (TPP) cationic group and a fluorescent dye dansyl (DAN) group. The TPP/DAN functionalized PCDA was incorporated into the 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) phospholipid to generate the functionalized PCDA/DMPC-based liposome (Lip-DT). Here we describe the ability of Lip-DT to convey the chemotherapeutic agent doxorubicin (Dox) into the mitochondria of cancer cells and trigger apoptosis.
Results and discussion
Herein, we have developed a PDA based, mitochondria-specific fluorescent drug delivery platform. As outlined in Scheme 1, we reacted the PCDA (1) monomer with N-hydroxy succinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) followed by ethylene diamine to synthesize the PCDA–ethylene diamine conjugate (3). We further functionalized this conjugate with a mitochondriotropic unit, (4-carboxybutyl) triphenyl phosphoniumbromide, (4) using HBTU and DIPEA as coupling reagents to obtain a triphenylphosphonium PCDA–ethylenediamine conjugate (PCDA-TPP) (7). Similarly, to monitor the cellular uptake and drug release, 3 is functionalized with a fluorophore, dansyl chloride, (5) to give the PCDA–ethylenediamine–dansyl conjugate (PCDA-DAN) (6). We have characterized all the synthesized compounds using standard analytical techniques such as 1H NMR, 13C NMR, ESI-Mass spectroscopy and 31P NMR (Fig. S1–S9, ESI†).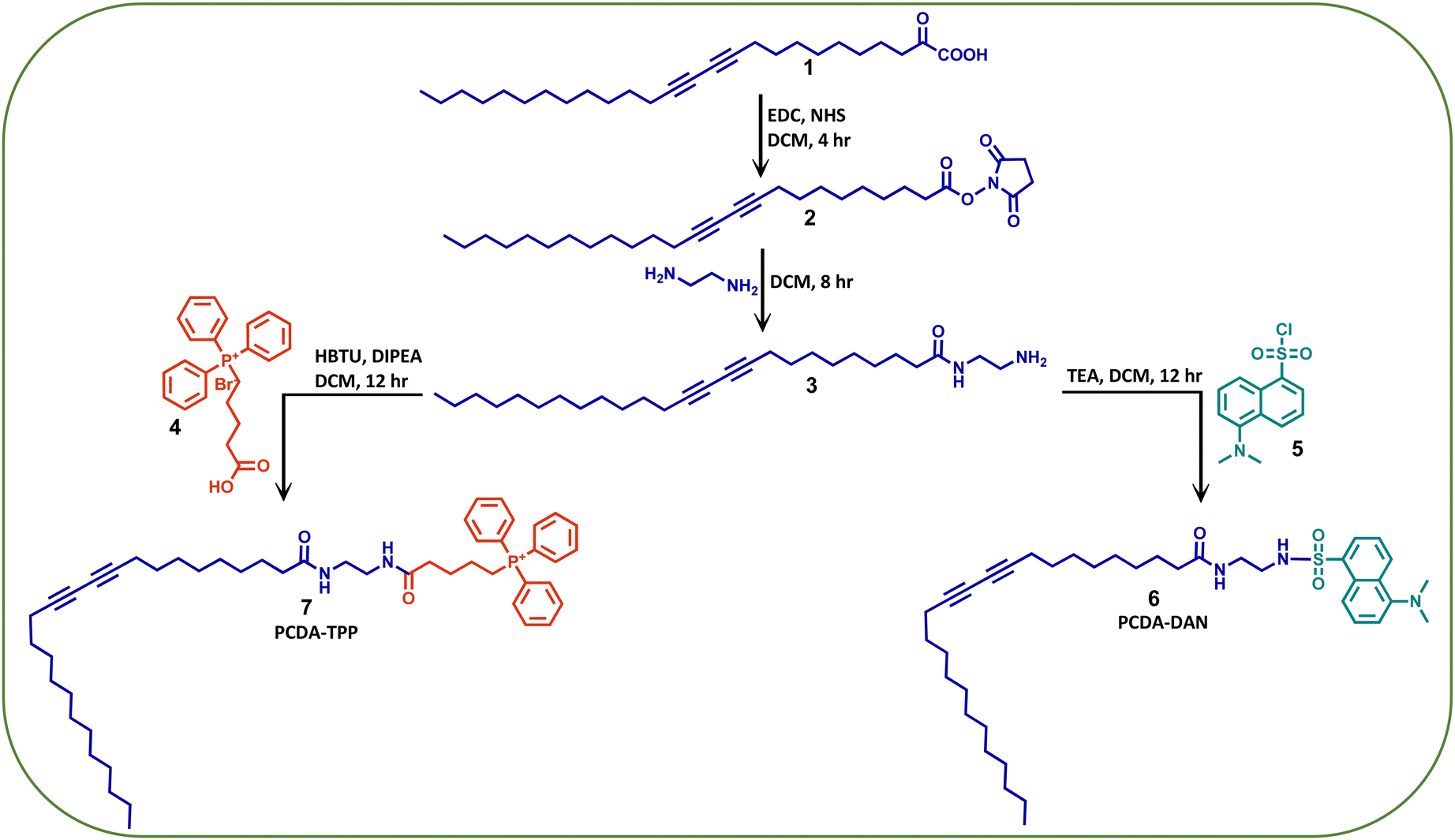 | ||
| Scheme 1 The synthetic methodology adopted for the synthesis of the dansyl (DAN) and triphenylphonium (TPP) conjugates of PCDA. | ||
Our next aim is the employment of PCDA-TPP and PCDA-DAN in the formulation of a stable liposomes through the co-assembly with the phospholipid DMPC. Morphology, size, and charge of the liposomes can significantly affect their drug delivery performance including circulation time and cellular uptake. To generate liposomes with the desired morphology, size, charge, stability and drug encapsulation efficiency, preparation of the PDA and DMPC based liposome with an optimized ratio of PDA to phospholipid and appropriate formulation parameters is crucial.35 Using thin-film hydration followed by extrusion, the most common method of liposome generation,35 we successfully prepared liposomes containing PCDA-TPP, PCDA-DAN and DMPC through co-assembly in 1X PBS buffer medium (pH = 7.4). PCDA-TPP, PCDA-DAN and DMPC were individually dissolved in chloroform, then blended at 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:1 ratio (PCDA-TPP:PCDA-DAN:DMPC). The total lipid concentration of the prepared liposome (Lip-DT) is 1 mM. The schematic representation of the formation of Lip-DT liposome is shown in Scheme 2. A more detailed synthesis protocol is provided in the materials and methods section.
:4:1 ratio (PCDA-TPP:PCDA-DAN:DMPC). The total lipid concentration of the prepared liposome (Lip-DT) is 1 mM. The schematic representation of the formation of Lip-DT liposome is shown in Scheme 2. A more detailed synthesis protocol is provided in the materials and methods section.
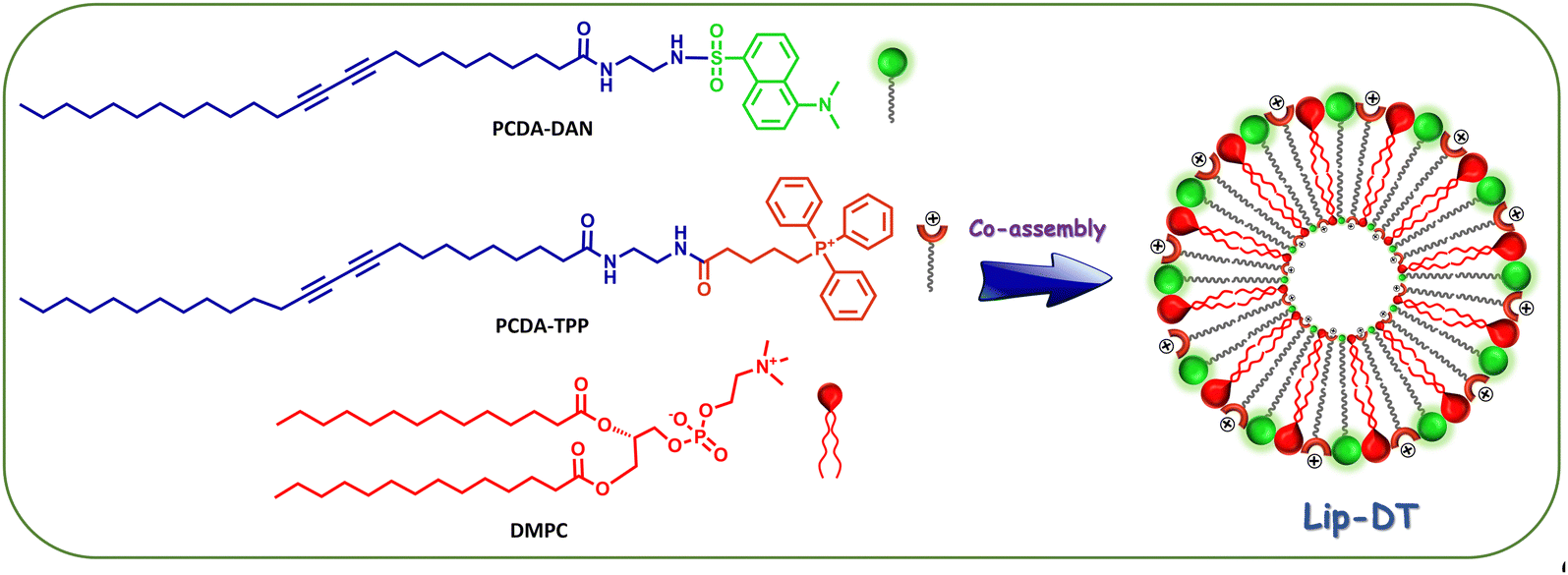 | ||
| Scheme 2 Schematic representation of the formation of PCDA-DAN, PCDA-TPP and DMPC-based liposome (Lip-DT) via co-assembly. | ||
The morphology of these newly prepared liposomes was confirmed by high-resolution scanning electron microscopy (HR-SEM) (Fig. 1(A) and (B)) and high-resolution transmission electron microscopy (HR-TEM) (Fig. 1(C) and (D)) analysis. The average size of the Lip-DT vesicles obtained from HR-TEM micrographs (Fig. 1(E)) is ∼225.47 ± 4.5 nm, which is in well agreement with the average size (∼235.2 ± 5.7 nm) obtained from dynamic light scattering (DLS) (Fig. S10, ESI†). The effective surface charge or the zeta potential value of this liposome was found to be 4.3 ± 1.6 mV (Fig. 1(F)). Literature reports show that liposomes, with average size ranging from 50–100 nm exhibit minimal interaction with plasma proteins and have a prolonged lifespan in the bloodstream. In contrast, liposomes with a diameter up to 400 nm effectively penetrate and accumulate in tumor tissues.36 Our results suggest that the liposomes are well dispersed and within the appropriate size range for penetration into tumor cells with enhanced permeability and retention (EPR).37
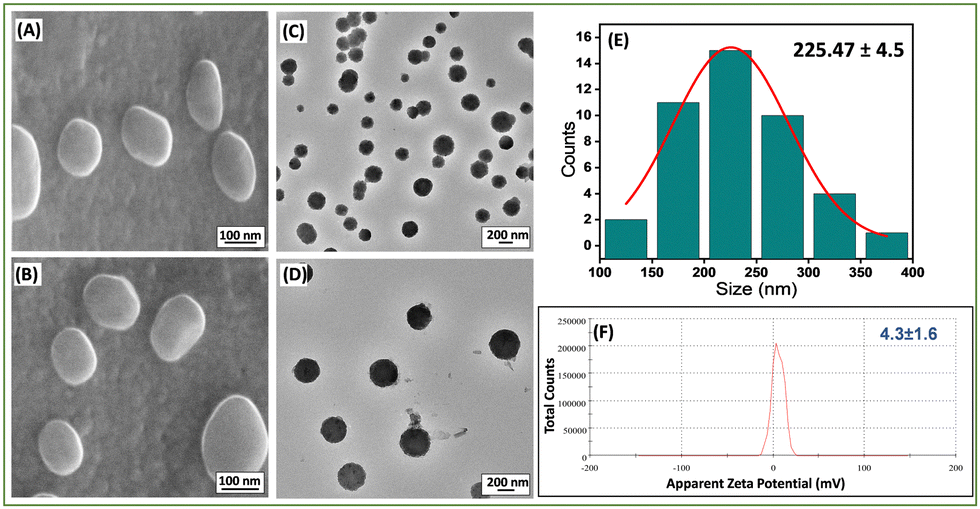 | ||
| Fig. 1 (A), (B) HR-SEM and (C), (D) HR-TEM images of the Lip-DT. (E) Size distribution of Lip-DT obtained from HR-TEM micrographs. (F) Zeta potential analysis of Lip-DT vesicles obtained from DLS measurement. | ||
To investigate the impact of functionalization of PCDA with mitochondriotropic (TPP) and fluorophore (dansyl) groups on the average size and charge of newly prepared liposome (Lip-DT), we compared to Lip-DT with conventional PCDA/DMPC-based liposome without any functionalization. DLS and zeta potential analysis displayed that the average size and charge of the PCDA/DMPC-based liposome (without functionalization) were found to be 379.2 ± 6.5 nm and −14.5 ± 2.6 mV (Fig. S11, ESI†) respectively. Interestingly, it was observed that the average size of the Lip-DT is lower compared to conventional PCDA/DMPC-based liposome. This is most probably due to the effective π–π stacking interactions between the aromatic moieties of the dansyl and TPP groups during vesicle formation leading to the formation of a more compact liposome (Lip-DT) with smaller size.38 Furthermore, the effective positive surface charge of Lip-DT clearly indicates its cationic nature due to the presence of triphenyl phosphonium ion (TPP). More importantly, functionalization by TPP group enhances the lipophilicity as well as tunes the potential surface charge of the liposome. These results clearly showed that the functionalization of the PCDA with TPP and dansyl groups significantly influences the characteristic properties of the liposome including the size, shape, and effective charge, which play a crucial role in the drug delivery process.
The optical properties of the Lip-DT were analysed using UV-Vis and a fluorescence spectrophotometer. First, we checked the photoinduced polymerization of the prepared liposome (Lip-DT) by irradiating the solution with 254 nm UV light for 5 min. After the UV irradiation process the color of the Lip-DT was changed from colorless to a violet color (Fig. 2(A) and (B)). This visible color change of the Lip-DT solution upon UV irradiation clearly indicates the successful polymerization with the formation of a stable π-conjugated PDA backbone. We then recorded the UV-Vis absorbance spectra of the PCDA-TPP, PCDA-DAN in PBS buffer medium (pH 7.4). UV-Vis absorption spectrum of PCDA-TPP displayed two absorption peaks at 271 and 303 nm (Fig. 2(C)). The absorbance band at 303 nm can be attributed to intermolecular charge transfer (ICT) band, whereas the shorter wavelength absorption peak at 271 nm is associated with the high energy π–π*electronic transition. Similarly, the absorption spectrum of the PCDA-DAN exhibited two absorption peaks: one at 280 nm, preliminary ascribed to the π–π* transition, and the second one is the characteristic ICT band at 390 nm.39 We also recorded the UV-Vis absorption spectrum of liposomes (Lip-DT) containing both PCDA-DAN and PCDA-TPP before and after UV irradiation at 254 nm. The absorption spectrum of the liposome after UV irradiation displayed a characteristic absorption peak at 630 nm along with a shoulder peak at 540 nm (Fig. 2(C)). The absorption maxima at 630 nm corresponds to the π–π* electronic transition of the π-conjugated PDA backbone, which indicates polymerization of the Lip-DT with a stable ene–yne-conjugated backbone.16 On the other hand, the UV-Vis absorption spectra of Lip-DT without UV irradiation (without polymerization) did not exhibit the characteristic absorption band associated with π–π* transition of the polymerized π-conjugated PDA backbone (Fig. 2(B)). We then recorded the steady sate fluorescence spectra of PCDA-TPP, PCDA-DAN and Lip-DT in PBS buffer medium (pH 7.4).
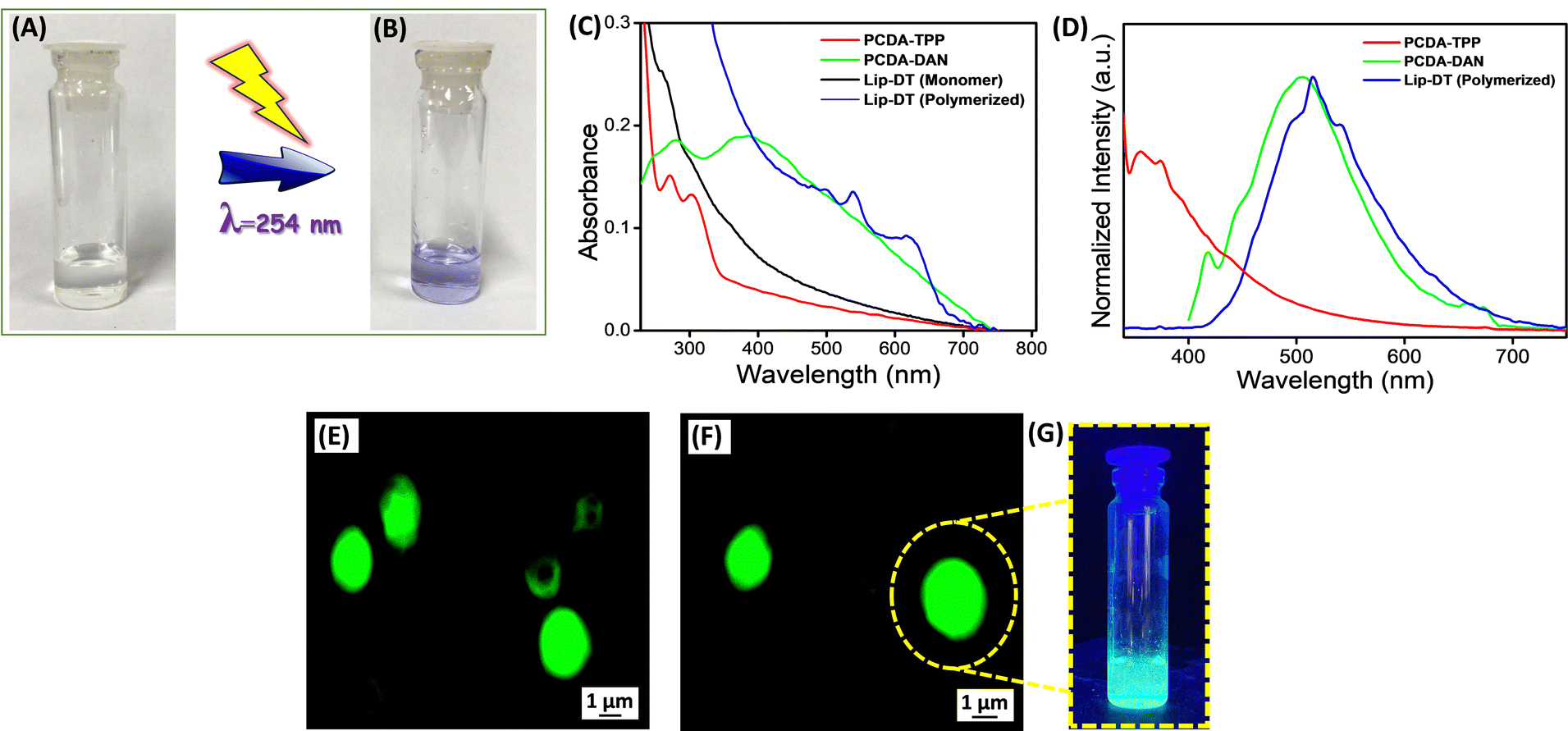 | ||
| Fig. 2 Photographs of the Lip-DT solution (A) before and (B) after UV irradiation (λ = 254 nm). (C) UV-Vis absorption and (D) Steady state emission spectrum of PCDA-TPP, PCDA-DAN, Lip-DT after UV irradiation (λ = 254 nm) in PBS buffer medium (pH 7.4). (E) and (F) Fluorescence microscopic images of Lip-DT vesicles. (G) Photograph showing the fluorescence color of the Lip-DT solution upon irradiation with 365 nm light. | ||
The steady-state fluorescence spectrum of the PCDA-TPP and PCDA-DAN displayed the characteristic emission maximum at 373 nm (λExt = 303 nm) and 505 nm (λExt = 390 nm), respectively (Fig. 2(D)). In contrast, the emission spectra of Lip-DT (after UV irradiation) exhibited an emission maximum at 515 nm along with two shoulder bands at 496 nm and 541 nm (Fig. 2(D)). The emission maximum at 515 nm (λExt = 300 nm) is due to presence of the dansyl moiety in the liposome backbone and the two shoulder bands at 496 and 541 nm may be attributed to the ene–yne-conjugated polymeric backbone of the Lip-DT vesicles.40 However, any significant or characteristic emission at 630 nm was not observed for the Lip-DT (after UV irradiation or polymerization). The intrinsic fluorescent properties of the Lip-DT comprised with the dansyl fluorophore was further examined using fluorescence microscopy. Fig. 2(E) and (F) clearly shows the characteristic green fluorescence signal due to the presence of dansyl group in the liposome (Lip-DT). We have also checked the fluorescence color of the Lip-DT solution upon irradiation with 365 nm light. Fig. 2(G) clearly displays the characteristic green fluorescence of the Lip-DT solution due to the presence of the dansyl moiety, which also confirms the formation of the stable liposome with intrinsic fluorescent properties required for imaging as well as real-time monitoring of drug release.
To determine the potential utility of this mitochondriotropic unit and fluorophore functionalized liposome (Lip-DT) as a mitochondria-targeting drug delivery vehicle, we have incorporated the anticancer drug doxorubicin (Dox) during the co-assembly process (Fig. 3(A)). Liposomes have been identified as promising drug delivery platforms for developing clinical nanomedicines such as Doxil™/Caelyx, a liposomal Dox preparation.41 As liposomes are unable to penetrate through the tight junctions present in the heart's vasculature, this design successfully address the dose-limiting and occasionally life-threatening cardiotoxicity associated with Dox exposure.41 Although Doxil exhibited enhanced cardiac safety, still this provide few side effects such as myelosuppression, alopecia, and nausea/vomiting compared to the unencapsulated Dox.41,42 The polyethylene glycol coating present in Doxil™ also causes preferential accumulation in the skin, leading to palmar plantar erythrodysesthesia (PPE) in 50% of patients.42 Therefore, still there is a need for an efficient drug delivery system that can allow site specific delivery of Dox without causing any adverse side effects. In this regard, we have incorporated the Dox into the liposome during the thin film rehydration process, which formed Lip-DT-Dox, where the drug molecules are encapsulated within the Lip-DT liposome. Fluorescence microscopic analysis clearly confirms the successful encapsulation of fluorescent Dox molecules into the vesicle structures (Fig. 3(B) and (C)).
 | ||
| Fig. 3 (A) Schematic representation of the encapsulation of Doxorubicin (Dox) into Lip-DT vesicles via co-assembly. (B) and (C) Fluorescence microscopy image of the Doxorubicin (Dox)-encapsulated Lip-DT. | ||
The average size of the Lip-DT-Dox vesicles obtained from HR-TEM micrographs (Fig. 4(A) and (B)) is ∼310.83 ± 4.9 nm, higher than Lip-DT without Dox. This increase (∼85 nm) in the average size of the Lip-DT-Dox clearly suggests the successful incorporation of the Dox molecules within the liposome. The calculated encapsulation efficiency (EE%) of the Lip-DT was found to be 66.09%. The in vitro drug release profile of Dox from Lip-DT-Dox was then evaluated, which was obtained by monitoring the fluorescence intensity overtime, as described in our earlier work.43 The florescence intensity of the PBS buffer solution outside of the dialysis bag showed a gradual increase over time due to the release of the encapsulated Dox molecule from the Lip-DT-Dox (Fig. 4(D) and (E)). The fluorescence intensity of the buffer solution outside the dialysis bag steadily increased up to 17 h and after that there was no significant increase in the emission intensity for the next 7 h (up to 24 h). This plateau suggests that the drug release process was completed, and equilibrium was achieved. To investigate the mechanism behind the burst release of the encapsulated Dox from Lip-DT-Dox, we compared the morphological variation of the vesicles before and after completion the drug release process. For this purpose, we have recorded the TEM images of the Lip-DT-Dox before (T = 0 h) and after the drug release process (T = 24 h). These TEM micrographs clearly displayed the rupture and breakage of the liposome vesicles (Fig. 4(C)). This result clearly indicates the Lip-DT-Dox underwent burst release, resulting in the gradual release of the encapsulated Dox (Fig. 4(C)–(E)). The salts present in the buffer medium are believed to play a vital role in triggering the burst release of encapsulated drug molecules from liposomes by modulating osmotic pressure and facilitating interactions with the encapsulated drug molecules.44 Though DLCs like triphenylphononium (TPP) cations are considered ideal mitochondriotropic agents due to their lipophilicity and stable delocalized positive charge,23 direct functionalization of drug molecules with TPP may significantly reduce the potency of the drug molecule.23 Instead, we functionalized the drug-encapsulating liposomes with TPP to preclude loss of drug potency.45 To confirm the utility of this Lip-DT-Dox as a mitochondria-targeting drug delivery vehicle, we first checked the cellular toxicity in both human embryonic kidney HEK293 (Fig. 5(A)) cells and the human cardiomyocyte cell line AC-16 (Fig. S12A, ESI†). There was no significant toxicity found for Lip-DT with up to 50 μM concentration and Lip-DT-Dox at 25 μM concentration. However, Lip-DT-Dox demonstrated increased cytotoxicity in glycolytic cancer cells MCF7 (Fig. 5(B)), HCT116 (Fig. S12B, ESI†), and triple negative breast cancer cells MDAMB231 (Fig. S12C, ESI†) from concentrations as low as 2.5 μM. Importantly, in all cell lines tested the cytotoxic concentrations of the Lip-DT alone were at least 10-fold higher than for the encapsulated drug.
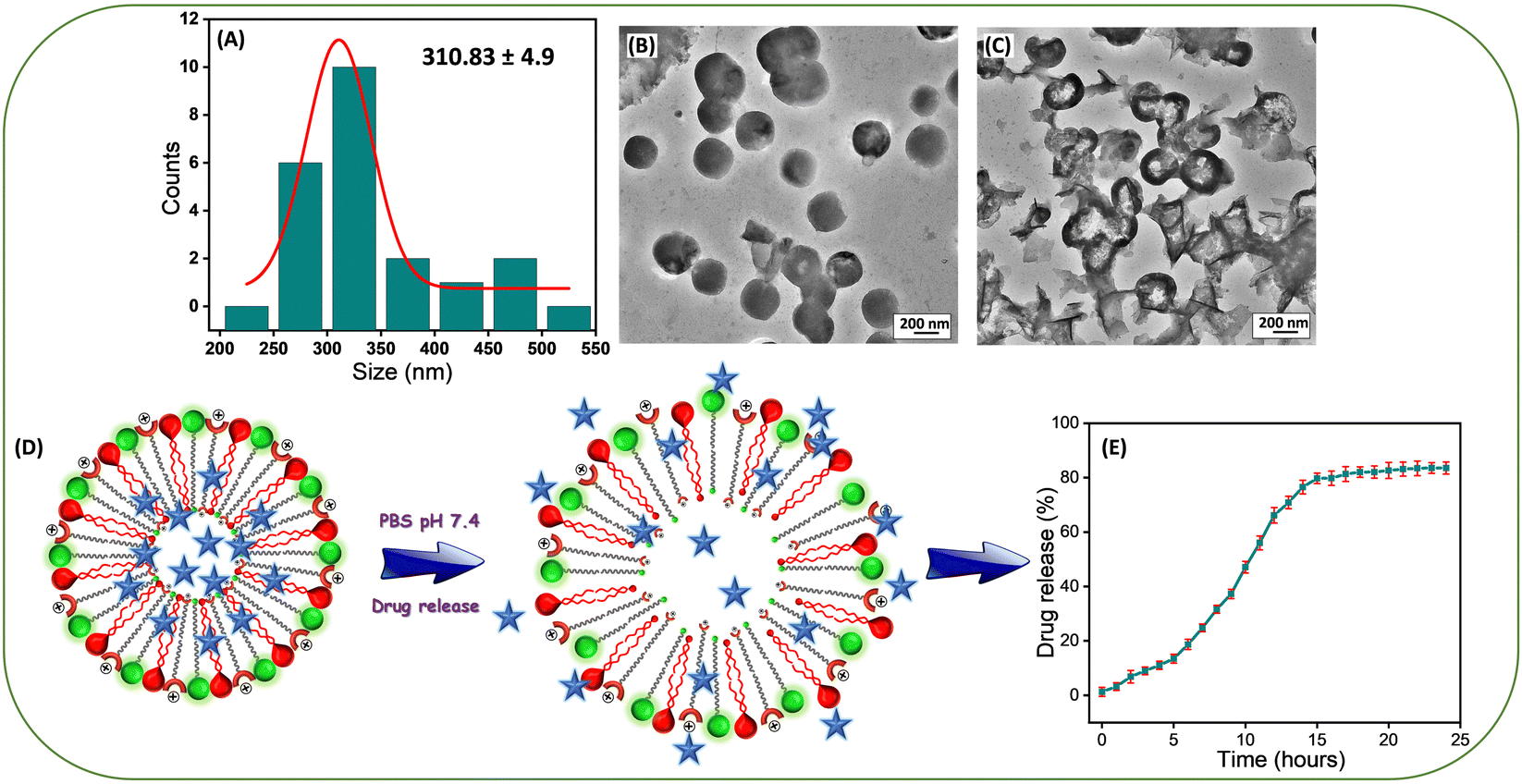 | ||
| Fig. 4 (A) The size distribution change of Lip-DT vesicles after Doxorubicin (Dox) incorporation. HR-TEM images show the changes of the vesicle morphology of Lip-DT-Dox (B) before and (C) after the burst release of encapsulated Dox. (D) Schematic representation of burst release of the encapsulated Dox, and (E) The plot of % of drug release from Lip-DT-Dox with time (λMon 590 nm, λExt 490 nm). | ||
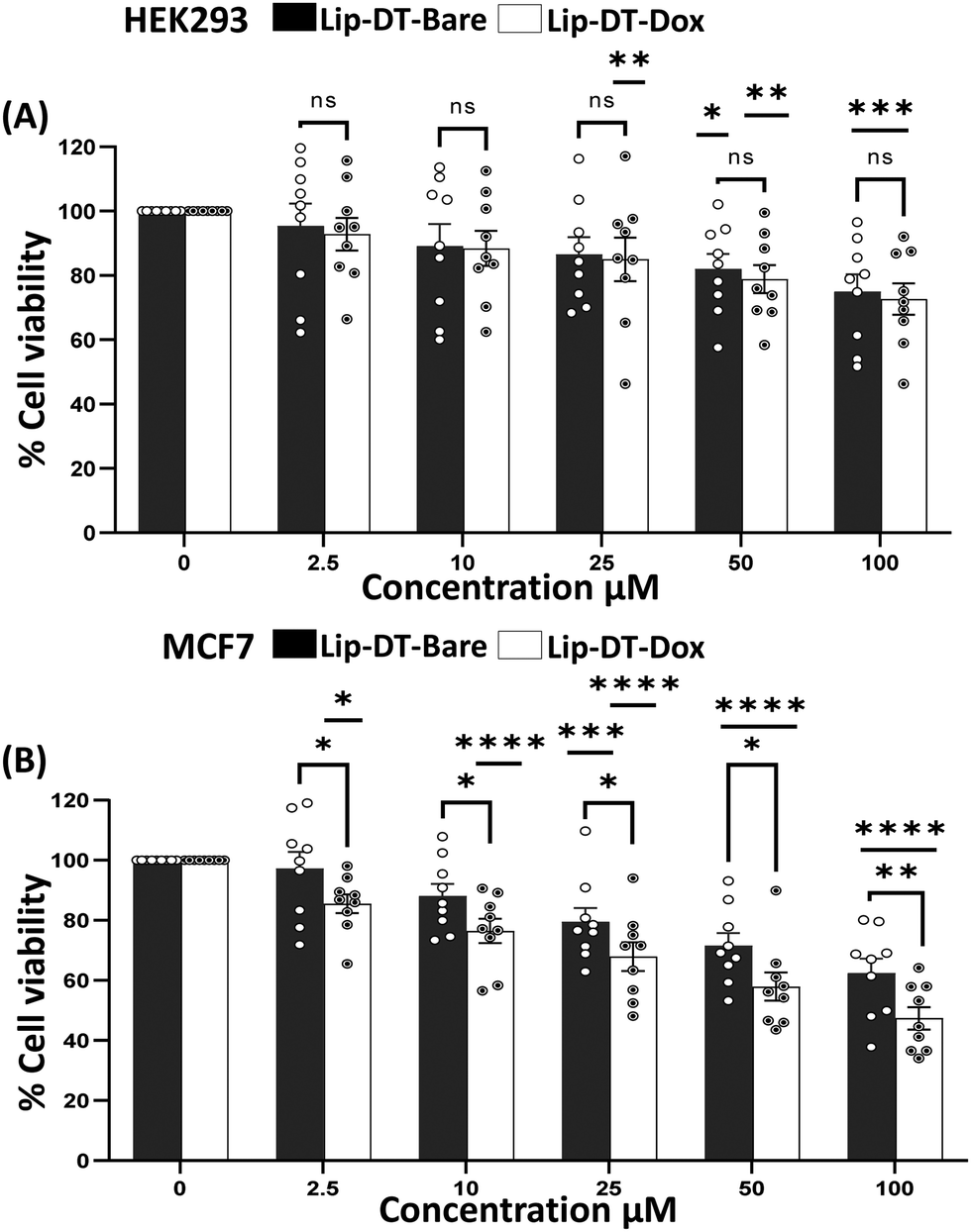 | ||
| Fig. 5 Cytotoxicity and incorporation of the Lip-DT compound in cell. The MTT assay was used to measure the cellular toxicity following incubation with Lip-DT or Lip-DT-Dox (1 μM) at 36 h (A) The survival of human embryonic kidney HEK293 cells (B) Cytotoxicity of cancer cell MCF7. All results are presented as means + S.E. of multiple experiments (n = 9, *p < 0.05; **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to control group). | ||
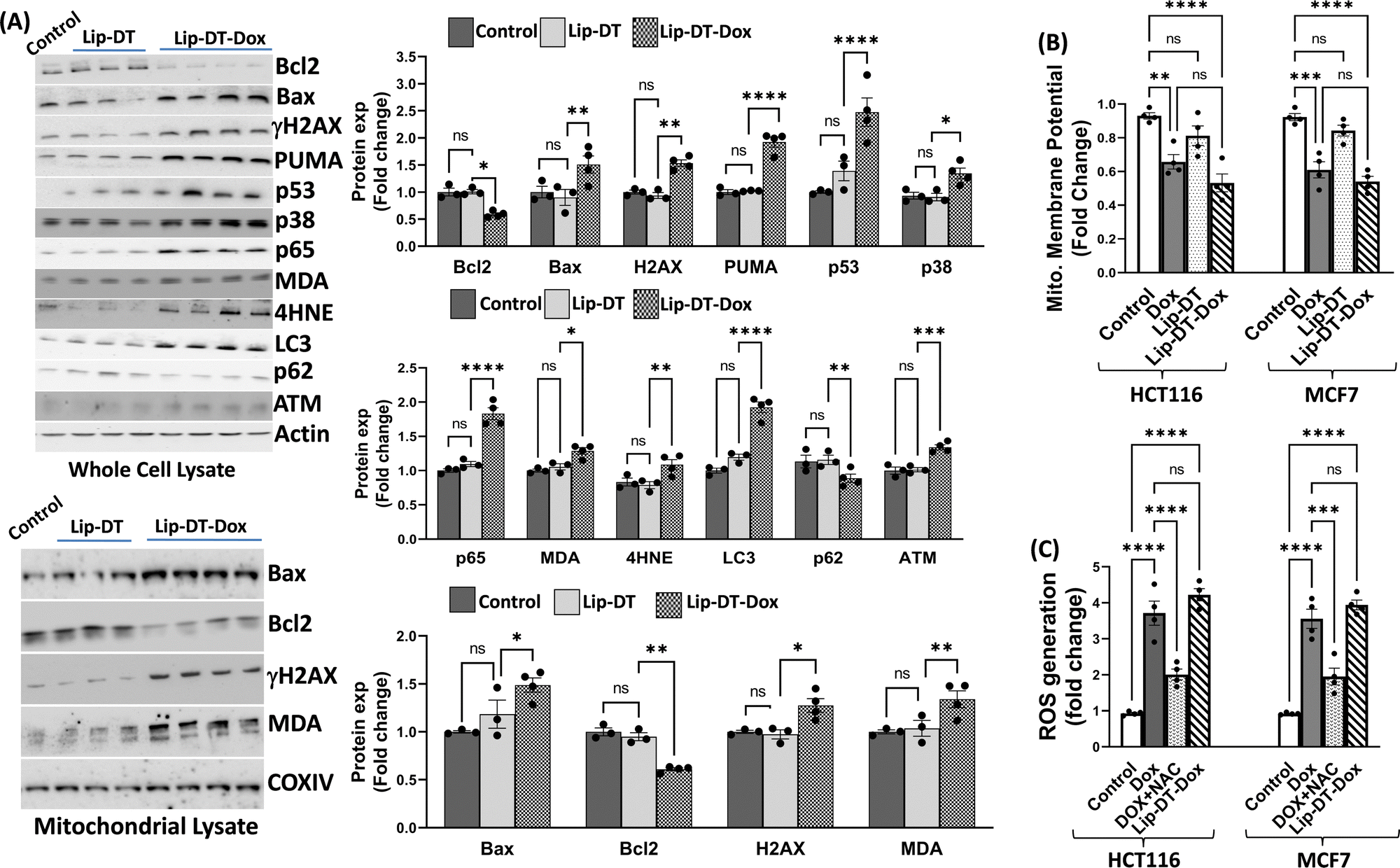
Next, the bioactivity of Lip-DT-Dox was measured. The ability of Lip-DT-Dox to trigger cellular toxicity (Fig. 6(A)) and apoptosis (Fig. 6(B)) in both HCT116 and MCF7 cancer cells was comparable to the chemotherapeutic drug doxorubicin. These data collectively demonstrate that the ability of Dox to kill the cancer cells is not lost when Lip-DT encapsulates Dox, shuttles Dox into cells, and releases the drug. Dox-dependent cytotoxicity results from DNA intercalation and damage initiating the DNA damage signalling cascade that begins with phosphorylation of histone H2AX (γH2AX) at the site of double-strand breaks and leads to recruitment of ATM, which can either facilitate DNA repair or trigger cell death via up-regulation of proteins such as p38, p53 and p65 (Fig. 8(A)).46 p53 interacts with PUMA which ultimately neutralizes anti-apoptotic protein Bcl-2, or p53 can directly interact with pro-apoptotic Bax.
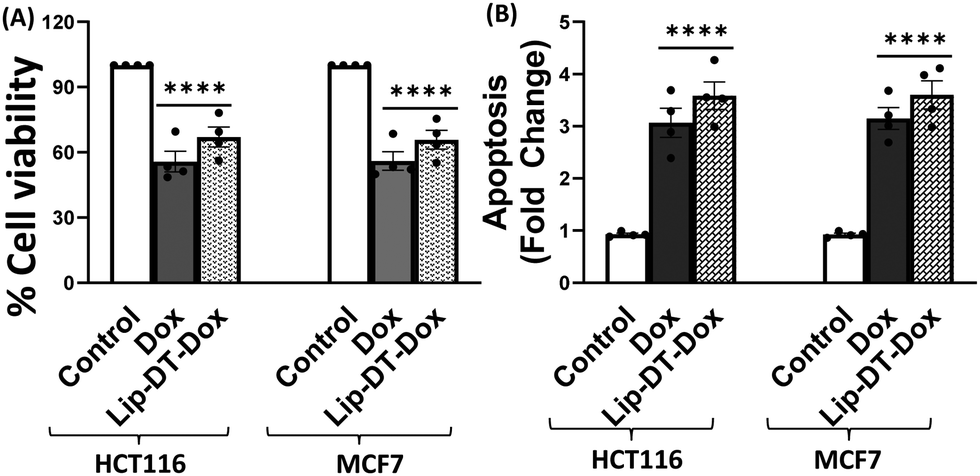 | ||
| Fig. 6 Lip-DT encapsulated Dox (Lip-DT-Dox) promotes apoptotic cell death. (A) Cellular toxicity of cancer cells (HCT116 and MCF7) after incubation with Dox or Lip-DT-Dox (1 μM) at 36 h as measured by MTT assay. (B) Apoptosis was considered as the rise in fold change in enrichment factor by formation of the cytoplasmic histones associated DNA fragments. Data are shown as means + S.E. of multiple experiments (n = 4, *p < 0.05; **p < 0.01, ***p < 0.001, ****p < 0.0001 compared with control group and Dox, and Lip-DT-Dox). | ||
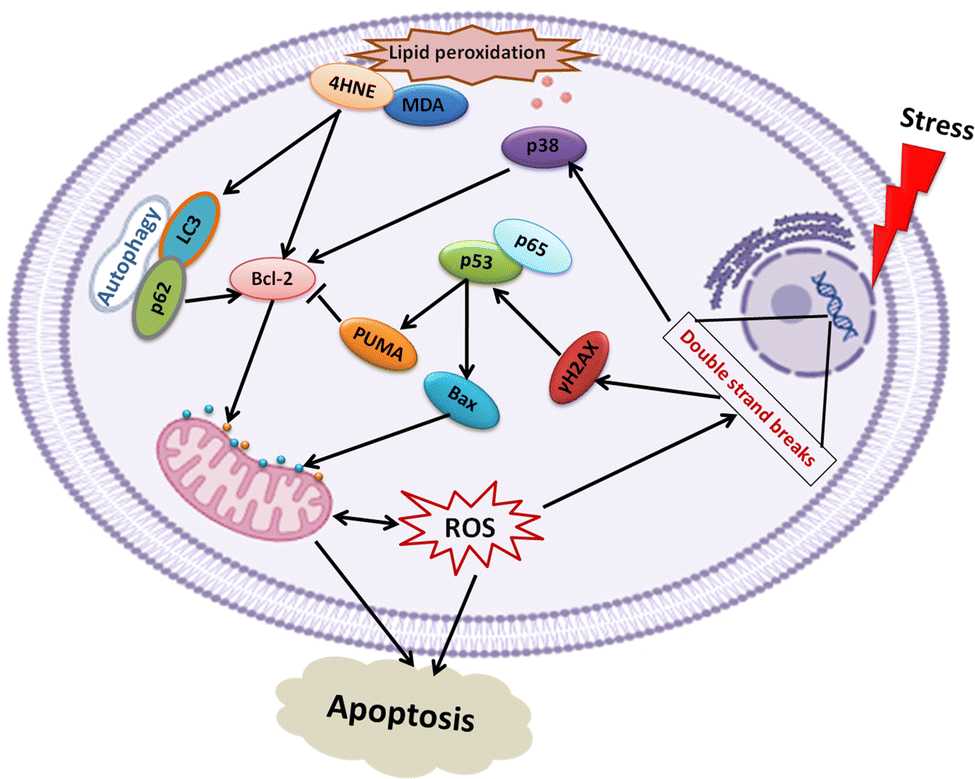
Bcl-2 family pro-apoptotic proteins, such as Bax, function at the mitochondrial membrane to facilitate permeabilization and the subsequent release of cytochrome c and ROS initiating the apoptosis cascade, actions counterbalanced by Bcl-2 action (Fig. 7). Although Lip-DT alone exhibited minimal impact on components of the DNA damage response, Dox and Lip-DT-Dox resulted in a comparable increase in γH2AX, p53, p65, and the Bax/Bcl-2 ratio within MCF7 cancer cells (Fig. 8(A)). We observed that the treatment of Dox and Lip-DT-Dox, but not Lip-DT alone, in the cells decreases mitochondrial membrane potential (ΔΨm) leading to opening of the mitochondrial permeability transition pore (Fig. 8(B)).
 | ||
| Fig. 7 Schematic outlining apoptotic signalling cascades activated by Dox and Lip-DT-Dox in cancer cells. | ||
 | ||
| Fig. 8 Lip-DT-Dox triggers the DNA damage response in MCF7 cancer cells. (A) Expression of various apoptosis-related proteins was documented in MCF7 cells treated with Lip-DT or Lip-DT-Dox. For all the western blots β-actin was used as a loading control. (B) Loss of mitochondrial membrane potential (ΔΨm) was analysed in control cells or those treated with Dox, Lip-DT o Lip-DT-Dox. (C) Generation of ROS in HCT116 and MCF7 cancer cells induced by Dox and Lip-DT-Dox in the presence or absence of glutathione donor N-acetyl cysteine (NAC). CM-H2DCFDA was used to measure the ROS generation by fluorescence at 24 h. All results are expressed as means ± S.E. (n = 4, *p < 0.05; **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to control group). | ||
The increase in intracellular reactive oxygen species (ROS) (Fig. 8(C)) as well as increase immunoreactivity for markers of lipid peroxidation malondialdehyde (MDA) and 4-hydroxynonenal (4HNE) (Fig. 8(A)) in cells treated with either Dox or Lip-DT-Dox also results from mitochondrial dysfunction and indicates cellular oxidative damage. MDA and 4HNE can also suppress Bcl-2 or trigger autophagy by increasing LC3 (Fig. 8(A)), which interacts with the ubiquitin-binding protein p62, facilitates the clearance of polyubiquitinated protein aggregates, and triggers apoptosis by activating caspase-8, a process recruited to a similar extent in cells treated with either Dox or Lip-DT-Dox. The ability of Dox and our encapsulated drug to facilitate death of cancer cells thus relies both on initiation of the pro-apoptotic DNA damage signalling cascade, mitochondrial dysfunction and a breakdown in cellular energy production, oxidative damage, and suppression of intracellular mechanisms (e.g., autophagy) designed to clear damaged proteins and organelles. Our data demonstrating that Dox or Lip-DT-Dox modulate these processes to a similar degree confirms that Dox is released into the cell from the liposomal NDDS and that incorporation of Dox into this system fails to impact drug potency.
To investigate the cellular uptake and intracellular distribution of Lip-DT, MCF7 cells were incubated with the dansyl fluorophore attached Lip-DT liposome. The compound Lip-DT can be detected by green fluorescence following rapid uptake into MCF7 cells (Fig. 9(A)). Cells were treated with 100 nM of Lip-DT for a period of 2 h with MitoTracker™ (red fluorescence) used as a marker of mitochondria. Colocalization of both signals indicates delivery of the Lip-DT molecule to the mitochondria (Fig. 9(A)). Lip-DT-Dox also co-localizes with Lip-DT in MCF7 cell (Fig. 9(B)) suggesting that Lip-DT is capable of directing Dox to the mitochondria, a function likely attributed to the lipophilicity and delocalized positive charge of Lip-DT that results in a strong negative electric potential and mitochondrial membrane partitioning ability.47
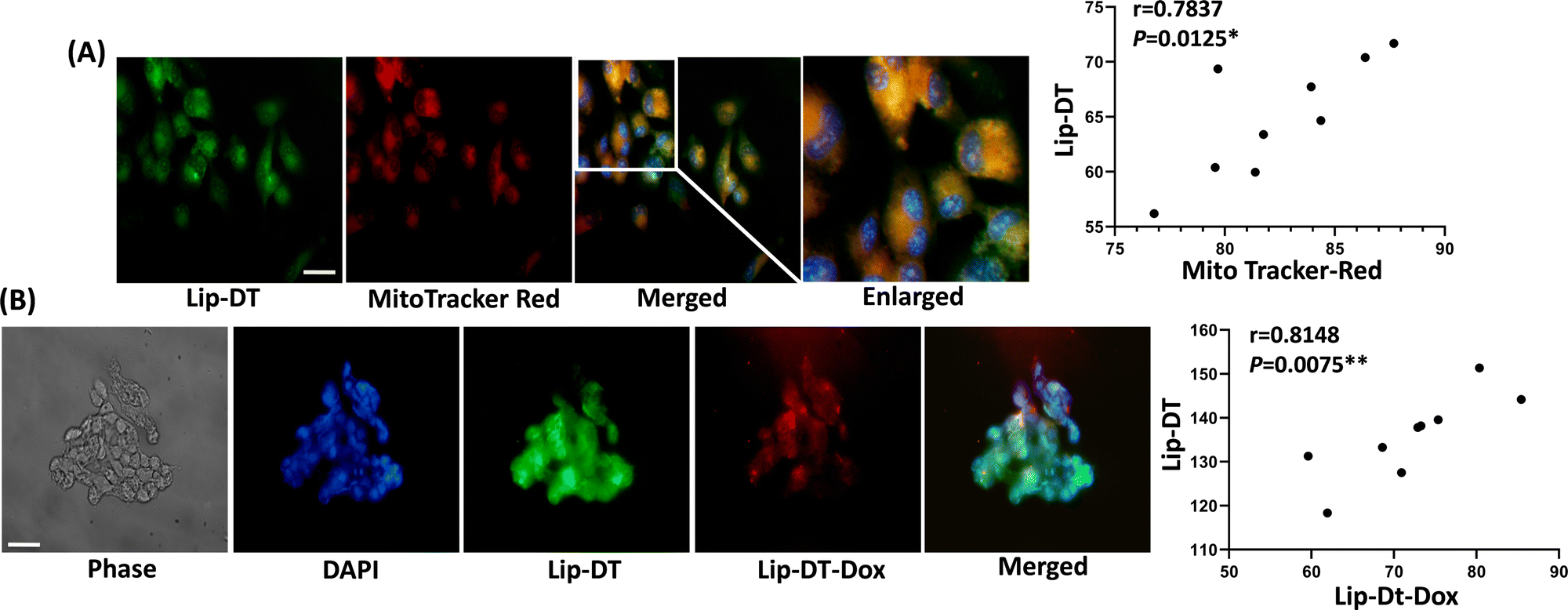 | ||
| Fig. 9 Effect of Lip-DT on Dox delivery into MCF7 cells. (A) In vitro fluorescence imaging of MCF7 cells with Lip-DT and MitoTracker™ red. The green colour represents the compound whereas the red colour marks mitochondria (scale bar = 100 μm). (B) Incorporation of fluorescent Lip-DT and Lip-DT-Dox in MCF7 cells and visualized with a fluorescence microscope. MCF7 cells were incubated with Lip-DT and Lip-DT-Dox (10 nM) for 8 h at 37 °C and visualized with a fluorescence microscope. The green color represents the Lip-DT and red color represents the Doxorubicin (Dox) (scale bar = 100 μm). Pearson correlation coefficient between Lip-DT and MitoTracker™ red, and Lip-DT and Lip-DT-Dox was assessed and represented (n = 9, *p < 0.05; **p < 0.01). | ||
Conclusions
In summary, we have described the design, synthesis, and functionalization of the conventional PCDA/DMPC based liposome into a mitochondria-targeting drug delivery system. The mitochondria targeting ability was achieved by the conjugation of Triphenylphosphonium (TPP) ligand on the surface of liposomes. The ability of the Lip-DT to target the mitochondria was successfully demonstrated in MCF7 cancer cell lines using co-localization study with MitoTracker™ red. The visible green fluorescence of the dansyl (DAN) group present in the Lip-DT allowed monitoring the cellular uptake and the presence of Lip-DT inside the mitochondria. Lip-DT-Dox displayed similar activity to unencapsulated Dox but without impacting or causing any adverse effect to the normal human embryonic kidney (HEK293) and cardiomyocytes (AC-16) cell lines, representing the biocompatibility of this drug delivery system. Notably, mitochondria-mediated apoptosis of the cancer cell (HCT116 and MCF7) was initiated to a similar degree by both unencapsulated Dox and Lip-DT-Dox. Based on these results, we believe this strategy is desirable and would provide a promising platform to develop mitochondria-targeted drug delivery systems.Materials and methods
All the chemicals and solvents are commercially available and were used as received. 10,12-Pentacosadiynoic acid (PCDA), 2′,7′-dichlorofluorescin diacetate (CM-H2DCFDA), dimethyl sulfoxide (DMSO), (4-carboxybutyl)triphenylphosphonium bromide, and 2-(1h-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate were purchased from Sigma Aldrich (St. Louis, MO, USA. Dansyl chloride and 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) was purchased from Tokyo Chemical Industry Co. Ltd. Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), N-hydroxysuccinimide (NHS), triethylamine (TEA), N,N-diisopropylethylamine (DIPEA) and phosphate buffer saline (PBS) were purchased from Sisco Research Laboratories (SRL) Pvt. Ltd, India. Dulbecco's Modified Eagle Medium (DMEM) and Trypsin EDTA 0.05% were procured from Thermo Fisher Scientific (Waltham, MA, USA). MitoTracker Red was obtained from Invitrogen (Carlsbad, CA, USA). Fetal bovine serum (FBS) was purchased from Himedia (Mumbai, India). Other standard laboratory chemicals and reagents were purchased from Sisco Research Laboratories (SRL) Pvt. Ltd, India.Preparation of Lip-DT and PCDA/DMPC based Liposomes
Chloroform solutions of the PCDA-DAN, PCDA-TPP and DMPC were prepared separately in amber vials and then mixed at a certain molar ratio (4:4:1) to give a total lipid concentration of 1.0 mM. The mixture of lipid solution in chloroform was slowly dried under gentle nitrogen flow. Then phosphate-buffered saline (PBS) solution (10 mM, pH 7.4) was added and the solution was sonicated in an ultra-sonicator for about 20 min. Finally, a semi-transparent solution was obtained. It was then filtered with a 0.45 μm syringe filter and the resultant solution was cooled to room temperature and then stored overnight at 4 °C to induce crystallization of lipid membranes. PCDA/DMPC-based liposomes were also prepared by the above-mentioned procedure.17
For the preparation of Doxorubicin (Dox) loaded Lip-DT-Dox, a Dox stock solution in chloroform was added to the lipid mixture in chloroform at the concentration of 3 × 10−5 M and the liposome (Lip-DT-Dox) was prepared following the above-mentioned procedure. The non-incorporated Dox was removed by centrifugation of Lip-DT-Dox at 14000 rpm for 15 min, which precipitated the Dox incorporated Lip-DT-Dox.
Structural characterization of the Lip-DT and PCDA/DMPC-based liposomes
The vesicle morphology of the Lip-DT was characterized with a HR-SEM (Thermo scientific Apreo S) operating at 15.00 kV and HR-TEM (JEOL-JEM-2100 Plus) operating at 200 kV. For HR-SEM analysis 20 μL of the Lip-DT was drop casted on a clean glass coverslip and dried at RT. For HR-TEM analysis a 10 μL drop of a Lip-DT was placed on a 200-mesh copper grid and covered by carbon-stabilized Formvar® film. After 1 min, excess fluid was removed from the grid. The morphology and fluorescent nature of the vesicles were visualized using a Leica DM6 Fluorescent Microscope with Cryostat. Dynamic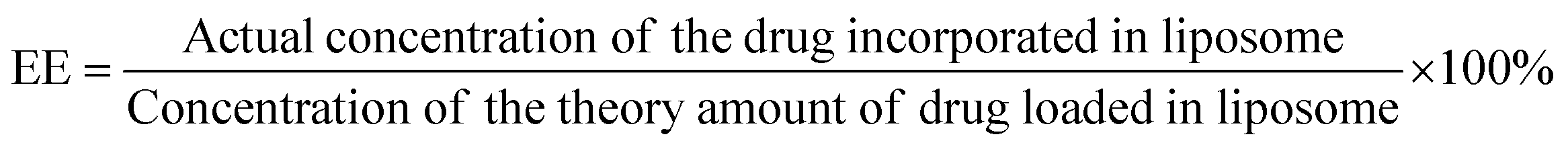 | (1) |
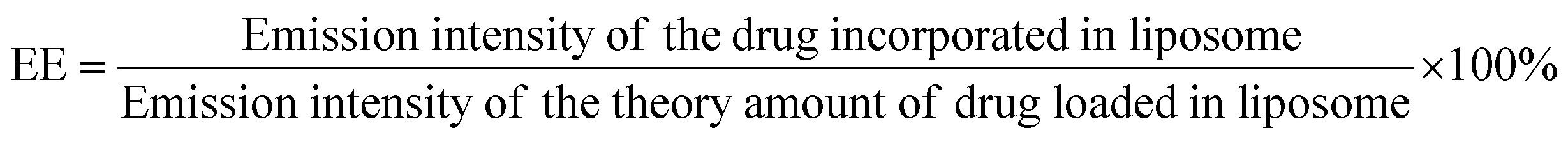 | (2) |
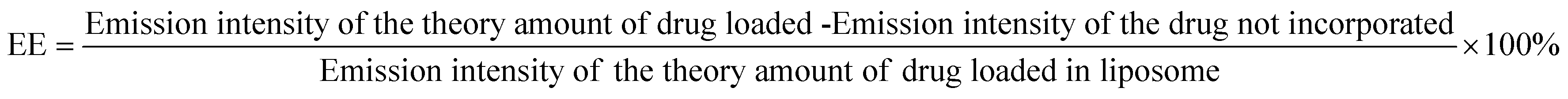 | (3) |
UV-Vis spectroscopy
UV-Vis absorption spectra of the synthesized PCDA-DAN, PCDA-TPP, and Lip-DT (liposome) were recorded in a PBS (pH 7.4) buffer medium using a UV/Vis spectrophotometer (Thermo scientific, Nanodrop 2000c UV-Vis absorption spectrometer).Fluorescence spectroscopy
Fluorescence measurements were performed at RT using a photoluminescence spectrophotometer (Edinburgh Instruments, FLS 1000). The emission spectra of the synthesized molecules were recorded in an aqueous medium using proper excitation wavelengths.Microanalysis
C, H, and N analysis was performed using a Vario Micro Cube (Elementar) instrument.Drug release of Lip-DT-Dox
The incorporation of Doxorubicin was conducted during the preparation of Lip-DT-Dox as mentioned above. The drug-incorporated Lip-DT-Dox was prepared by the above-mentioned protocol and dispersed in PBS (10 mM NaCl pH = 7.4, 150 mM). After that, this suspension (3 mL) was transferred into a dialysis bag (MWCO 3 kDa), and the bag was dipped in 40 mL of PBS at RT. The emission intensity of the buffer solution outside the dialysis bag was measured at different time intervals for 24 hours. The volume of the solution was kept constant by adding 1 mL of the original PBS solution after each measurement. The emission intensities were measured at RT using a fluorescence spectrophotometer. The emission spectra were recorded from 500 nm to 750 nm for the % drug release vs. time plot (λExt = 490 nm and λMon = 590 nm).43Drug-encapsulation efficiency (EE)
Lip-DT-Dox was prepared as reported above, and the aqueous medium was decanted and the emission intensity at the desired wavelength was measured. The drug-encapsulation efficiency (EE), which is correlated with the concentration of the drug not incorporated or the free unentrapped drug molecule, can be expressed by eqn (1).48As the concentration of the drug is directly proportional to the emission intensity, eqn (2), the emission of the drug incorporated in nanoparticles is equal to the total emission subtracted by the emission intensity of the drug not incorporated; EE can be calculated using eqn (3).
The drug encapsulation efficiency calculated for the Lip-DT 66.09%.
Cell culture
Cells used in our experiments were procured from the Cell Repository of National Centre for Cell Sciences (NCCS), Pune India. The National Centre for Cell Science (NCCS), Pune, India provided us with the human embryonic kidney (HEK293), breast cancer (MCF7), and colon carcinoma (HCT116) cell lines. The human cardiac myocyte cell line (AC16, SCC109) was acquired from Merck, Millipore, USA. As specified in the given manual, the cells were cultured in DMEM media with 10% FBS, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin. The cells were cultured at a temperature of 37 °C within a 5% CO2 incubation environment.49 The synthesized compounds were dissolved in DMSO for cell-based experiments and to minimize the toxicity of the DMSO itself, the concentration of DMSO was kept at 0.1%. To rule out potential impacts of DMSO on cellular viability and other endpoints, control cells were treated with a 0.1% DMSO-containing vehicle control.MTT assay
The cellular toxicity of the synthesized compounds was assessed using the MTT reduction assay. To investigate the impact of the Lip-DT compound on cell growth and proliferation, various cells such as HEK293, AC16, HCT116, and MCF7 were exposed to varying concentrations of the compound for a period of 40 hours. Following the seeding of cells at a density of 6 × 104 cells per well in a 96-well plate, they were incubated in a CO2 incubator at 37 °C and 5% CO2, with the culture medium consisting of DMEM supplemented with 10% FCS (Gibco). After 24 hours, the culture medium was replaced, and the cells were subsequently incubated with various concentrations of the synthesized compounds for an additional 40 hours.50 The incubation was carried out using a culture medium without phenol red. Then a solution of MTT (Sigma) at a concentration of 0.5 mg mL−1 was added to each well, and the cells were incubated for 3 hours. Once the incubation period was complete, 150 μL of DMSO was introduced into each well to facilitate the solubilization of the formazan crystals. Lastly, the optical density (OD) was assessed at a wavelength of 570 nm with a plate reader (Biotek Instrument).Fluorometric analysis
HEK293 and AC16 cell lines were seeded at a density of 3 × 105 cells in 35 mm dishes containing DMEM supplemented with 10% FCS to initiate the culture. After a 30-hour incubation period, varying concentrations of the Lip-DT compound were introduced, and the cells were allowed to incubate undisturbed for an additional 24 hours. Afterward, the cells were harvested, subjected to rinsing with chilled PBS, and eventually lysed in PBS supplemented with 1% Tween 20.50 Upon the centrifugation of the cell lysates, the supernatant was collected, and the fluorescence level was determined using an excitation wavelength of 560 nm and an emission wavelength of 633 nm.Immunofluorescence study
AC16 and MCF7 cells were grown on cover slips sited in six-well plates filled with DMEM (Gibco) supplemented with 10% FBS (Invitrogen), 100 units per ml penicillin (Sigma-Aldrich), 100 μg mL−1 streptomycin (Sigma-Aldrich), and 2mM L-glutamine (Himedia) at 37°C and 5% carbon dioxide. Following a 30-hour incubation period, the Lip-DT and Lip-DT-Dox compounds were introduced to the cells at a concentration of 100 nM in DMEM. After an 8-hour incubation period with the compounds, the cells were subjected to three consecutive rinses with chilled PBS solution, each lasting 5 minutes. Following this, the cells were fixed using 4% paraformaldehyde at room temperature for 10 minutes and washed twice with cold PBS.51 The cells were then subjected to examination using fluorescence microscopy (Optika B-100FL HBO, Italy) after being mounted with Vectashield mounting media with DAPI (Invitrogen). This process facilitated the visualization of the labelled fluorescence.
ROS generation study
To determine the intracellular levels of reactive oxygen species (ROS), the cell-permeable oxidation-sensitive probe, CM-H2DCFDA, was employed. HCT116 or MCF7 cells were treated with Dox and Lip-DT-Dox for 24 hours. Following the incubation period, the cells were detached by scraping, collected through centrifugation, and subsequently washed twice with cold PBS for 5 minutes each. Afterward, the cells were resuspended in PBS and subjected to a 20-minute incubation in the dark at 37 °C with CM-H2DCFDA (5 μM).50 Then the cells were washed with chilled PBS and lysed using PBS containing 1% Tween 20. The fluorescence of dichlorofluorescein (DCF) was measured at an excitation wavelength of 480 nm and an emission wavelength of 530 nm to determine the generation of intracellular ROS.Measurement of mitochondrial membrane potential
The evaluation of mitochondrial membrane potential (MMP, ΨM) involved monitoring the interaction of the fluorescent dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl benzimidazolylcarbocyanine iodide) with mitochondria, following a standard protocol. MCF7 and HCT116 cells were initially cultured in DMEM medium supplemented with 10% FCS. Following the treatment with Dox and Lip-DT-Dox, the cells were subsequently incubated for a duration of 30 hours. After the incubation period, the cells were harvested to measure the levels of reactive oxygen species (ROS). The cell lysates were incubated with JC-1 (2.5 μg mL−1) in PBS solution at 37 °C for approximately 30–35 minutes with continuous shaking.50 Then the cells were washed thrice with chilled PBS for 5 minutes each, and resuspended in PBS. To assess the mitochondrial membrane potential (MMP ΨM), the ratio of fluorescence intensity at 590 nm to 530 nm was measured.Apoptosis assay
The Roche cell death detection kit was utilized to quantify the occurrence of apoptosis in MCF7 and HCT116 cancer cells treated with Dox (positive control) and Lip-DT-Dox. The quantification of cytoplasmic histone-associated DNA fragments was performed using an ELISA kit. The results are presented as the fold increase in the enrichment factor of cytoplasmic nucleosomes.50Mitochondrial isolation from cultured cell
Cells post-treatment or control were centrifuged at 300 × g for 10 minutes and the pellet was collected. The cell pellet was gently re-suspended in 1–2 mL of ice-cold homogenization buffer containing tris–HCl or HEPES, salts (e.g., potassium chloride), and protease inhibitors. It was then homogenized by gently grinding or shearing, taking care not to generate excessive heat. The homogenate solution was transferred to a 15 mL centrifuge tube and centrifuged at a low speed (e.g., 800 × g) for 5 minutes. This step removes unbroken cells, nuclei, and cellular debris, leaving the mitochondria in the supernatant. The supernatant was taken and centrifuged at a higher speed (e.g., 10000 × g) for about 10–15 minutes. The supernatant having the cytosol and other soluble components was discarded while the pellet at the bottom with mitochondria was collected for lysate preparation and further processing.52
Immunoblotting
After being exposed to liquid nitrogen for rapid freezing, the treated cells were subsequently lysed in a RIPA buffer containing premixed protease and phosphatase inhibitor cocktails (Abcam). The cell lysates were prepared and quantified using the same procedure as previously performed.53 Each protein sample, consisting of 20 μg, was subjected to SDS-PAGE for separation and then transferred onto nitrocellulose membranes. Following the transfer, the membranes were blocked in a 3% BSA solution in 1X TBST for 1 hour. Subsequently, the membranes were immunoblotted with the appropriate primary antibodies and left overnight for incubation. After rinsing the immunoblots with 1X TBST, they were incubated with secondary antibodies labelled with horseradish peroxidase. The detection of immunoreactivity was achieved by employing chemiluminescence. To quantify the densitometry of western blots, Image J software (NIH) was utilized. The protein expression levels were normalized to β-Actin and then presented as a ratio relative to the control conditions for all experiments.Statistical analysis
All the data were analyzed by Student's t test or one- or two-way ANOVA with the Bonferroni post hoc interpretation. GraphPad Prism software (San Diego, CA, USA) was used for statistical analyses. Results were considered significantly different at P < 0.05 and the values are shown as means ± SEM.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. D. acknowledges the Board of Research in Nuclear Sciences (BRNS), India, for research funding (grant no.: 58/14/14/2021-BRNS/37219) and the support of the Interdisciplinary Institute of Indian System of Medicine (IIISM) for providing NMR and ESI mass spectrometry facility, Nano Research Centre (NRC), Department of Biotechnology (DBT), and Department of Chemistry of the SRM Institute of Science and Technology for several characterization studies. B. M. acknowledges the fund from the Indian Council of Medical Research (ICMR – 5/4/1-26/2020-NCD-I and 5/4/1-22/CVD/2022-NCD-I). We also thank CBMR for the instruments and laboratory facilities for performing the work.Notes and references
- F. Grizzi and M. Chiriva-Internati, Cancer: looking for simplicity and finding complexity, Cancer Cell Int., 2006, 6, 4 CrossRef PubMed.
- H. Nagai and Y. H. Kim, Cancer prevention from the perspective of global cancer burden patterns, J. Thorac. Dis., 2017, 9, 448–451 CrossRef PubMed.
- M. Abotaleb, P. Kubatka, M. Caprnda, E. Varghese, B. Zolakova, P. Zubor, R. Opatrilova, P. Kruzliak, P. Stefanicka and D. Büsselberg, Chemotherapeutic agents for the treatment of metastatic breast cancer: An update, Biomed. Pharmacother., 2018, 101, 458–477 CrossRef CAS PubMed.
- J. H. Kim, P. Verwilst, M. Won, J. Lee, J. L. Sessler, J. Han and J. S. Kim, A Small Molecule Strategy for Targeting Cancer Stem Cells in Hypoxic Microenvironments and Preventing Tumorigenesis, J. Am. Chem. Soc., 2021, 143, 14115–14124 CrossRef CAS PubMed.
- Z. Chen, L. Zhang, Y. Song, J. He, L. Wu, C. Zhao, Y. Xiao, W. Li, B. Cai, H. Cheng and W. Li, Hierarchical targeted hepatocyte mitochondrial multifunctional chitosan nanoparticles for anticancer drug delivery, Biomaterials, 2015, 52, 240–250 CrossRef CAS PubMed.
- R. H. Kang, Y. Kim, J. H. Kim, N. H. Kim, H. M. Ko, S.-H. Lee, I. Shim, J. S. Kim, H.-J. Jang and D. Kim, Self-Activating Therapeutic Nanoparticle: A Targeted Tumor Therapy Using Reactive Oxygen Species Self-Generation and Switch-on Drug Release, ACS Appl. Mater. Interfaces, 2021, 13, 30359–30372 CrossRef CAS PubMed.
- S. Biswas, N. S. Dodwadkar, P. P. Deshpande and V. P. Torchilin, Liposomes loaded with paclitaxel and modified with novel triphenylphosphonium–PEG–PE conjugate possess low toxicity, target mitochondria and demonstrate enhanced antitumor effects in vitro and in vivo, J. Controlled Release, 2012, 159, 393–402 CrossRef CAS PubMed.
- Z. Ahmad, A. Shah, M. Siddiq and H.-B. Kraatz, Polymeric micelles as drug delivery vehicles, RSC Adv., 2014, 4, 17028–17038 RSC.
- H. Cho, Y.-Y. Cho, M. S. Shim, J. Y. Lee, H. S. Lee and H. C. Kang, Mitochondria-targeted drug delivery in cancers, Biochim. Biophys. Acta, Mol. Basis Dis., 2020, 1866, 165808 CrossRef CAS PubMed.
- A. Khosa, S. Reddi and R. N. Saha, Nanostructured lipid carriers for site-specific drug delivery, Biomed. Pharmacother., 2018, 103, 598–613 CrossRef CAS PubMed.
- P. Mahato, A. Ghosh, S. K. Mishra, A. Shrivastav, S. Mishra and A. Das, Zn(II) based colorimetric sensor for ATP and its use as a viable staining agent in pure aqueous media of pH 7.2, Chem. Commun., 2010, 46, 9134–9136 RSC.
- W. Mu, Q. Chu, Y. Liu and N. Zhang, A Review on Nano-Based Drug Delivery System for Cancer Chemoimmunotherapy, Nano-Micro Lett., 2020, 12, 142 CrossRef CAS PubMed.
- D. Guimarães, A. Cavaco-Paulo and E. Nogueira, Design of liposomes as drug delivery system for therapeutic applications, Int. J. Pharm., 2021, 601, 120571 CrossRef PubMed.
- M. Alavi, N. Karimi and M. Safaei, Application of Various Types of Liposomes in Drug Delivery Systems, Adv. Pharm. Bull., 2017, 7, 3–9 CrossRef CAS PubMed.
- A. Sharma, Liposomes in drug delivery: Progress and limitations, Int. J. Pharm., 1997, 154, 123–140 CrossRef CAS.
- S. Okada, S. Peng, W. Spevak and D. Charych, Color and Chromism of Polydiacetylene Vesicles, Acc. Chem. Res., 1998, 31, 229–239 CrossRef CAS.
- C. Kim and K. Lee, Polydiacetylene (PDA) Liposome-Based Immunosensor for the Detection of Exosomes, Biomacromolecules, 2019, 20, 3392–3398 CrossRef CAS PubMed.
- Q. Tran, H. Lee, C. Kim, G. Kong, N. Gong, S. H. Kwon, J. Park, S.-H. Kim and J. Park, Revisiting the Warburg Effect: Diet-Based Strategies for Cancer Prevention, BioMed Res. Int., 2020, 2020, 1–9 Search PubMed.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS PubMed.
- X. Luo, I. Budihardjo, H. Zou, C. Slaughter and X. Wang, Bid, a Bcl2 Interacting Protein, Mediates Cytochrome c Release from Mitochondria in Response to Activation of Cell Surface Death Receptors, Cell, 1998, 94, 481–490 CrossRef CAS PubMed.
- A. Letai, M. C. Bassik, L. D. Walensky, M. D. Sorcinelli, S. Weiler and S. J. Korsmeyer, Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics, Cancer Cell, 2002, 2, 183–192 CrossRef CAS PubMed.
- S. Tiwari, M. Gupta and S. P. Vyas, Nanocarrier Mediated Cytosolic Delivery of Drug, DNA and Proteins, Proc. Natl. Acad. Sci., India, Sect. B, 2012, 82, 127–150 CrossRef CAS.
- Z.-P. Chen, M. Li, L.-J. Zhang, J.-Y. He, L. Wu, Y.-Y. Xiao, J.-A. Duan, T. Cai and W.-D. Li, Mitochondria-targeted drug delivery system for cancer treatment, J. Drug Target., 2016, 24, 492–502 CrossRef CAS PubMed.
- H. S. Jung, J.-H. Lee, K. Kim, S. Koo, P. Verwilst, J. L. Sessler, C. Kang and J. S. Kim, A Mitochondria-Targeted Cryptocyanine-Based Photothermogenic Photosensitizer, J. Am. Chem. Soc., 2017, 139, 9972–9978 CrossRef CAS PubMed.
- L. B. Chen, Mitochondrial Membrane Potential in Living Cells, Annu. Rev. Cell Biol., 1988, 4, 155–181 CrossRef CAS PubMed.
- L. V. Johnson, M. L. Walsh, B. J. Bockus and L. B. Chen, Monitoring of relative mitochondrial membrane potential in living cells by fluorescence microscopy, J. Cell Biol., 1981, 88, 526–535 CrossRef CAS PubMed.
- H. Agarwalla, P. S. Mahajan, D. Sahu, N. Taye, B. Ganguly, S. B. Mhaske, S. Chattopadhyay and A. Das, A Switch-On NIR Probe for Specific Detection of Hg2+ Ion in Aqueous Medium and in Mitochondria, Inorg. Chem., 2016, 55, 12052–12060 CrossRef CAS PubMed.
- M. P. Murphy, Selective targeting of bioactive compounds to mitochondria, Trends Biotechnol., 1997, 15, 326–330 CrossRef CAS PubMed.
- M. P. Murphy, Targeting lipophilic cations to mitochondria, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 1028–1031 CrossRef CAS PubMed.
- K. Sunwoo, M. Won, K.-P. Ko, M. Choi, J. F. Arambula, S.-G. Chi, J. L. Sessler, P. Verwilst and J. S. Kim, Mitochondrial Relocation of a Common Synthetic Antibiotic: A Non-genotoxic Approach to Cancer Therapy, Chem, 2020, 6, 1408–1419 CAS.
- S. K. Pramanik and A. Das, Fluorescent probes for imaging bioactive species in subcellular organelles, Chem. Commun., 2021, 57, 12058–12073 RSC.
- H. Agarwalla, S. Pal, A. Paul, Y. W. Jun, J. Bae, K. H. Ahn, D. N. Srivastava and A. Das, A fluorescent probe for bisulfite ions: its application to two-photon tissue imaging, J. Mater. Chem. B, 2016, 4, 7888–7894 RSC.
- F. Ali, A. H. A, N. Taye, R. G. Gonnade, S. Chattopadhyay and A. Das, A fluorescent probe for specific detection of cysteine in the lipid dense region of cells, Chem. Commun., 2015, 51, 16932–16935 RSC.
- A. H. Ashoka, F. Ali, R. Tiwari, R. Kumari, S. K. Pramanik and A. Das, Recent Advances in Fluorescent Probes for Detection of HOCl and HNO, ACS Omega, 2020, 5, 1730–1742 CrossRef CAS PubMed.
- H. Zhang, Thin-Film Hydration Followed by Extrusion Method for Liposome Preparation, Methods Mol. Biol., 2017, 1522, 17–22 CrossRef CAS PubMed.
- G. Bozzuto and A. Molinari, Liposomes as nanomedical devices, Int. J. Nanomed., 2015, 10, 975–999 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- S. Pandey, S. Patil, N. Ballav and S. Basu, Spatial targeting of Bcl-2 on endoplasmic reticulum and mitochondria in cancer cells by lipid nanoparticles, J. Mater. Chem. B, 2020, 8, 4259–4266 RSC.
- P. Xie, F. Guo, R. Xia, Y. Wang, D. Yao, G. Yang and L. Xie, A rhodamine–dansyl conjugate as a FRET based sensor for Fe3+ in the red spectral region, J. Lumin., 2014, 145, 849–854 CrossRef CAS.
- X. Li, S. Matthews and P. Kohli, Fluorescence Resonance Energy Transfer in Polydiacetylene Liposomes, J. Phys. Chem. B, 2008, 112, 13263–13272 CrossRef CAS PubMed.
- S. M. Rafiyath, M. Rasul, B. Lee, G. Wei, G. Lamba and D. Liu, Comparison of safety and toxicity of liposomal doxorubicin vs. conventional anthracyclines: a meta-analysis, Exp. Hematol. Oncol., 2012, 1, 1–9 CrossRef PubMed.
- J. J. M. Kwakman, Y. S. Elshot, C. J. A. Punt and M. Koopman, Management of cytotoxic chemotherapy-induced hand-foot syndrome, Oncol. Rev., 2020, 14, 442 CAS.
- S. Sivagnanam, M. Basak, A. Kumar, K. Das, T. Mahata, P. Rana, A. S. Sengar, S. Ghosh, M. Subramanian, A. Stewart, B. Maity and P. Das, Supramolecular Structures Generated via Self-Assembly of a Cell Penetrating Tetrapeptide Facilitate Intracellular Delivery of a Pro-apoptotic Chemotherapeutic Drug, ACS Appl. Bio Mater., 2021, 4, 6807–6820 CrossRef CAS PubMed.
- X. Ji, Y. Yan, T. Sun, Q. Zhang, Y. Wang, M. Zhang, H. Zhang and X. Zhao, Glucosamine sulphate-loaded distearoyl phosphocholine liposomes for osteoarthritis treatment: combination of sustained drug release and improved lubrication, Biomater. Sci., 2019, 7, 2716–2728 RSC.
- S. V. Boddapati, G. G. M. D’Souza, S. Erdogan, V. P. Torchilin and V. Weissig, Organelle-Targeted Nanocarriers: Specific Delivery of Liposomal Ceramide to Mitochondria Enhances Its Cytotoxicity in Vitro and in Vivo, Nano Lett., 2008, 8, 2559–2563 CrossRef CAS PubMed.
- Z. Guo, S. Kozlov, M. F. Lavin, M. D. Person and T. T. Paull, ATM Activation by Oxidative Stress, Science, 2010, 330, 517–521 CrossRef CAS PubMed.
- R. W. Horobin, S. Trapp and V. Weissig, Mitochondriotropics: A review of their mode of action, and their applications for drug and DNA delivery to mammalian mitochondria, J. Controlled Release, 2007, 121, 125–136 CrossRef CAS PubMed.
- Z. Zhang and S.-S. Feng, The drug encapsulation efficiency, in vitro drug release, cellular uptake and cytotoxicity of paclitaxel-loaded poly(lactide)–tocopheryl polyethylene glycol succinate nanoparticles, Biomaterials, 2006, 27, 4025–4033 CrossRef CAS PubMed.
- K. Das, M. Basak, T. Mahata, M. Kumar, D. Kumar, S. Biswas, S. Chatterjee, M. Moniruzzaman, N. C. Saha, K. Mondal, P. Kumar, P. Das, A. Stewart and B. Maity, RGS11-CaMKII complex mediated redox control attenuates chemotherapy-induced cardiac fibrosis, Redox Biol., 2022, 57, 102487 CrossRef CAS PubMed.
- S. Sivagnanam, K. Das, M. Basak, T. Mahata, A. Stewart, B. Maity and P. Das, Self-assembled dipeptide based fluorescent nanoparticles as a platform for developing cellular imaging probes and targeted drug delivery chaperones, Nanoscale Adv., 2022, 4, 1694–1706 RSC.
- S. Sivagnanam, K. Das, V. Sivakadatcham, T. Mahata, M. Basak, I. Pan, A. Stewart, B. Maity and P. Das, Generation of Self-Assembled Structures Composed of Amphipathic, Charged Tripeptides for Intracellular Delivery of Pro-Apoptotic Chemotherapeutics, Isr. J. Chem., 2022, 62, e202200001 CrossRef CAS.
- J. M. Suárez-Rivero, C. J. Pastor-Maldonado, A. Romero-González, D. Gómez-Fernandez, S. Povea-Cabello, M. Álvarez-Córdoba, I. Villalón-García, M. Talaverón-Rey, A. Suárez-Carrillo, M. Munuera-Cabeza and J. A. Sánchez-Alcázar, Pterostilbene in Combination With Mitochondrial Cofactors Improve Mitochondrial Function in Cellular Models of Mitochondrial Diseases, Front. Pharmacol., 2022, 13, 862085 CrossRef PubMed.
- M. Basak, K. Das, T. Mahata, A. S. Sengar, S. K. Verma, S. Biswas, K. Bhadra, A. Stewart and B. Maity, RGS7-ATF3-Tip60 Complex Promotes Hepatic Steatosis and Fibrosis by Directly Inducing TNFα, Antioxid. Redox Signaling, 2023, 38, 137–159 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of experimental details including synthesis and characterization, zeta potential, DLS measurement, MTT assay. See DOI: https://doi.org/10.1039/d3cb00219e |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |