
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA toolbox for enzymatic modification of nucleic acids with photosensitizers for photodynamic therapy†
Germain
Niogret
ab,
Camille
Chériaux
c,
Frédéric
Bonhomme
d,
Fabienne
Levi-Acobas
a,
Carlotta
Figliola
 c,
Gilles
Ulrich
c,
Gilles
Gasser
*b and
Marcel
Hollenstein
*a
c,
Gilles
Ulrich
c,
Gilles
Gasser
*b and
Marcel
Hollenstein
*a
aInstitut Pasteur, Université Paris Cité, CNRS UMR3523, Department of Structural Biology and Chemistry, Laboratory for Bioorganic Chemistry of Nucleic Acids, 28, rue du Docteur Roux, 75724 Paris Cedex 15, France. E-mail: marcel.hollenstein@pasteur.fr
bChimie ParisTech, PSL University, CNRS, Institute of Chemistry for Life and Health Sciences, Laboratory for Inorganic Chemical Biology, 75005 Paris, France. E-mail: gilles.gasser@chimieparistech.psl.eu
cInstitut de Chimie et Procédés pour L'Energie, L'Environnement et La Santé (ICPEES), Groupe de Chimie Organique pour Les Matériaux, La Biologie et L’Optique (COMBO), CNRS UMR 7515, École de Chimie, Polymères, Matériaux de Strasbourg (ECPM), 25, Rue Becquerel, 67087 Strasbourg Cedex 02, France
dInstitut Pasteur, Université Paris Cité, CNRS UMR3523, Department of Structural Biology and Chemistry, Unité de Chimie Biologique Epigénétique, 28, rue du Docteur Roux, 75724 Paris Cedex 15, France
First published on 8th July 2024
Abstract
Photodynamic therapy (PDT) is an approved cancer treatment modality. Despite its high efficiency, PDT is limited in terms of specificity and by the poor solubility of the rather lipophilic photosensitizers (PSs). In order to alleviate these limitations, PSs can be conjugated to oligonucleotides. However, most conjugation methods often involve complex organic synthesis and result in the appendage of single modifications at the 3′/5′ termini of oligonucleotides. Here, we have investigated the possibility of bioconjugating a range of known PSs by polymerase-mediated synthesis. We have prepared a range of modified nucleoside triphosphates by different conjugation methods and investigated the substrate tolerance of these nucleotides for template-dependent and -independent DNA polymerases. This method represents a mild and versatile approach for the conjugation of single or multiple PSs onto oligonucleotides and can be useful to further improve the efficiency of the PDT treatment.
Introduction
Cancer is the second leading cause of death worldwide, and its incidence is predicted to rise to over 20 million new cases annually by 2025.4 Although chemotherapy and radiotherapy are currently the main methods of treatment, they are usually limited by their inability to specifically target tumor cells, leading to toxic side effects.5 Hence, alternative, highly efficient and personalized treatment modalities are in dire need to effectively combat cancers.6–10 In this context, photodynamic therapy (PDT) has advanced as a promising non-invasive strategy for combating cancer11–16 and antimicrobial resistance.17–20 This approved treatment modality is based on the combined action of a photosensitizer (PS), light of a specific wavelength, and endogenous oxygen. Initially, the PS which should be non-toxic in the absence of light, is administered either systemically or locally to a patient. After photoexcitation to an excited singlet state, the PS will transition to a different, mainly triplet, state by intersystem crossing instead of returning directly back to the ground state. Following these photochemical processes, the PS will generate reactive oxygen species (ROS) or reactive singlet oxygen 1O2 depending on the mechanism of action.13,21–23 These reactive species in turn create an oxidative stress which eventually leads to cell death. PDT offers many advantages, including the selective activation of the PSs by light with a defined wavelength, reduced systemic toxicity, a degree of spatio-temporal control, the possibility of targeting a wide range of cells, a non- or minimally-invasive approach, combination with other treatments, combating drug resistance, and blending with nanomaterials.20,24 Nonetheless, PDT is still afflicted by a number of limitations including lack of or low tumor/cancer cell specificity, difficulties in reaching deep tumors due to low penetration of light, and poor water solubility of hydrophobic PSs.24–26 The potency of the PDT treatment modality can be improved by conjugating PSs to biomolecules, particularly oligonucleotides.27–29 Indeed, equipping oligonucleotides with PSs has been shown to be a versatile method to induce and investigate site-directed DNA damage,30,31 to improve the bioavailability of PSs,32 or to develop combination therapies.33–35 Importantly, oligonucleotides can mediate a certain degree of specificity to PSs in order to suppress off-target activity.36–38 In this context, aptamers have advanced as popular vectors for PSs due to their ease of production and their high target specificity and affinity.39–45 Conjugation of single PS moieties to oligonucleotides typically involves chemical reactions such as amide bond formation32,43,45–47 or click chemistry-based approaches36,40,48 on pre-functionalized oligonucleotides. While these approaches are robust, they only permit the addition of a single PS and require chemically modified oligonucleotides which are expensive or need specialized experimental set-ups for their production. Here, we present an enzymatic method for the labelling of oligonucleotides with a variety of PSs that is readily applicable to any DNA sequence without the need for any additional chemical reactions. This method is based on the polymerase-mediated incorporation of nucleoside triphosphates equipped with PSs. Importantly, we also demonstrate that PS-modified nucleotides are compatible with aptamer isolation via the mod-SELEX (systematic evolution of ligands by eXponential enrichment) approach.Results
Design and synthesis of nucleotides and PS precursors
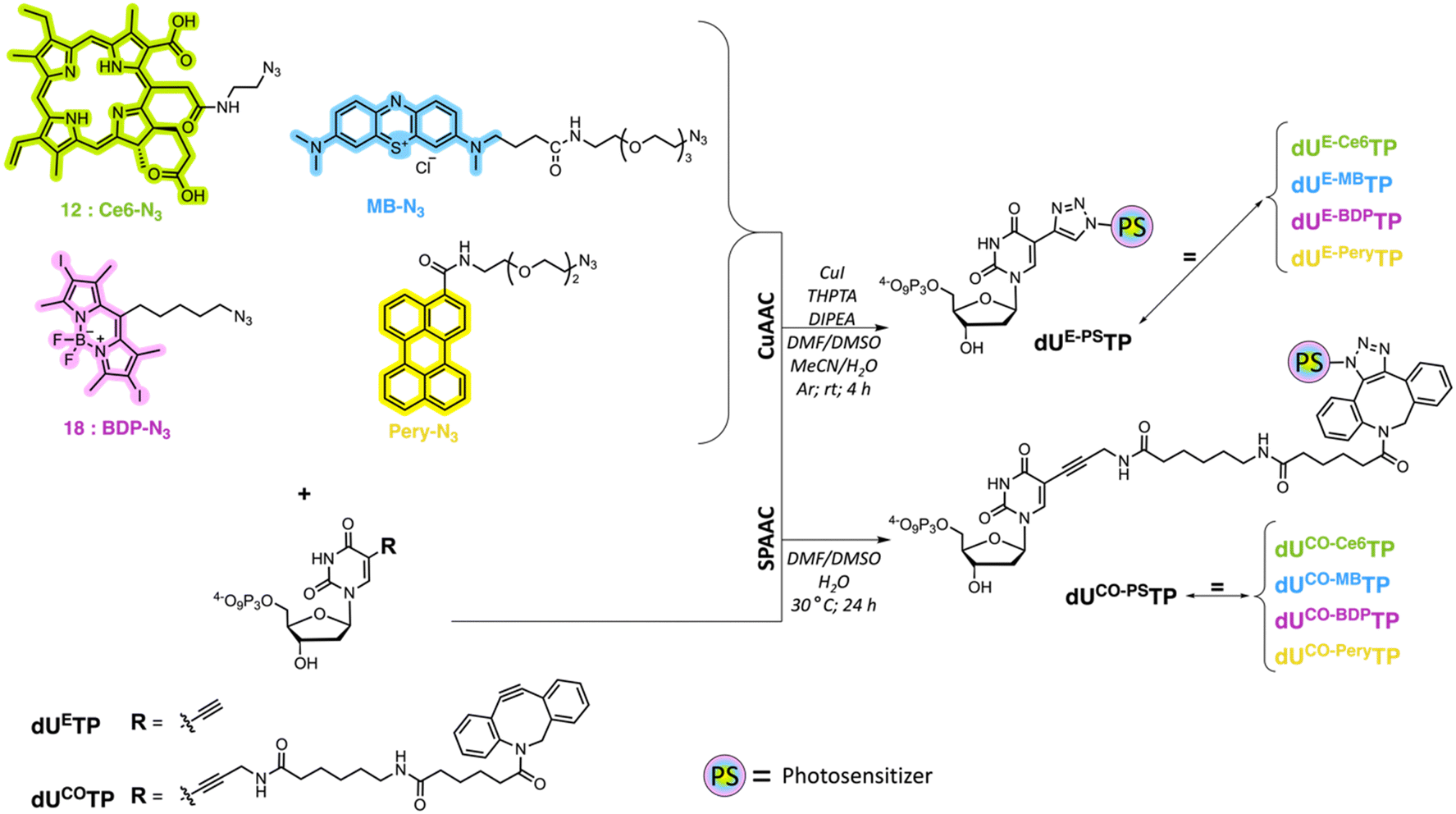
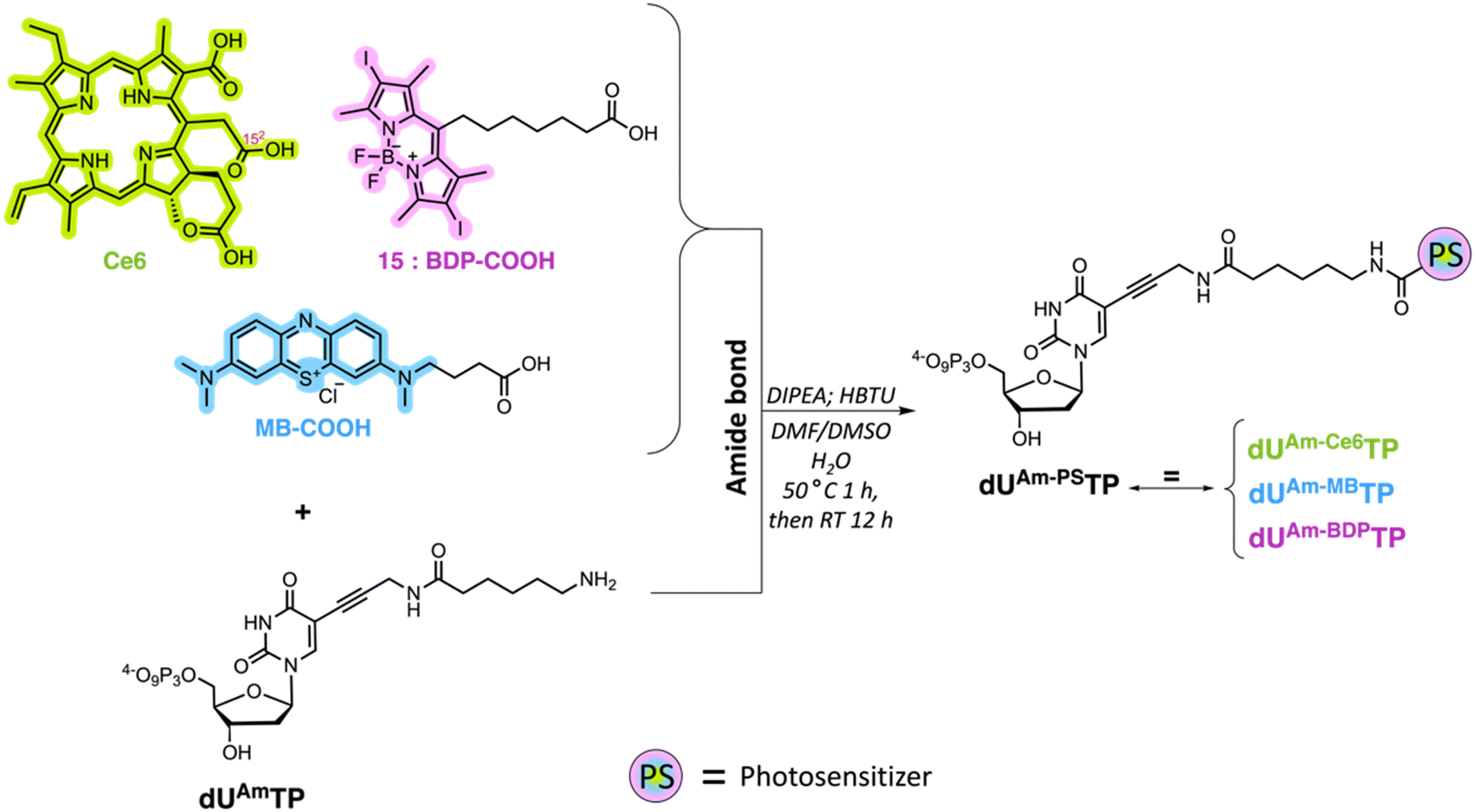
We surmised that using nucleoside triphosphates equipped with PSs connected to the nucleobases combined with polymerase-mediated DNA synthesis would represent a versatile and robust method for labelling of oligonucleotides with PSs. Indeed, nucleobase-modified nucleotides (dN*TPs) are usually well-tolerated by DNA polymerases, especially when attached to the C5 position of pyrimidines.49–60 Hence, using template-independent or template-dependent polymerases, DNA oligonucleotides could easily be decorated with one or multiple PSs. In order to explore this possibility, we set out to prepare dU*TPs equipped with the most commonly used PS in PDT applications, i.e. chlorin e6, methylene blue, BODIPY moieties, and perylene (Fig. 1).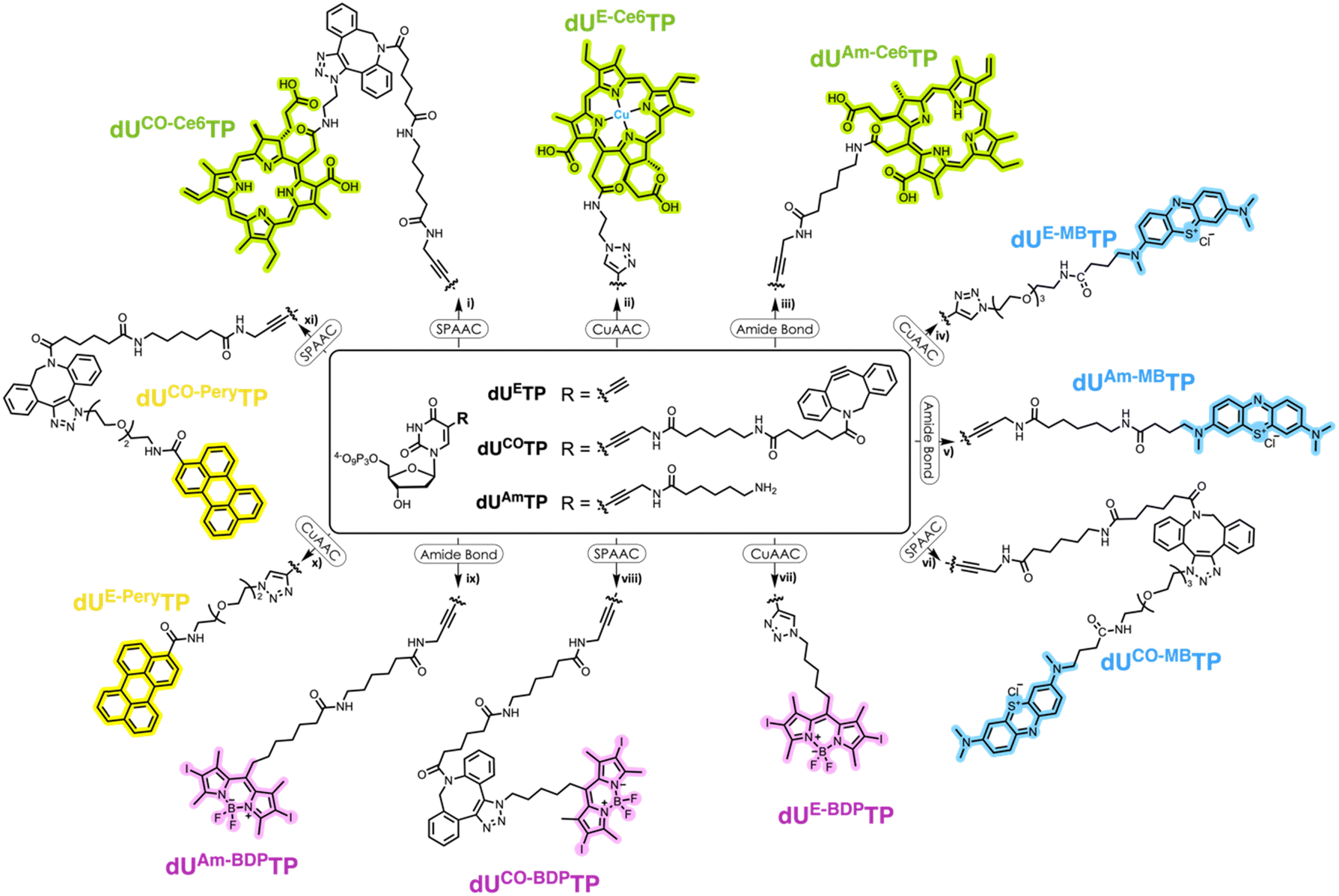 | ||
| Fig. 1 Chemical structure of all PS-modified nucleotides that were considered and produced. | ||
In addition to common PSs, we also considered three typical reactions which are used for the preparation of oligonucleotide-PS bioconjugates, namely copper-catalysed alkyne–azide cycloaddition (CuAAC), strain-promoted alkyne–azide cycloaddition (SPAAC), and amide bond formation (vide infra). Choosing different conjugation strategies also permitted the exploration and direct comparison of the effect of linker length and composition on the substrate acceptance by polymerases. The synthesis of the modified nucleotides commenced with the preparation of suitable PS building blocks equipped with azide and/or carboxylic acid moieties.
Porphyrins are popular PDT agents due to their favorable photophysical properties.61 Chlorin e6 is a porphyrin-based PSs that displays high reactive oxygen species production efficiencies and is integrated in the scaffold of approved drugs such as talaporfin.62 The structural key feature of chlorin e6 resides in the presence of three carboxylic acid functional groups which display different reactivities.63 In order to prepare a suitable chlorin e6 building block for click reactions, we first prepared the seven-membered anhydride ring between carboxylic acids 131 and 152 by dehydration mediated by the addition of EDC (Scheme 1).63,64 The resulting anhydride was then further reacted with 2-azidoethylamine which yielded the azide-modified chlorin e6 analog 1 in moderate yields (50%).
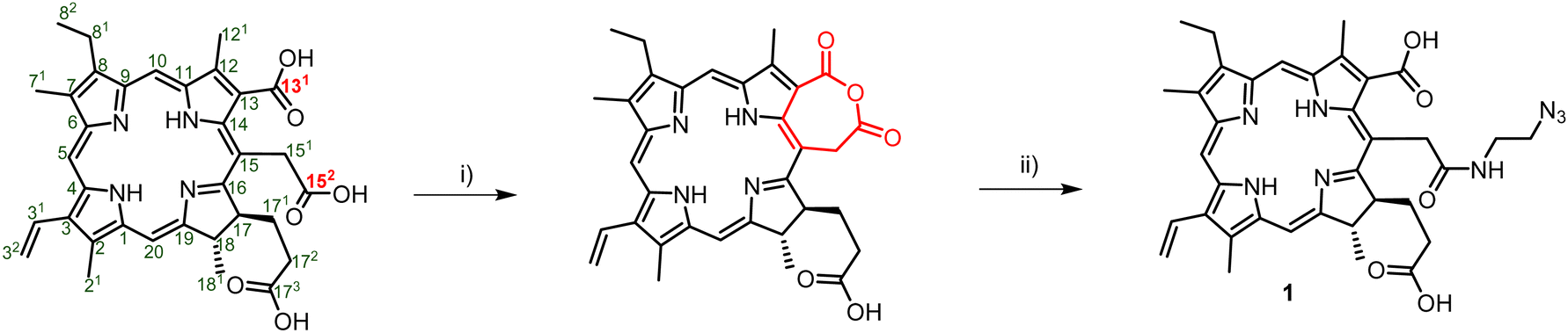 | ||
| Scheme 1 Synthetic pathway for the generation of Ce6–N31 and numbering of the porphyrin system. Reagents and conditions: (i) EDC, DIPEA, DCM, rt, 1 h; (ii) 2-azidoethylamine, rt, 12 h, 50%. | ||
Next, we set out to prepare iodinated BODIPY (boron-dipyrromethene) building blocks compatible with click reactions and amide bond formation. BODIPY derivatives have emerged as a promising new class of PDT agents and many analogues, particularly bis-iodinated-BODIPY, display favorable photophysical properties such as high extinction coefficients, resistance to photobleaching, and higher light-dark toxicity ratios.65,66 The synthesis of suitable BODIPY derivatives begins with the condensation43,67 of 2,4-dimethylpyrrole with 8-chloro-8-oxo-octanoate (Scheme 2A) or 6-bromohexanoyl chloride (Scheme 2B). The resulting intermediates were then subjected to a cyclization reaction in the presence of BF3·Et2O to form the BODIPY core, leading to compounds 2 and 5, respectively. Ester 2 was then saponified with LiOH to generate the carboxylic acid 3 in near quantitative yields. Compound 3 was then subjected to oxidative iodination in the presence of I2 and HIO3 to give the desired compound 4 in moderate yields (51%). For azide containing BODIPY derivative 7, the synthetic route involved an SN2 reaction after the initial condensation reaction. Indeed, BODIPY 5 was treated with NaN3 to afford derivative 6 in moderate yields (47%). Finally, oxidative iodination was carried out in the presence of I2 and HIO3, resulting in target compound 7 in excellent yields (87%).
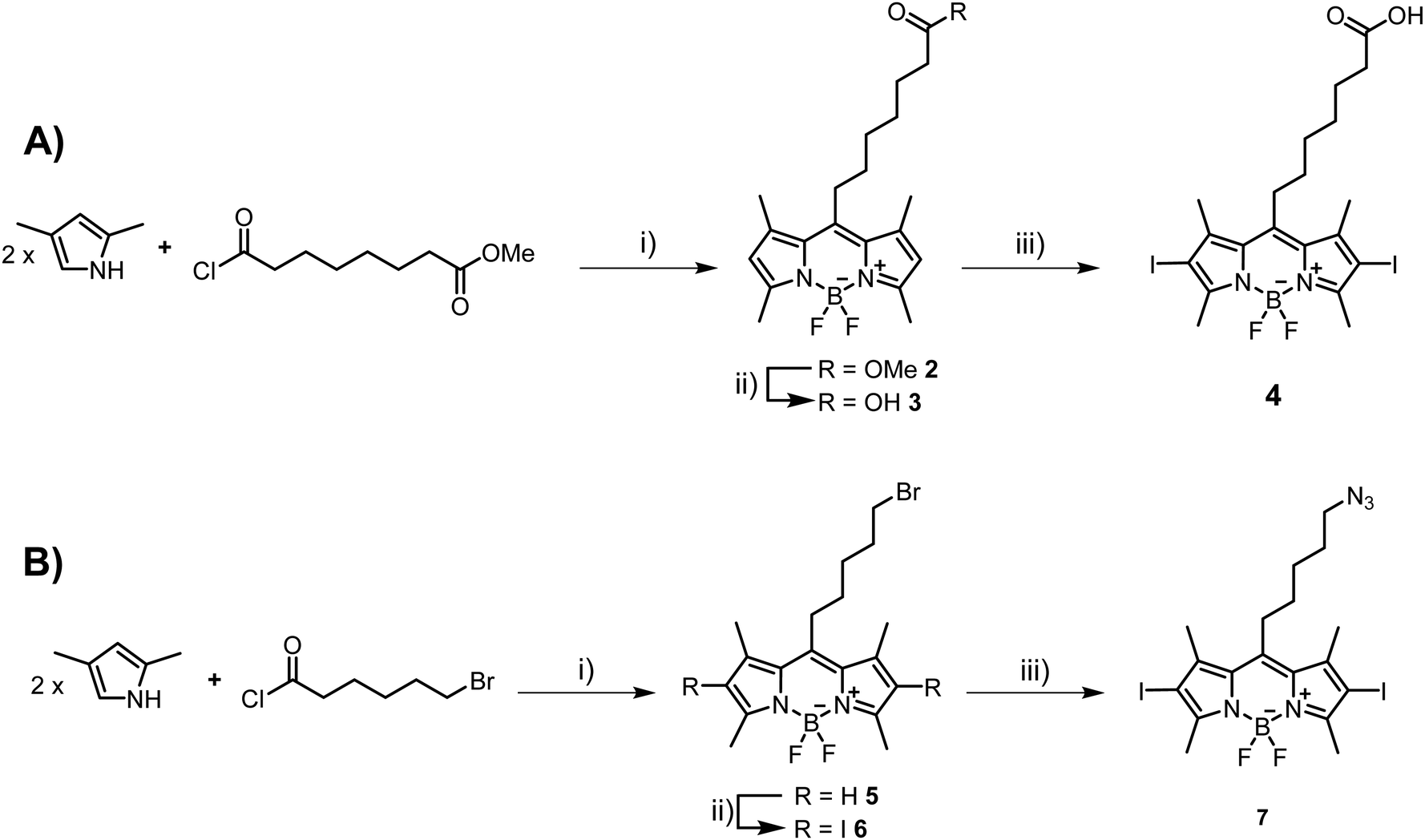 | ||
| Scheme 2 Synthetic routes to BODIPY building blocks. (A) Synthesis of carboxylic acid derivative 4. Reagents and conditions: (i) (a) DCM, reflux, 30 min; (b) NEt3, BF3·Et2O, reflux, 2 h, 41%; (ii) LiOH, THF/H2O, 40 °C, 4 h, 97%; (iii) MeOH, I2, HIO3, H2O, rt, 30 min, 51%. (B) Synthesis of azide 7. Reagents and conditions: (i) (a) DCM, reflux, 30 min; (b) NEt3, BF3·Et2O, reflux, 2 h, 47%; (ii) MeOH, I2, HIO3, H2O, rt, 30 min, 62%; (iii) NaN3, ether crown 18-C-6, THF, Ar, on, rt, 87%. | ||
Suitable precursors for click and amide bond formation for the synthesis of modified nucleotides equipped with perylene and methylene blue moieties were obtained from commercial suppliers (see ESI†).
With all azide and carboxylic acid modified PS building blocks at hand, we next set out to synthesize a subset library of modified nucleotides (Scheme 3). To do so, we prepared the modified nucleotides 5-ethynyl-dUTP (dUETP)58 and aza-dibenzocyclooctyne (ADIBO)-modified dUTP (dUCOTP)51 by application of published protocols, while amino-11-dUTP (dUAmTP) was commercially available.
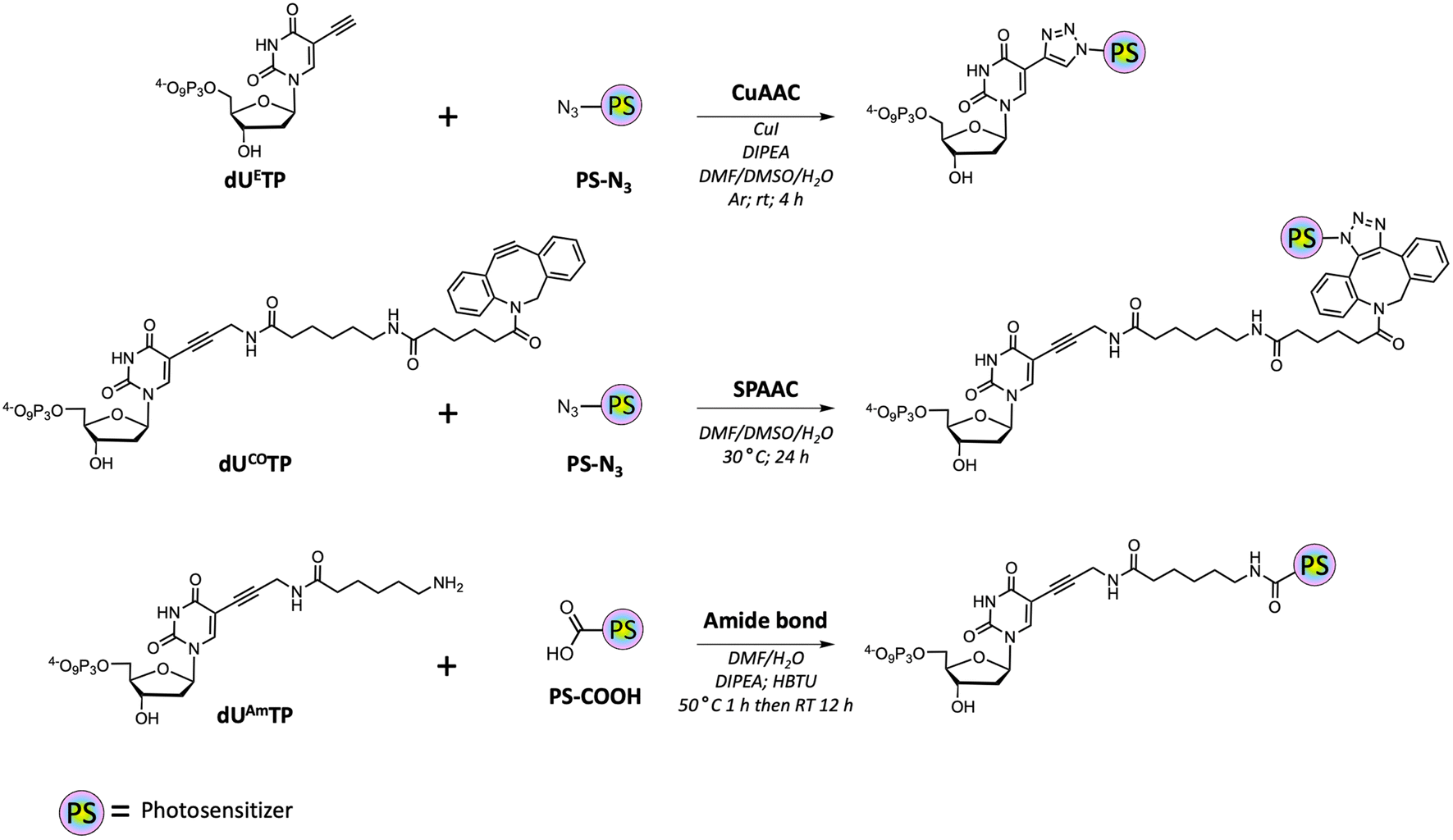 | ||
| Scheme 3 Overview of the different conjugation approaches for the synthesis of PS-modified dUTPs. | ||
We first carried out CuAAC reactions with azide-modified PS and dUETP under mild conditions (Scheme 4). The yields of isolated PS-modified nucleotides after thorough HPLC-purification were low (∼20%) when chlorin e6 azide 1 and BODIPY analogue 7 were employed and moderate (∼50%) with perylene- and methylene blue-azide derivatives. In addition, it is noteworthy mentioning that CuAAC reactions carried out with chlorin e6 azide 1 resulted in the formation of the copper complex despite a prolonged EDTA treatment prior and during HPLC purification.68 The presence of Cu2+ will alter the spectral properties of the PS and hence affect PDT efficiency of the resulting PS-modified oligonucleotides,69 but alternative synthetic methods could be applied to avoid chelation of the metal cation.70,71
 | ||
| Scheme 4 Synthetic pathways for the synthesis of dUE-PSTP and dUCO-PSTP derivatives. General reagents and conditions used for CuAAC reactions: PS-N3, dUETP, CuI, DIPEA, DMF/DMSO/MeCN/H2O, ar, rt, 4 h; 0.5 M EDTA to chelate residual copper then purification on RP-HPLC (20 mM TEAA/MeCN). Yields of isolated nucleotides: dUE-Ce6TP: 19%, dUE-MBTP: 58%, dUE-BDPTP: 22%, dUE-PeryTP: 43%. General reagent and conditions for SPAAC reactions: PS-N3, dUCOTP, DMF/DMSO/H2O, 30 °C, 24 h, purification on RP-HPLC (20 mM TEAA/MeCN). Yields of isolated nucleotides: dUCO-Ce6TP: 17%, dUCO-MBTP: 67%, dUCO-BDPTP: 18%, dUCO-PeryTP: 22%. | ||
For SPAAC reactions, we incubated dUCOTP with azide-modified PS for 24h at 30 °C in a mixture of solvents consisting of different ratios of DMF, DMSO, and H2O. As for the copper-catalyzed reactions, the yields varied between low (∼20%) and moderate (>60%) depending on the nature of the azide precursor. Nonetheless, all target nucleotides could also be obtained by application of SPAAC reactions.
Lastly, we carried out amide bond formation reactions with three PS agents equipped with carboxylic acid moieties and dUAmTP under standard conditions (Scheme 5). Conjugation to chlorin e6 revealed to be the most challenging reaction due to the formation of doubly substituted porphyrin. Due to the occurrence of this side-product, a thorough purification was required, and the yields remained modest for the formation of the desired nucleotide dUAm-Ce6TP (17%). The BODIPY and methylene blue containing nucleotides dUAm-BDPTP and dUAm-MBTP on the other hand were obtained in moderate to good yields, respectively.
 | ||
| Scheme 5 General synthetic pathway for the synthesis of dUAm-PSTP derivatives. General reagents and conditions: PS-COOH, DMF/DMSO, 40 °C, 1 h followed by addition of dUAmTP in H2O, rt, 12 h. Yields of isolated nucleotides: dUAm-Ce6TP 17%, dUAm-MBTP 73%, dUAm-BDPTP 42%. | ||
Biochemical characterization with the terminal deoxynucleotidyl transferase (TdT)
With a subset library of PS-modified dUTPs at hand, we sought to determine their compatibility with enzymatic DNA synthesis. To do so, we first evaluated whether the modified nucleotides could be used in template-independent DNA synthesis mediated by the terminal deoxynucleotidyl transferase (TdT). The TdT is an X family of polymerase and is capable of incorporating nucleotides randomly at the 3′-termini of ssDNA and ssRNA primers.72–74 Importantly, the TdT has rather lax nucleotide substrate requirements which propelled this polymerase in the forefront of numerous practical applications such as de novo enzymatic synthesis,75–80 aptamer selection,81–83 functional tagging,84–86 and construction of DNA nanostructures.87 Given these alluring features, combining TdT-mediated tailing reactions with PS-modified nucleotides could be employed for the post-SELEX functionalization of aptamers or in enzymatic aptamer generation. A first step towards these applications is the evaluation of the substrate tolerance of the TdT for the PS-modified dUTPs. To do so, we carried out primer extension (PEX) reactions with a 19 nucleotide long, 5′-FAM-labelled primer P1 (see the ESI† for sequence composition) in the presence of three metal cofactors (Co2+, Mn2+, and Mg2+). Nucleotides equipped with methylene blue acted as the best substrates for TdT-mediated reactions (Fig. 2). Indeed, tailing reactions conducted with dUAm-MBTP and Co2+ led to product distributions comparable to those obtained with natural dTTP. The tailing reactions with dUCO-MBTP and Co2+ were also highly efficient albeit products with faster gel mobilities could be also observed suggesting a somewhat reduced substrate tolerance. When the metal cofactor was changed to Mn2+ or Mg2+, the efficiency of the tailing reaction markedly dropped. PEX reactions carried out with dUE-MBTP only led to primers extended by one or two modified nucleotides, regardless of the cofactor that was employed. While nucleotides equipped with methylene blue acted as good substrates for the polymerase, nucleotides equipped with Ce6 and BODIPY were not readily tolerated by the TdT since no or very little extended primer products could be observed (Fig. S1 and S2, respectively, ESI†). Tailing reactions with perylene-modified dUTPs led mainly to products corresponding to primer extended by one to two modified nucleotides, albeit in low yields (Fig. S3, ESI†). | ||
| Fig. 2 PAGE gel (20%) analysis of tailing reactions with methylene blue containing nucleotides. Reactions were conducted with TdT (20 U) using primer P1 (2 pmol) with 1 μL TdT 10× buffer, with 200 μM of either dUE-MBTP, dUCO-MBTP, or dUAm-MBTP in the presence of either 1 mM Mn2+, 0.25 mM Co2+, or 1 mM Mg2+. The reactions with modified nucleotides were incubated for 5 min, 15 min, 30 min and 1 h at 37 °C. Controls: (T−1) negative control without TdT; (T−2) negative control without dTTP; (T+Mn; T+Co; T+Mg) positive control with a final concentration of 200 μM dTTP and 1 mM Mn2+, 0.25 mM Co2+, or 1 mM Mg2+ and 1 h incubation. | ||
Biochemical characterization of modified nucleotides with template-dependent polymerases
Enzymatic synthesis with template-dependent polymerases and the PS-modified nucleotides represents the first step towards the generation of modified libraries for SELEX experiments but also permits the introduction of one or multiple PS agents at any position of a DNA sequence. Hence, we continued our biochemical characterization by carrying out PEX reactions with the family B polymerase Vent (exo−) along with a rather simple primer/template system (P1/T1, see ESI†) which allows for the incorporation of a single, modified, 3′-terminal nucleotide. This analysis revealed that all PS-modified nucleotides were tolerated as substrates by the DNA polymerase and resulted in extended primer products in high yields (see Fig. S3, ESI†). Only the reactions conducted with dUE-Ce6TP (lane 3, Fig. S3, ESI†), dUE-MBTP (lane 6, Fig. S3, ESI†), and dUE-BDPTP (lane 9, Fig. S3, ESI†) led to ∼60–80% conversion of the primer to extended products, suggesting that the triazole linker arm obtained after CuAAC reactions was less tolerated by the polymerase. It is also noteworthy mentioning that in some cases, n + 2 products stemming from untemplated incorporation events could be observed.88–90 Next, we wished to further demonstrate the compatibility of the PS-modified nucleotides with polymerase-mediated DNA synthesis by digestion-LC–MS analysis experiments. To do so, we carried out large-scale PEX reactions with modified nucleotides and Vent (exo−) on the P1/T1 primer/template system. The resulting dsDNA products were then digested by the collective action of nucleases and the shrimp alkaline phosphatase down to single nucleosides which were then analysed by LC-MS as described previously.51,58,91 All LC-MS profiles displayed the four characteristic peaks corresponding to the canonical nucleosides along with additional peaks corresponding to the expected PS-modified nucleosides (Fig. S12–S17 (ESI†) and Experimental section). Collectively, these experiments confirm that all the modified nucleotides were readily tolerated by polymerases and incorporated into DNA.Encouraged by these results, we next set out to explore the possibility of synthesizing longer DNA sequences equipped with multiple PS units. To do so, we carried out PEX reactions with the 31-nt long template T2, the 15-nt long primer P2 (ESI†), the PS-modified dUTPs, and a series of family A (Taq, Hemo KlenTaq, Bst) and family B (Phusion, Vent (exo−), deep Vent (exo−), Therminator, Q5) DNA polymerases. PEX reactions carried out with the BODIPY-modified nucleotides dUE-BDPTP, dUCO-BDPTP, and dUAm-BDPTP yielded full length products with all polymerases that were tested with complete consumption of primer P2 (Fig. 3). In addition, a slight change in electrophoretic mobility could be observed depending on the chemical nature of the linker arm. Indeed, DNA containing nucleotides equipped with a short triazole moiety displayed faster running bands than those modified with a bulky ADIBO unit. When the methylene-blue containing nucleotides were engaged in similar PEX reactions, full length product formation could be observed with dUCO-MBTP and dUAm-MBTP (Fig. S4, ESI†). Consumption of the product was complete when Hemo KlenTaq, Taq, Bst, Vent (exo−) and deep Vent (exo−) (lanes 2, 4, 5, 7 and 8 in Fig. S4, ESI†) were used as polymerases whereas the reactions with the remaining polymerases produced the expected product in ∼70% yield. On the other hand, dUE-MBTP acted as a rather poor substrate for the polymerases since essentially truncated products could be observed in all reactions. Similar results were obtained when Ce6-modified dUTPs were engaged in PEX reactions (Fig. S5, ESI†). Finally, successful incorporation of the perylene-containing dUE-PeryTP and dUCO-PeryTP could be achieved with several polymerases especially Phusion and Q5 which readily tolerated both nucleotides as substrates (Fig. S6, ESI†).
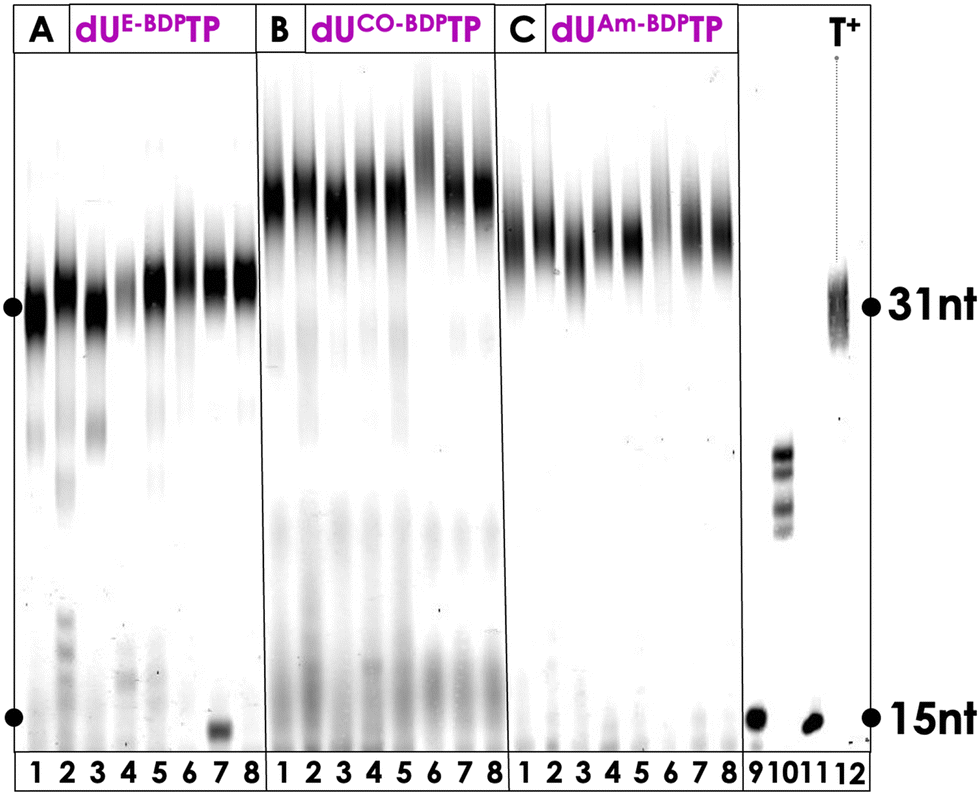 | ||
| Fig. 3 PAGE gel (20%) analysis of PEX reactions (3 hour) using primer P2 (10 pmol) and template T2 (15 pmol) with natural dNTPs (200 μM) and the modified triphosphates (200 μM) dUE-BDPTP, dUCO-BDPTP, and dUAm-BDPTP. Polymerases used: lane (1) Phusion (2 U); lane (2) Hemo KlenTaq (8 U); lane (3) Q5 (2 U); lane (4) Taq (5 U); lane (5) Bst (8 U); lane (6) Therminator (2 U); lane (7) Vent (exo−) (2 U); lane (8) deep Vent (exo−) (2 U); lane (9) negative control reaction without polymerase; (10) negative control reaction with only dATP, dGTP, and dCTP; lane (11) primer P2 (12) positive control reaction with natural dNTPs and Vent (exo−) (2 U). 15 nucleotides (nt) correspond to P2 and 31nt to full-length product. | ||
Next, we evaluated the possibility of modifying yet longer DNA oligonucleotides with PS moieties. To do so, we carried out PEX reactions with the P3/T3 primer/template system which allows for the incorporation of up to thirteen modified nucleotides (ESI†). These PEX reactions were carried out under similar conditions as for the P2/T2 system and the gel images are depicted in Fig. S7–S10 (ESI†). This analysis reveals that (1) dUCO-MBTP, dUAm-MBTP, dUCO-BDPTP, dUAm-BDPTP, and dUCO-PeryTP all act as excellent substrates for most or all polymerases that were considered and produce the expected full-length products, (2) dU*TPs obtained by CuAAC reactions (i.e.dUE-BDPTP, dUE-MBTP, and dUE-PeryTP) were moderate substrates but at least Vent (exo−) and deep Vent (exo−) readily incorporated these modified nucleotides and produced the expected full-length products with high (>80%) conversion yields, and (3) reactions with Ce6-modified nucleotides produce either rather undefined, smeared product bands or high molecular products with little electrophoretic mobility. Based on this analysis, we identified for each modified nucleotide the best experimental conditions to produce heavily modified dsDNA which are highlighted in Fig. 4.
 | ||
| Fig. 4 Gel (PAGE 20%) analysis of the PEX reactions with primer P3 (10 pmol), template T3 (15 pmol), dNTPs and dUPSTPs (200 μM) at 60 °C for 3 h. Lane 1: dUE-PeryTP with Bst (8 U); lane 2: dUCO-PeryTP with Vent (exo−) (2 U); lane 3: dUE-Ce6TP with Therminator (2 U); lane 4: dUCO-Ce6TP with Hemo Klen Taq (8 U); lane 5: dUAm-Ce6TP with Hemo Klen Taq (8 U); lane 6: dUE-MBTP with deep Vent (exo−) (2 U); lane 7: dUCO-MBTP with deep Vent (exo−); lane 8: dUAm-MBTP with deep Vent (exo−); lane 9: dUE-BDPTP with deep Vent (exo−); lane 10: dUCO-BDPTP with deep Vent (exo−); lane 11: dUAm-BDPTP with deep Vent (exo−); lane 12: negative control reactions without polymerase; lane (13) positive control reaction with natural dTTP. 18 nt corresponds to the length of P3, 71 nt correspond to full-length product. | ||
Finally, we evaluated these eleven PS-modified nucleotides for their capacity at serving as substrates for polymerases under PCR conditions. To do so, we used a 79-nt long template (T4) flanked by the primers P4 and P5 and evaluated the possibility of amplifying T4 with different polymerases along with the PS-modified dUTPs. Even though experimental conditions could be identified that permitted efficient PEX reactions on long template strands (Fig. 4), the modified nucleotides were rather reluctant at serving as substrates for polymerases under PCR conditions (Fig. S11, ESI†). Low yielding amplification was observed only with dUE-PeryTP and only in the presence of deep Vent (exo−).
Discussion
PDT is a highly efficient and recognized tumor treatment modality. The major limitations in PDT reside in the lack of tumor specificity due to their passive cellular uptake as well as the limited water solubility of the rather hydrophobic photosensitizers.7 Bioconjugation of pPSs represents an alluring strategy to alleviate these limitations. In this context, DNA and RNA oligonucleotides represent rather suitable platforms to improve the efficiency of PDT via coupling to PS agents. However, most conjugation methods involve chemical steps that allow for the incorporation of single photosensitizing units mainly located at the 3′- and 5′-termini of oligonucleotides. Here, we have evaluated the possibility of introducing one or multiple PS units by enzymatic DNA synthesis at any position of the sequence.We have synthesized nucleoside triphosphates modified with a small variety of PSs at the level of the nucleobase. We have then evaluated the capacity of these PS-modified nucleotides to act as substrates for template-dependent and template-independent polymerases. For the template-independent polymerase TdT, highly efficient tailing reactions could be obtained with nucleotides equipped with methylene blue, particularly those prepared by SPAAC and amide-bond coupling reactions. Single incorporation events could be obtained with perylene-modified nucleotides, while modifying nucleotides with BODIPY or chlorin e6 appeared to be deleterious to enzymatic synthesis. On the other hand, various template-dependent polymerases readily tolerated the modified nucleotides and could be used to produce short as well as long DNA sequences equipped with PSs. As for template-independent DNA synthesis, nucleotides equipped with methylene blue appeared to be the best while chlorin e6-modified nucleotides appeared to be the worst substrates for polymerases. Even though most modified nucleotides are excellent substrates for DNA polymerases under PEX reaction conditions they are refractory to PCR amplification, which has previously been observed for other modification patterns.49,91,92 This study also allowed to investigate the effect of the nature of the linker arm on the substrate tolerance. Overall, it appears that nucleotides bearing a rigid and short triazole connector at position C5 of the nucleotide are the least tolerated by all DNA polymerases. Nonetheless, the efficacy of incorporation of such nucleotides also depends on the nature of the PS modification attached to the nucleobase since dUE-BDPTP acts as a very good substrate for all polymerases that were evaluated (Fig. 3).
Lastly, nucleotides with similar linker arms (yet different modification patterns) have been shown to be fully compatible with the mod-SELEX approach even though they are not necessarily substrates for polymerases under PCR conditions.93–97 Particularly, libraries for SELEX prepared with nucleotides containing propargylamino- and triazole-based linker arms can efficiently be converted into unmodified DNA by standard PCR.91,98–106 We are thus confident that these modified nucleotides could be used for the identification of modified aptamers and DNAzymes and on-going work in the laboratory on an aptamer SELEX with dUAm-BDPTP seem to confirm this statement.
Conclusions
Here, we have demonstrated that modified nucleotides represent alluring tools for bioconjugating DNA oligonucleotides with photosensitizing agents under mild and efficient, polymerase-catalyzed reaction conditions. This method can be applied to virtually any type of oligonucleotides and permits the incorporation of one or multiple PSs. The modified nucleotides are well-tolerated by various template-dependent DNA polymerases and the template-independent TdT polymerase can be used to add single PS-modified nucleotides at the 3′-end of oligonucleotides. Overall, this facile conjugation method could be applied in SELEX to identify PS-modified aptamers (on-going work in our laboratory), the production of spherical nucleic acids equipped with multiple PS,107 for the post-SELEX modification of existing aptamers, or to improve ferroptosis during PDT treatment.108Methods
General protocol for CuAAC reactions
PS-N3 and dUETP (stock solution at 34.5 mM) were dissolved in 300 μL of DMF and 300 μL H2O in a 1.5 mL Eppendorf tube and degassed under Argon. In a separated Eppendorf tube, CuI (1.5 eq.) and THPTA (2 eq.) are suspended in DIPEA (6 eq.), 20 μL H2O and 100 μL MeCN and degassed with Argon. The two fractions were merged and 100 μL to 2 mL of H2O/DMF/DMSO were added until complete dissolution. The reaction mixture was degassed with Argon and left to shake at 1000 rpm, at r.t. for 4 hours. The reaction mixture was evaporated in vacuo and resuspended in 300 μL H2O and centrifuged at 4000 rpm for 15 minutes, and the supernatant was isolated and purified. Prior to each HPLC injection, 50 μL EDTA 0.5 M in H2O (pH = 8).General protocol for SPAAC reactions
PS-N3 was dissolved in 300 μL DMF and 100 μL of dUCOTP (34.2 mM stock solution) in an Eppendorf. 100 μL to 2 mL H2O/DMF/DMSO were added until complete dissolution. The reaction mixture was shaken at 1000 rpm at 30 °C for 24 hours. DMF was then evaporated in vacuo and the crude resuspended in 200 μL H2O. The mixture was centrifuged at 4000 rpm for 15 minutes, and the supernatant was purified by semi-prep reverse phase HPLC.General protocol for amide bond formation
PS-COOH was dissolved in DMF or DMSO and HBTU was then added. The resulting mixture was stirred and kept at 40 °C for 1 hour. After this period, dUAmTP (100 mM in H2O) was introduced and the reaction mixture was left under continuous stirring at room temperature for given reaction times. After reaction, the solvent was carefully removed under reduced pressure and the remaining residue redissolved in deionized water. The mixture was centrifuged and the supernatant decanted, leaving behind the precipitate. The isolated material was then dissolved in deionized water for further purification.HPLC purification of modified nucleotides
The HPLC purification is common to the 3 procedures described above. They were done with a semi-preparative reverse-phase column (Kinetex 5 μm C18 100 Å LC column). Solvent. A 0–100% B elution gradient over 45 minutes was used, with buffer A: 20 mM TEAA in water, buffer B MeCN. Before proceeding with the full purification, an analytical injection was performed to evaluate the chromatographic profile and check the retention time of the compounds in the reaction mixture. Based on this preliminary analysis, gradient adjustments were made, introducing a plateau of 5 to 10 minutes centered around the retention times of interest. Detector wavelengths on the HPLC were set at 260 nm, 280 nm and the specific absorption maximum of the photosensitizers, facilitating a purification process (i.e. Ce6: 405 nm, MB: 664 nm, BODIPY: 525 nm; perylene: 435 nm). After HPLC purification, the collected fractions were analyzed by MALDI-TOF to ensure successful production of the modified triphosphates.General method for TdT-mediated extension reactions
In a 10 μL reaction volume, the following components were combined: a 5′-FAM-labelled primer (2 pmol; see the ESI† for sequence composition), the modified nucleotide, the selected metal ion co-factor (1 mM Mn2+, 0.25 mM Co2+, and 1 mM Mg2+ final concentration), 1 μL of TdT polymerase buffer (10× concentration) and 1 μL of TdT polymerase. The reaction mixture was incubated at 37 °C, with the reaction times ranging from 5 minutes to 1 hour, depending on the specific requirements. To stop the reaction, 10 μL of a quenching solution (70% formamide, 50 mM EDTA, 0.1% bromophenol, and 0.1% xylene cyanol) was added. The resulting reaction mixtures were then analyzed using denaturing polyacrylamide gel electrophoresis (PAGE 20%). Gel electrophoresis was run with a trisborate–EDTA (TBE) buffer at a 1× concentration with a pH of 8 and 7 M urea.General method for primer extension reactions with template-dependent polymerases
A 5′-FAM-labelled primer (10 pmol) was hybridized with the corresponding template (15 pmol) in DNase/RNase-free ultrapure water. This was achieved by elevating the temperature to 95 °C and then allowing it to gradually cool down to room temperature over an hour. Subsequently, DNA polymerase (0.5 to 1 μL), suitable reaction buffer, and the required dNTP(s) were added to yield a 10 μL reaction mixture. This mixture underwent incubation at the polymerase-specific optimal temperature for given times. The reactions were quenched by adding 10 μL of a solution containing formamide (70%), EDTA (50 mM), bromophenol (0.1%), and xylene cyanol (0.1%). The resulting reaction mixtures were analyzed by gel electrophoresis in a denaturing 20% polyacrylamide gel, complemented with 1× TBE buffer (pH 8) and urea (7 M).General protocol for PCR reactions
The PCR mixtures were obtained by adding primers P4/P5 (6 μM each), template T4 (0.1 μM), dNTPs (200 μM), Mg2+ (2 mM), polymerase (0.4 μL), and polymerase buffer (either 2 μL of 10× or 4 μL of 5×), in a total volume of 20 μL. The PCR amplification protocol commenced with an initial 5-minute denaturation step at 95 °C. This was succeeded by amplification (25 cycles for modified nucleotides and 10 to 15 for canonical nucleotides), each consisting of a 1-minute denaturation at 95 °C, annealing for 1 minute at 52 °C, and elongation for 2 minutes at 72 °C. The cycles finished with a final extension period of 5 minutes at 72 °C. After PCR amplification, the reaction products were evaluated using 4% agarose gels, supplemented with 1× E-GEL sample loading buffer (loading: 1 to 5 pmol).General protocol for digestion-LC-MS analysis of modified DNA
Primer P1 (100 pmol) was annealed with template T1 (150 pmol) in DNase/RNase-free ultrapure water. This was carried out by first raising the temperature to 95 °C, then gradually cooling down to room temperature over a period of one hour. 1 μL of Vent (exo−) polymerase, 1 μL of 10× Thermopol reaction buffer, and 200 μM of modified dUTP or canonical dTTP for the positive control were successively added to the annealed duplex. Next, the combined mixture was left to incubate for a period of four hours at 60 °C. 1 μL of the resulting reaction mixtures were diluted in 9 μL H2O and quenched by adding the stop solution (vide supra). The reaction mixtures were subjected to gel electrophoresis in denaturing polyacrylamide gel (20%) containing trisborate-EDTA (TBE) 1× buffer (pH 8) and urea (7 M) to confirm efficient extension reactions. After evaluation of the efficiency of the PEX reactions by gel electrophoresis, reaction products were purified using Monarch DNA Cleanup columns (5 μg), each column processing a maximum of 250 pmol of product. The purified products (around 100 pmol in 10 μL) were then combined with nucleoside digestion mix buffer (2 μL of 10×) and nucleoside digestion mix (1 μL) in a final volume of 20 μL. The solution was left to incubate at 37 °C for one hour. Finally, the resulting products were subjected to LC-MS analysis without further purification.A solution of digested dsDNA was introduced into a ThermoFisher Hypersil Gold aQ chromatography column (100× 2.1 mm, with a particle size of 1.9 μm), maintained at a temperature of 30 °C. Flow rate was set at 0.3 mL min−1, and isocratic elution was performed at 1% MeCN in H2O with 0.1% formic acid for 8 minutes, then at 100% CH3CN from the 9th to the 11th minute. In positive ion mode, parent ions were fragmented using a normalized collision energy of 10% in PRM (parallel reaction monitoring) mode. MS2 resolution was set at 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 with an AGC target of 2e5, a maximum injection time of 50 ms and an isolation window of 1.0 m/z. The inclusion list contained the following masses: dC (228.1), dA (252.1), dG (268.1), dT dT (243.1), dUAm-Ce6 (973.41; calcd 972.44), dUE-MB (809.35; calcd 972.44), dUAm-MB (733.32; calcd 732.32), dUE-BDP (864.08; calcd 863.08), dUAm-BDP (1005.19; calcd 1004.18), and dUE-Pery (705.27; calcd 704.26).
500 with an AGC target of 2e5, a maximum injection time of 50 ms and an isolation window of 1.0 m/z. The inclusion list contained the following masses: dC (228.1), dA (252.1), dG (268.1), dT dT (243.1), dUAm-Ce6 (973.41; calcd 972.44), dUE-MB (809.35; calcd 972.44), dUAm-MB (733.32; calcd 732.32), dUE-BDP (864.08; calcd 863.08), dUAm-BDP (1005.19; calcd 1004.18), and dUE-Pery (705.27; calcd 704.26).
Author contributions
M. H., G. N., and G. G. conceived and designed the study. G. N. performed the chemical synthesis of all nucleosides and nucleotides, and all the biochemical characterization of the modified nucleotides together with F. L.-A. C. C. and C. F. contributed to the chemical synthesis. M. H., G. G., and G. U. supervised the project. G. N. and M. H. wrote the manuscript and all authors edited the article. All authors approved the final version.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and/or its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. N. gratefully acknowledges a fellowship from the doctoral school MTCI from Université Paris Cité. The authors gratefully acknowledge financial support from Institut Pasteur. M. H. acknowledges financial support from the ANR grant PEPR MolecularXiv (ANR-22-PEXM-0002) and the Joe W. and Dorothy Doresett Brown Foundation (exploratory and advanced grants). C. F. and C. C. thank the Interdisciplinary Institute HiFunMat for support of the SMARTY starting grant. This work of the Interdisciplinary Institute HiFunMat, as part of the ITI 2021–2028 program of the University of Strasbourg, CNRS and Inserm, was supported by IdEx Unistra (ANR-10-IDEX-0002) and SFRI (STRAT’US project, ANR-20-SFRI-0012) under the framework of the French Investments for the Future Program.References
- Q. Guan, L.-L. Zhou, Y.-A. Li and Y.-B. Dong, Chem. Commun., 2019, 55, 14898–14901 RSC.
- M. Macias-Contreras, K. L. Daykin, J. T. Simmons, J. R. Allen, Z. S. Hooper, M. W. Davidson and L. Zhu, Org. Biomol. Chem., 2017, 15, 9139–9148 RSC.
- N. Elumalai, A. Berg, K. Natarajan, A. Scharow and T. Berg, Angew. Chem., Int. Ed., 2015, 54, 4758–4763 CrossRef CAS PubMed.
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394–424 CrossRef PubMed.
- K. D. Miller, L. Nogueira, A. B. Mariotto, J. H. Rowland, K. R. Yabroff, C. M. Alfano, A. Jemal, J. L. Kramer and R. L. Siegel, Ca-Cancer J. Clin., 2019, 69, 363–385 CrossRef PubMed.
- B. K. Kashyap, V. V. Singh, M. K. Solanki, A. Kumar, J. Ruokolainen and K. K. Kesari, ACS Omega, 2023, 8, 14290–14320 CrossRef CAS PubMed.
- Y. C. Wang, J. N. Staudinger, T. L. Mindt and G. Gasser, Theranostics, 2023, 13, 5501–5544 CrossRef CAS PubMed.
- R. Luo, H. Liu and Z. Cheng, RSC Chem. Biol., 2022, 3, 830–847 RSC.
- O. Khorkova, J. Stahl, A. Joji, C.-H. Volmar and C. Wahlestedt, Nat. Rev. Drug Discovery, 2023, 22, 539–561 CrossRef CAS PubMed.
- A. Mantovani, P. Allavena, F. Marchesi and C. Garlanda, Nat. Rev. Drug Discovery, 2022, 21, 799–820 CrossRef CAS PubMed.
- Y. Zhang, B.-T. Doan and G. Gasser, Chem. Rev., 2023, 123, 10135–10155 CrossRef CAS PubMed.
- J. Karges, Angew. Chem., Int. Ed., 2022, 61, e202112236 CrossRef CAS PubMed.
- T. C. Pham, V.-N. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 121, 13454–13619 CrossRef CAS PubMed.
- S. Pratihar, K. K. Bhagavath and T. Govindaraju, RSC Chem. Biol., 2023, 4, 826–849 RSC.
- A. Sharma, P. Verwilst, M. Li, D. Ma, N. Singh, J. Yoo, Y. Kim, Y. Yang, J.-H. Zhu, H. Huang, X.-L. Hu, X.-P. He, L. Zeng, T. D. James, X. Peng, J. L. Sessler and J. S. Kim, Chem. Rev., 2024, 124, 2699–2804 CrossRef CAS PubMed.
- S. Zeng, Z. Guo, Y. Hao, Y. S. Kafuti, Z. Yang, Q. Yao, J. Wang, X. Peng and H. Li, Coord. Chem. Rev., 2024, 509, 215786 CrossRef CAS.
- C. Gourlot, A. Gosset, E. Glattard, C. Aisenbrey, S. Rangasamy, M. Rabineau, T.-S. Ouk, V. Sol, P. Lavalle, C. Gourlaouen, B. Ventura, B. Bechinger and V. Heitz, ACS Infect. Dis., 2022, 8, 1509–1520 CrossRef CAS PubMed.
- M. Piksa, C. Lian, I. C. Samuel, K. J. Pawlik, I. D. W. Samuel and K. Matczyszyn, Chem. Soc. Rev., 2023, 52, 1697–1722 RSC.
- M. A. Munegowda, A. Manalac, M. Weersink, S. A. McFarland and L. Lilge, Coord. Chem. Rev., 2022, 470, 214712 CrossRef PubMed.
- T. W. Rees, P.-Y. Ho and J. Hess, ChemBioChem, 2023, 24, e202200796 CrossRef CAS PubMed.
- L. Gourdon, K. Cariou and G. Gasser, Chem. Soc. Rev., 2022, 51, 1167–1195 RSC.
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS PubMed.
- M. D. Pozza, P. Mesdom, A. Abdullrahman, T. D. Prieto Otoya, P. Arnoux, C. Frochot, G. Niogret, B. Saubaméa, P. Burckel, J. P. Hall, M. Hollenstein, C. J. Cardin and G. Gasser, Inorg. Chem., 2023, 62, 18510–18523 CrossRef CAS PubMed.
- J. H. Correia, J. A. Rodrigues, S. Pimenta, T. Dong and Z. Yang, Pharmaceutics, 2021, 13, 1332 CrossRef CAS PubMed.
- G. Gunaydin, M. E. Gedik and S. Ayan, Front. Chem., 2021, 9, 691697 CrossRef CAS PubMed.
- B. M. Luby, C. D. Walsh and G. Zheng, Angew. Chem., Int. Ed., 2019, 58, 2558–2569 CrossRef CAS PubMed.
- L. K. B. Tam and D. K. P. Ng, Mater. Chem. Front., 2023, 7, 3184–3193 RSC.
- H. Kashida, H. Azuma, H. Sotome, H. Miyasaka and H. Asanuma, Angew. Chem., Int. Ed., 2024, 63, e202319516 CrossRef CAS PubMed.
- M. Martínez-Alonso and G. Gasser, Coord. Chem. Rev., 2021, 434, 213736 CrossRef.
- A. S. Boutorine, D. Brault, M. Takasugi, O. Delgado and C. Hélène, J. Am. Chem. Soc., 1996, 118, 9469–9476 CrossRef CAS.
- T. Kanamori, S. Kaneko, K. Hamamoto and H. Yuasa, Sci. Rep., 2023, 13, 288 CrossRef CAS PubMed.
- A. Yuan, B. Laing, Y. Hu and X. Ming, Chem. Commun., 2015, 51, 6678–6680 RSC.
- L. Chen, G. Li, X. Wang, J. Li and Y. Zhang, ACS Nano, 2021, 15, 11929–11939 CrossRef CAS PubMed.
- Y. Wu, L. Ding, C. Zheng, H. Li, M. Wu, Y. Sun, X. Liu, X. Zhang and Y. Zeng, Acta Biomater., 2022, 153, 419–430 CrossRef CAS PubMed.
- L. Shi, W. Wu, Y. Duan, L. Xu, Y. Xu, L. Hou, X. Meng, X. Zhu and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 19168–19174 CrossRef CAS PubMed.
- E. Kozma, M. Bojtár and P. Kele, Angew. Chem., Int. Ed., 2023, 62, e202303198 CrossRef CAS PubMed.
- M. Shang, Y. Wu, Y. Wang, Y. Cai, J. Jin and Z. Yang, Biomed. Pharmacother., 2022, 146, 112564 CrossRef CAS PubMed.
- C. Vever-Bizet, A. S. Boutorine, O. Delgado, D. Brault and C. Hélène, FEBS Lett., 1999, 462, 467–471 CrossRef CAS PubMed.
- L. K. McKenzie, M. Flamme, P. S. Felder, J. Karges, F. Bonhomme, A. Gandioso, C. Malosse, G. Gasser and M. Hollenstein, RSC Chem. Biol., 2022, 3, 85–95 RSC.
- R. J. Zhang, D. K. Ji, D. F. Yao, Z. Y. Xiao and W. H. Tan, CCS Chem., 2024, 6, 1240–1254 CrossRef CAS.
- Y.-A. Shieh, S.-J. Yang, M.-F. Wei and M.-J. Shieh, ACS Nano, 2010, 4, 1433–1442 CrossRef CAS PubMed.
- W. Hou, Y. Liu, Y. Jiang, Y. Wu, C. Cui, Y. Wang, L. Zhang, I. T. Teng and W. Tan, Nanoscale, 2018, 10, 10986–10990 RSC.
- J. Wang, G. Zhu, M. You, E. Song, M. I. Shukoor, K. Zhang, M. B. Altman, Y. Chen, Z. Zhu, C. Z. Huang and W. Tan, ACS Nano, 2012, 6, 5070–5077 CrossRef CAS PubMed.
- P. Mallikaratchy, Z. Tang and W. Tan, ChemMedChem, 2008, 3, 425–428 CrossRef CAS PubMed.
- J. Kim, W. Park, D. Kim, E. S. Lee, D. H. Lee, S. Jeong, J. M. Park and K. Na, Adv. Funct. Mater., 2019, 29, 1900084 CrossRef.
- N. De Laet and A. Madder, J. Photochem. Photobiol., A, 2016, 318, 64–70 CrossRef CAS.
- C. G. Pheeney and J. K. Barton, Langmuir, 2012, 28, 7063–7070 CrossRef CAS PubMed.
- G. Linden, L. Zhang, F. Pieck, U. Linne, D. Kosenkov, R. Tonner and O. Vázquez, Angew. Chem., Int. Ed., 2019, 58, 12868–12873 CrossRef CAS PubMed.
- N. Kuprikova, M. Ondruš, L. Bednárová, M. Riopedre-Fernandez, L. P. Slavětínská, V. Sýkorová and M. Hocek, Nucleic Acids Res., 2023, 51, 11428–11438 CrossRef CAS PubMed.
- M. Kuba, P. Khoroshyy, M. Lepšík, E. Kužmová, D. Kodr, T. Kraus and M. Hocek, Angew. Chem., Int. Ed., 2023, 62, e202307548 CrossRef CAS PubMed.
- G. Niogret, P. Röthlisberger, F. Levi-Acobas, F. Bonhomme, G. Gasser and M. Hollenstein, Synlett, 2024, 677–683 CrossRef CAS.
- D. Kodr, E. Kužmová, R. Pohl, T. Kraus and M. Hocek, Chem. Sci., 2023, 14, 4059–4069 RSC.
- P. Ghosh, H. M. Kropp, K. Betz, S. Ludmann, K. Diederichs, A. Marx and S. G. Srivatsan, J. Am. Chem. Soc., 2022, 144, 10556–10569 CrossRef CAS PubMed.
- A. Lozoya-Colinas, Y. Yu and J. C. Chaput, J. Am. Chem. Soc., 2023, 145, 25789–25796 CrossRef CAS PubMed.
- H. Sawai, A. Ozaki-Nakamura, M. Mine and H. Ozaki, Bioconjugate Chem., 2002, 13, 309–316 CrossRef CAS PubMed.
- M. Kuwahara, J.-i Nagashima, M. Hasegawa, T. Tamura, R. Kitagata, K. Hanawa, S.-i Hososhima, T. Kasamatsu, H. Ozaki and H. Sawai, Nucleic Acids Res., 2006, 34, 5383–5394 CrossRef CAS PubMed.
- J. Gierlich, K. Gutsmiedl, P. M. E. Gramlich, A. Schmidt, G. A. Burley and T. Carell, Chem. – Eur. J., 2007, 13, 9486–9494 CrossRef CAS PubMed.
- G. Niogret, A. Bouvier-Müller, C. Figazzolo, J. M. Joyce, F. Bonhomme, P. England, O. Mayboroda, R. Pellarin, G. Gasser, J. H. R. Tucker, J. A. Tanner, G. P. Savage and M. Hollenstein, ChemBioChem, 2024, 25, e202300539 CrossRef CAS PubMed.
- M. Welter, D. Verga and A. Marx, Angew. Chem., Int. Ed., 2016, 55, 10131–10135 CrossRef CAS PubMed.
- J. Balintová, M. Welter and A. Marx, Chem. Sci., 2018, 9, 7122–7125 RSC.
- F. Yang, M. Xu, X. Chen and Y. Luo, Biomed. Pharmacother., 2023, 164, 114933 CrossRef CAS PubMed.
- A. Hak, M. S. Ali, S. A. Sankaranarayanan, V. R. Shinde and A. K. Rengan, ACS Appl. Bio Mater., 2023, 6, 349–364 CrossRef CAS PubMed.
- H. Chen, R. G. W. Jinadasa, L. Jiao, F. R. Fronczek, A. L. Nguyen and K. M. Smith, Eur. J. Org. Chem., 2015, 3661–3665 CrossRef CAS PubMed.
- X. Guo, L. Wang, S. Wang, Y. Li, L. Cao, R. Cai and W. Zhao, Bioorg. Med. Chem. Lett., 2017, 27, 4548–4551 CrossRef CAS PubMed.
- T. Yogo, Y. Urano, Y. Ishitsuka, F. Maniwa and T. Nagano, J. Am. Chem. Soc., 2005, 127, 12162–12163 CrossRef CAS PubMed.
- C. Figliola, H. Anton, C. Sutter, C. Chériaux, A. Sutter, V. Mazan, M. Elhabiri, P. Didier, D. Jacquemin and G. Ulrich, ChemBioChem, 2023, 24, e202300139 CrossRef CAS PubMed.
- A. Da Lama, J. Pérez Sestelo, L. A. Sarandeses and M. M. Martínez, Org. Biomol. Chem., 2022, 20, 9132–9137 RSC.
- I. A. Nikolaeva, A. Y. Misharin, G. V. Ponomarev, V. P. Timofeev and Y. V. Tkachev, Bioorg. Med. Chem. Lett., 2010, 20, 2872–2875 CrossRef CAS PubMed.
- K. A. Carter, S. Wang, J. Geng, D. Luo, S. Shao and J. F. Lovell, Mol. Pharmaceutics, 2016, 13, 420–427 CrossRef CAS PubMed.
- J. Gonzales, N. V. S. D. K. Bhupathiraju, W. Perea, H. Chu, N. Berisha, V. Bueno, N. Dodic, J. Rozenberg, N. L. Greenbaum and C. M. Drain, Chem. Commun., 2017, 53, 3773–3776 RSC.
- A. Narumi, R. Rachi, H. Yamazaki, S. Kawaguchi, M. Kikuchi, H. Konno, T. Osaki, Y. Okamoto, X. Shen, T. Kakuchi, H. Kataoka, A. Nomoto, T. Yoshimura and S. Yano, ACS Omega, 2021, 6, 7023–7033 CrossRef CAS PubMed.
- J. Ashley, I. M. Potts and F. J. Olorunniji, ChemBioChem, 2023, 24, e202200510 CrossRef CAS PubMed.
- I. Sarac and M. Hollenstein, ChemBioChem, 2019, 20, 860–871 CrossRef CAS PubMed.
- E. A. Motea and A. J. Berdis, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1151–1166 CrossRef CAS PubMed.
- M. Flamme, S. Hanlon, I. Marzuoli, K. Püntener, F. Sladojevich and M. Hollenstein, Commun. Chem., 2022, 5, 68 CrossRef CAS PubMed.
- M. Flamme, D. Katkevica, K. Pajuste, M. Katkevics, N. Sabat, S. Hanlon, I. Marzuoli, K. Püntener, F. Sladojevich and M. Hollenstein, Asian J. Org. Chem., 2022, 11, e202200384 CrossRef CAS.
- N. Sabat, D. Katkevica, K. Pajuste, M. Flamme, A. Stämpfli, M. Katkevics, S. Hanlon, S. Bisagni, K. Püntener, F. Sladojevich and M. Hollenstein, Front. Chem., 2023, 11, 1161462 CrossRef CAS PubMed.
- H. H. Lee, R. Kalhor, N. Goela, J. Bolot and G. M. Church, Nat. Commun., 2019, 10, 2383 CrossRef PubMed.
- J. Bowers, J. Mitchell, E. Beer, P. R. Buzby, M. Causey, J. W. Efcavitch, M. Jarosz, E. Krzymanska-Olejnik, L. Kung, D. Lipson, G. M. Lowman, S. Marappan, P. McInerney, A. Platt, A. Roy, S. M. Siddiqi, K. Steinmann and J. F. Thompson, Nat. Methods, 2009, 6, 593–595 CrossRef CAS PubMed.
- S. Palluk, D. H. Arlow, T. de Rond, S. Barthel, J. S. Kang, R. Bector, H. M. Baghdassarian, A. N. Truong, P. W. Kim, A. K. Singh, N. J. Hillson and J. D. Keasling, Nat. Biotechnol., 2018, 36, 645–650 CrossRef CAS PubMed.
- I. Randrianjatovo-Gbalou, S. Rosario, O. Sismeiro, H. Varet, R. Legendre, J.-Y. Coppée, V. Huteau, S. Pochet and M. Delarue, Nucleic Acids Res., 2018, 46, 6271–6284 CrossRef CAS PubMed.
- J. Vacacela, A.-L. Schaap-Johansen, P. Manikova, P. Marcatili, M. Prado, Y. Sun and J. Ashley, Angew. Chem., Int. Ed., 2022, 61, e202201061 CrossRef CAS PubMed.
- J. Ashley, A.-L. Schaap-Johansen, M. Mohammadniaei, M. Naseri, P. Marcatili, M. Prado and Y. Sun, Nucleic Acids Res., 2021, 49, 1065–1074 CrossRef CAS PubMed.
- P. Horáková, H. Macíčková-Cahová, H. Pivoňková, J. Špaček, L. Havran, M. Hocek and M. Fojta, Org. Biomol. Chem., 2011, 9, 1366–1371 RSC.
- P. Röthlisberger, F. Levi-Acobas, I. Sarac, B. Baron, P. England, P. Marlière, P. Herdewijn and M. Hollenstein, Chem. Commun., 2017, 53, 13031–13034 RSC.
- M. Ondruš, V. Sýkorová and M. Hocek, Chem. Commun., 2022, 58, 11248–11251 RSC.
- B. H. Qin, Q. Wang, Y. Wang, F. Han, H. Y. Wang, S. X. Jiang and H. Y. Yu, Angew. Chem., Int. Ed., 2024, 63, e202317334 CrossRef CAS PubMed.
- P. Güixens-Gallardo, M. Hocek and P. Perlíková, Bioorg. Med. Chem. Lett., 2016, 26, 288–291 CrossRef PubMed.
- A. V. Chudinov, V. A. Vasiliskov, V. E. Kuznetsova, S. A. Lapa, N. A. Kolganova and E. N. Timofeev, Sci. Rep., 2021, 11, 2423 CrossRef CAS PubMed.
- N. Sabat, A. Stämpfli, M. Flamme, S. Hanlon, S. Bisagni, F. Sladojevich, K. Püntener and M. Hollenstein, Chem. Commun., 2023, 59, 14547–14550 RSC.
- C. Figazzolo, F. Bonhomme, S. Saidjalolov, M. Ethève-Quelquejeu and M. Hollenstein, Molecules, 2022, 27, 8927 CrossRef CAS PubMed.
- M. Ondruš, V. Sýkorová, L. Bednárová, R. Pohl and M. Hocek, Nucleic Acids Res., 2020, 48, 11982–11993 CrossRef PubMed.
- D. M. Perrin, T. Garestier and C. Hélène, J. Am. Chem. Soc., 2001, 123, 1556–1563 CrossRef CAS PubMed.
- J. D. Vaught, C. Bock, J. Carter, T. Fitzwater, M. Otis, D. Schneider, J. Rolando, S. Waugh, S. K. Wilcox and B. E. Eaton, J. Am. Chem. Soc., 2010, 132, 4141–4151 CrossRef CAS PubMed.
- S. W. Santoro, G. F. Joyce, K. Sakthivel, S. Gramatikova and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2433–2439 CrossRef CAS PubMed.
- A. Hottin and A. Marx, Acc. Chem. Res., 2016, 49, 418–427 CrossRef CAS PubMed.
- B. N. Gawande, J. C. Rohloff, J. D. Carter, I. von Carlowitz, C. Zhang, D. J. Schneider and N. Janjic, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2898–2903 CrossRef CAS PubMed.
- T. R. Battersby, D. N. Ang, P. Burgstaller, S. C. Jurczyk, M. T. Bowser, D. D. Buchanan, R. T. Kennedy and S. A. Benner, J. Am. Chem. Soc., 1999, 121, 9781–9789 CrossRef CAS PubMed.
- A. V. Sidorov, J. A. Grasby and D. M. Williams, Nucleic Acids Res., 2004, 32, 1591–1601 CrossRef CAS PubMed.
- Y. W. Cheung, P. Röthlisberger, A. E. Mechaly, P. Weber, F. Levi-Acobas, Y. Lo, A. W. C. Wong, A. B. Kinghorn, A. Haouz, G. P. Savage, M. Hollenstein and J. A. Tanner, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 16790–16798 CrossRef CAS PubMed.
- S. Jäger, G. Rasched, H. Kornreich-Leshem, M. Engeser, O. Thum and M. Famulok, J. Am. Chem. Soc., 2005, 127, 15071–15082 CrossRef PubMed.
- J. Siegl, O. Plückthun and G. Mayer, RSC Chem. Biol., 2022, 3, 288–294 RSC.
- M. Hollenstein, Chem. – Eur. J., 2012, 18, 13320–13330 CrossRef CAS PubMed.
- F. Pfeiffer, F. Tolle, M. Rosenthal, G. M. Brändle, J. Ewers and G. Mayer, Nat. Protoc., 2018, 13, 1153–1180 CrossRef CAS PubMed.
- E. M. Kohn, K. Konovalov, C. A. Gomez, G. N. Hoover, A. K.-H. Yik, X. Huang and J. D. Martell, ACS Chem. Biol., 2023, 18, 1976–1984 CrossRef CAS PubMed.
- J. S. Temme, I. S. MacPherson, J. F. DeCourcey and I. J. Krauss, J. Am. Chem. Soc., 2014, 136, 1726–1729 CrossRef CAS PubMed.
- Z. Tao, H. Zhang, S. Wu, J. Zhang, Y. Cheng, L. Lei, Y. Qin, H. Wei and C.-Y. Yu, Nanoscale, 2024, 16, 4392–4406 RSC.
- M. J. S. A. Silva, Y. Zhang, R. Vinck, F. M. F. Santos, J. P. M. António, L. Gourdon-Grünewaldt, C. Zaouter, A. Castonguay, S. A. Patten, K. Cariou, F. Boscá, F. Nájera, J. F. Arteaga, G. Gasser, U. Pischel and P. M. P. Gois, Bioconjugate Chem., 2023, 34, 2337–2344 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All synthetic protocols, additional gel images, LCMS characterization of single incorporation events, and NMR spectra. In addition, the authors have cited additional references.1–3 See DOI: https://doi.org/10.1039/d4cb00103f |
| This journal is © The Royal Society of Chemistry 2024 |