
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of a new inhibitor for YTH domain-containing m6A RNA readers†
Chuan-Hui
Wang
 and
Huiqing
Zhou
*
and
Huiqing
Zhou
*
Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, MA 02467, USA. E-mail: zhouaf@bc.edu
First published on 25th July 2024
Abstract
N 6-methyladenosine (m6A) is an abundant modification in mammalian mRNAs and plays important regulatory roles in gene expression, primarily mediated through specific recognition by “reader” proteins. YTH family proteins are one major family of known m6A readers, which specifically recognize m6A-modified transcripts via the YTH domains. Despite the significant relevance of YTH-m6A recognition in biology and diseases, few small molecule inhibitors are available for specifically perturbing this interaction. Here we report the discovery of a new inhibitor (“N-7”) for YTH-m6A RNA recognition, from the screening of a nucleoside analogue library against the YTH domain of the YTHDF1 protein. N-7 is characterized to be a pan-inhibitor in vitro against five YTH domains from human YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2 proteins, with IC50 values in the range of 30–48 μM measured using a fluorescence polarization competition assay. We demonstrated that N-7 directly interacts with the YTH domain proteins via a thermal shift assay. N-7 expands the chemical structure landscape of the m6A YTH domain-containing reader inhibitors and potentiates future inhibitor development for reader functional studies and therapeutic efforts in targeting the epitranscriptome.
Introduction
N 6-methyladenosine (m6A) is one of the most abundant internal chemical modifications in eukaryotic messenger RNAs (mRNAs)1–4 and it exerts important regulatory functions in mRNA metabolism and gene expression, primarily mediated through direct or indirect recognition by m6A “readers”.5–13 The m6A “readers” refer to RNA binding proteins that bind to m6A-modified RNAs with much greater affinity than to unmodified RNAs.14,15 The YT521-B homology (YTH) family of proteins, including YTHDF1-3, YTHDC1, and YTHDC2, are the first reported and the most extensively studied m6A readers.3,7 YTH family proteins contain a YTH domain at or close to the C-termini of the full-length protein, which is responsible for m6A RNA recognition (Fig. 1A). In vitro binding studies showed that the purified YTH domains from YTHDF1-3 and YTHDC1 bind m6A-modified RNA oligonucleotides with micromolar binding affinities that are 10–100 fold tighter than binding with unmodified RNAs that carry the same sequences.16,17 High-resolution structures of the recognition complexes between m6A RNA and the YTH domains revealed that YTH domains directly recognize m6A through a hydrophobic binding pocket that contains two or three tryptophan residues (Fig. 1A); additionally, the YTH domain also presents a cleft that is rich in basic residues that present electrostatic interactions with the RNA backbone.14,17–23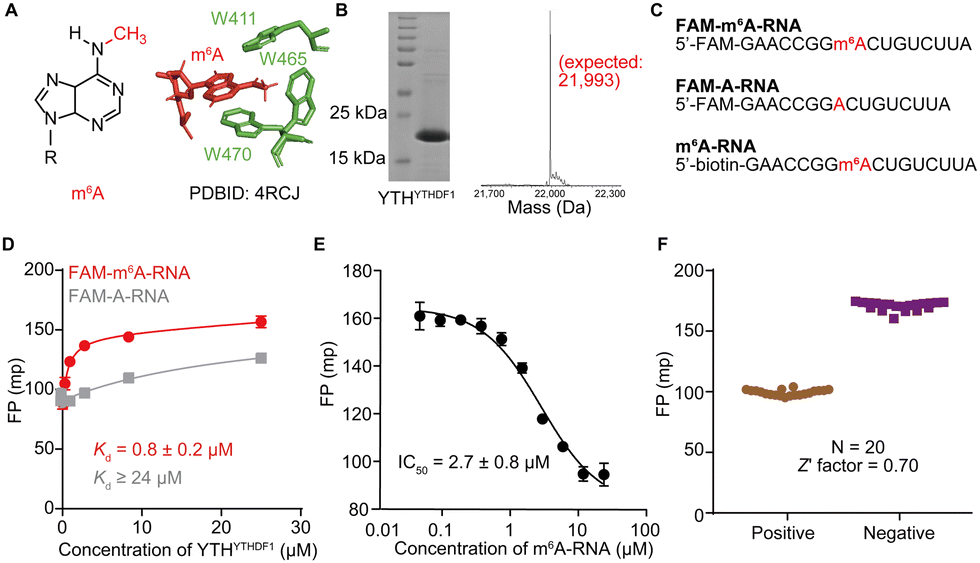 | ||
| Fig. 1 Screening YTHYTHDF1 inhibitors by FP competition assay. (A) The chemical structure of m6A (left) and hydrophobic binding pocket of YTHYTHDF1 (right). (B) SDS-PAGE and MS characterizations of the purified YTHYTHDF1 protein. The gel image of the purified protein with Coomassie Blue staining (left) and the deconvolution mass data (right) and the theoretical expected mass (in red) of the purified protein are shown. (C) Sequences of three RNA oligonucleotides: FAM-m6A-RNA, FAM-A-RNA, and m6A-RNA were used in the FP assay. (D) The FP binding data and fitted binding curves for YTHYTHDF1 and FAM-m6A-RNA or FAM-A-RNA. Data are presented as mean ± standard deviation (SD) with n = 3 biological replicates. (E) The displacement of FAM-m6A-RNA from YTHYTHDF1 by the non-fluorescent m6A-RNA by the FP competition assay with the fitted IC50 value. Data are presented as mean ± SD with n = 3 biological replicates. (F) The data for the determination of Z′ factor based on N = 20 repeated measurements of the FP competition assay for screening. | ||
Alongside the m6A recognition by the YTH domain, the rest of the regions in the YTH family proteins are involved in sophisticated protein–protein interactions in cells and exert diverse functional consequences of m6A RNA recognition by readers. For example, Du et al. showed that the N-terminal domain of human YTHDF2 protein directly interacts with the scaffolding subunit CNOT1 in the CCR4–NOT deadenylase complex via co-immunoprecipitation assays, which promotes the deadenylation-dependent mRNA decay.18 This interaction accelerates the decay of m6A-modified mRNAs. Meanwhile, the N-terminal domain of YTHDF2 was reported to interact with other proteins including the heat-responsive protein 12 (HRSP12) and the upstream frameshift 1 (UPF1) protein.19,20 Both interactions between YTHDF2 and HRSP12 or UPF1 promote the decay of m6A-modified mRNAs through internal cleavage or nonsense-mediated RNA decay, respectively.19,20 Besides YTHDF2, the functions of YTH family proteins are involved in many biological transactions, including RNA decay,10 nuclear export,8 RNA splicing,21 translation,10,11 transcription,13 and the formation and functions of large ribonucleoprotein complex assemblies such as stress granules and P bodies.18,22–24
In addition to the intricate protein–protein interaction network that YTH proteins are involved in, the YTH domains were reported to recognize substrates beyond m6A RNA in vitro and in cellulo, including m1A25,26 and N6-methyldeoxyadenosine (6mA) in DNA.27 All these factors complicate the studies of the precise functional roles of YTH family proteins and chemical modifications in physiology and diseases. Indeed, dysregulated expression levels of YTH proteins frequently occur in embryonic development,13 neuronal development,28 memory formation,29 multiple aspects of cancer biology,30,31 and viral infections.32 The current experimental strategies to study the biological consequences of YTH family proteins primarily rely on the knockdown, knockout, or overexpression of a YTH protein of interest.33,34 Such perturbations will concurrently interrupt all interactions the protein may have, making it inconclusive to reason whether the YTH–m6A recognition is causative for the observed biological consequence.
The development of small molecule inhibitors that specifically perturb the YTH–m6A recognition will provide invaluable insights to dissect the sophisticated interaction network YTH proteins involve and reveal m6A-causitive YTH functions. Indeed, a recent report by Zou et al. identified a natural product salvianolic acid C (SAC) as a selective inhibitor for m6A-RNA and YTHYTHDF1 with a 20-fold higher observed inhibitory effect compared to YTHYTHDF2via AlphaScreen assays in vitro,35 although the molecular mechanism of the observed selectivity remains to be characterized. SAC contributed to elucidating the function of YTHDF1 in the formation of ribonucleoprotein condensations and translation regulation in neurons.35 Aside from SAC, only a handful of other inhibitors have been reported for YTH and m6A recognition.36–42 It remains in great demand to explore broader chemical space for inhibitor discoveries and to carry out in-depth characterization of inhibitor selectivity against different YTH family proteins.
Here we report the discovery of an inhibitor for m6A–YTH recognition, resulting from the screening of a commercially available nucleoside analogue library against the YTH domain of YTHDF1 protein. Very interestingly, our screening data identified lead compound structures resembling adenosine, with modifications on the ribose moiety and the N6 position. We chose to further characterize a representative compound N-7, which showed a pan-inhibitory effect of m6A RNA–YTH recognition for five YTH domains of YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2 proteins with IC50 values ranging from 30 to 48 μM. We demonstrated that the inhibitory activity results from the direct interaction between N-7 and the YTH domain proteins via thermal shift assay. We systematically compared the inhibitory activity and selectivity against different YTH domains of N-7 to four reported YTH domain inhibitors (SAC,35m6A nucleoside,36,37 compound 6,36 and compound 2638). The comparison revealed that N-7 showed competitive inhibitory activity among the reported inhibitors and presented unique pan-inhibitory activity against the five YTH domains.
Results and discussion
Screening of inhibitors for YTHYTHDF1-m6A RNA recognition
Firstly, we confirmed that the prepared YTH domain of YTHDF1 (denoted as YTHYTHDF1) was an m6A reader by measuring the binding affinities of the protein with the synthetic oligonucleotides via the fluorescence polarization (FP) binding assay. We over-expressed and purified YTHYTHDF1 protein with an N-terminal His tag from E. coli cells, and validated the size of the purified protein via SDS-PAGE and mass spectrometry (MS) (Fig. 1B and Fig. S1, ESI†). The purified YTHYTHDF1 showed binding to the 16-nucleotide (nt) FAM-labeled m6A-containing oligonucleotide (“FAM-m6A-RNA”) with Kd = 0.8 ± 0.2 μM measured using the FP binding assay (Fig. 1C and D), consistent with the previously reported affinity (Kd = 1.0 ± 0.3 μM) measured using isothermal titration calorimetry (ITC).14 Meanwhile, we observed significantly weakened binding (Kd ≥ 24 μM) between the YTHYTHDF1 and unmodified RNA with the same sequence (“FAM-A-RNA”) by the FP assay (Fig. 1C and D). During the titration of the purified YTHYTHDF1 up to 24 μM in the FP binding assay, the total fluorescence intensities (i.e. the sum of the parallel and perpendicular fluorescence intensities) produced by the FAM-labeled RNA oligonucleotides remain stable (Fig. S2A, ESI†), suggesting the FP assay is a suitable assay to quantify the binding between the used RNA oligonucleotides and the YTH domain.To identify small molecule inhibitors of YTH-domain proteins, we set up an FP competition assay with the purified YTHYTHDF1 for the screening. Briefly, 25 nM of FAM-m6A-RNA was pre-incubated with 2 μM (greater than 2 times the measured Kd) YTHYTHDF1; an increasing concentration of a non-fluorescent “m6A-RNA” (Fig. 1C) was titrated into the pre-formed FAM-m6A-RNA:YTHYTHDF1 complex, to compete for binding. When the FAM-m6A-RNA was competed off the RNA–protein complex, it became free RNA with reduced FP. As expected, we observed decreased FP upon increasing concentrations of the non-fluorescent m6A-RNA, with an IC50 = 2.7 ± 0.8 μM (Fig. 1E). Next, we used the non-fluorescent m6A-RNA and blank binding buffer as the “positive control” and “negative control” competitors to perform the FP competition assay for 20 repeats and the repeated assays generated a Z′ factor = 0.7,43 demonstrating the excellent performance of the FP competition assay for screening (Fig. 1F). Additionally, we confirmed that the FP signal was not affected by the addition of 5% DMSO as small molecule libraries were dissolved in 100% DMSO (Fig. S2B, ESI†).
We conducted screening of 320 compounds from a commercially available nucleoside analogue library with the FP competition assay; excitingly, we identified several compounds showing reduced FP relative to the DMSO control when added to the pre-formed FAM-m6A-RNA:YTHYTHDF1 complex at the final compound concentration of 40 μM during the screening, suggesting them as potential binding inhibitors (Fig. 2). Next, we followed up with the top 13 compounds that showed at least a 35% reduction of the FP signal relative to the DMSO control (Fig. 2). Interestingly, the chemical structures of all 13 compounds showed as analogues of adenosine with chemical modifications at the N9 and the N6 of adenine (Fig. S3A, ESI†). Moreover, 7 out of the 13 compounds (N-7, H-14, D-20, D-15, B-16, H-15, N-12) shared the same bis-hydroxymethyl cyclobutyl group at the N9 position (Fig. S3A, ESI†), which increased our confidence in the relevance of the identified structures. We confirmed the reproducibility of the screening results by running the FP competition assay with higher compound dosages at 200 μM with replicates. The 13 compounds all showed a reduction of the FP signal in a dose–response manner (Fig. S3B, ESI†). We chose the compound “N-7”, one of the compounds that showed the most significant inhibitory effect and did not interfere with the total fluorescence intensity of the assay, for further characterizations (Fig. 3A and Fig. S3B, C, ESI†). Meanwhile, we chose the compound “D-6” in the screened library as a negative control. D-6 shares the same purine and the [1-(hydroxymethyl)cyclobutyl]methanol structures with N-7 but contains a 3-propan-2-yl-2-azaspiro[3.5]nonane group, rather than the 2,7-diazaspiro[4.4]nonan-8-one group in N-7 (Fig. 3A). We performed dose–response curve measurements to quantify the inhibitory activity (i.e. IC50) of N-7via the FP competition assay. N-7 (Fig. S4, ESI†) shows an inhibitory effect on the YTHYTHDF1 and FAM-m6A-RNA binding with an IC50 value of 39 ± 3 μM, whereas D-6 shows no detectable competition (Fig. 3B).
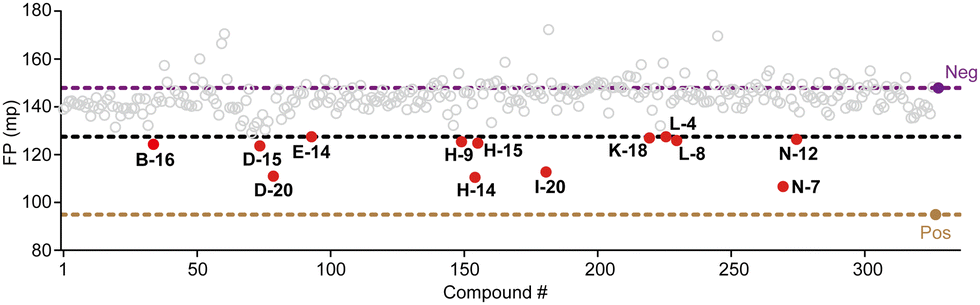 | ||
| Fig. 2 Screening YTHYTHDF1 inhibitors using the FP competition assay. The data of the screening against 320 compounds from the nucleoside mimetics library are shown. The concentration of each compound is 40 μM with two technical replicates. The FP values measured for the positive (“m6A-RNA”) and negative controls (“DMSO”) are shown in brown and purple. Compounds are shown in red if they show at least a 35% reduction of FP (cut-off indicated by the black dashed line) relative to the negative control. These compounds are labeled with their IDs in the original library. | ||
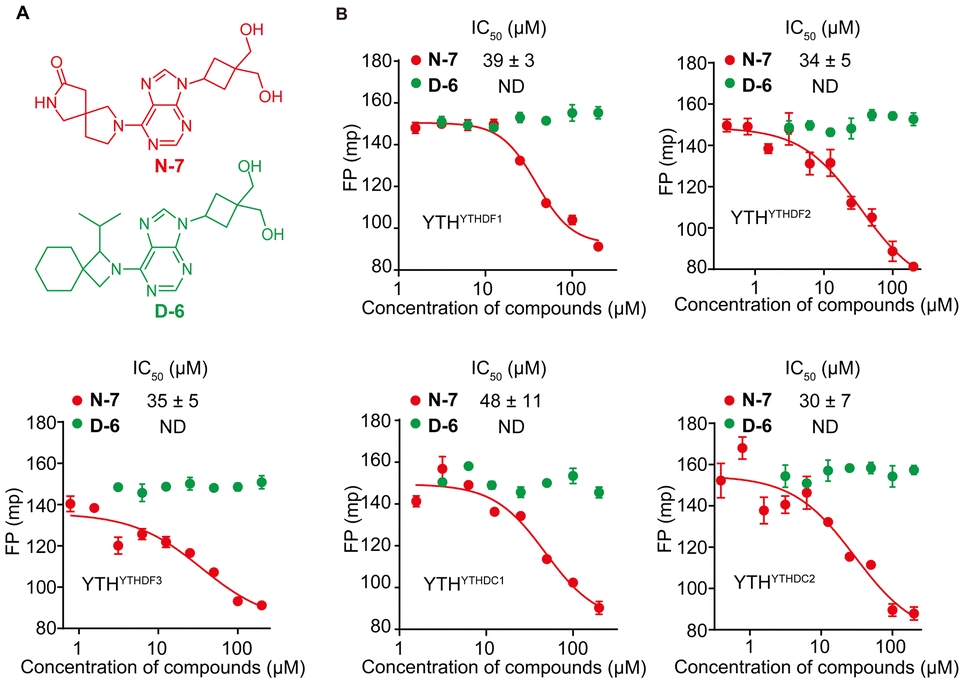 | ||
| Fig. 3 N-7 is a pan-YTH domain inhibitor. (A) The chemical structures of N-7 and D-6. (B) Dose–response curves for the inhibitory activities of N-7 and D-6 against YTHYTHDF1, YTHYTHDF2, YTHYTHDF3, YTHYTHDC1, and YTHYTHDC2 proteins, measured using the FP competition assay. Data are presented as mean ± SD (n = 3 replicates). “ND” denotes “not detected by FP competition assay”. | ||
N-7 is a pan-YTH-domain inhibitor
Next, we proceeded to examine the selectivity of N-7 against different YTH domains. We over-expressed and purified the YTH domains of human YTHDF2 (YTHYTHDF2), YTHDF3 (YTHYTHDF3), YTHDC1 (YTHYTHDC1), and YTHDC2 (YTHYTHDC2) proteins (Fig. S5–S8, ESI†). First, we confirmed the purified YTHYTHDF2, YTHYTHDF3, YTHYTHDC1, and YTHYTHDC2 as m6A readers in vitro by the FP binding assay. The measured binding affinities for all the YTH domains against the m6A-modified RNA agree with the reported values,7,14,17,44 and were significantly higher relative to the binding with the unmodified RNA (Fig. S9A, ESI†). We further benchmarked the FP competition assay for all purified YTH domain proteins with the non-fluorescent m6A-RNA as the positive control competitor. The results demonstrated that the non-fluorescent m6A-RNA competes effectively with the FAM-m6A-RNA in complex with each of the YTH proteins (Fig. S9B, ESI†). We proceeded to test the inhibitory activity for N-7 against m6A-RNA bound with different YTH domains. Interestingly, N-7 exhibited comparable inhibitory activities against all purified YTH domains, with IC50 = 39 ± 3 μM for YTHYTHDF1, 34 ± 5 μM for YTHYTHDF2, 35 ± 5 μM for YTHYTHDF3, 48 ± 11 μM for YTHYTHDC1, and 30 ± 7 μM for YTHYTHDC2 measured by the FP competition assay (Fig. 3B). The IC50 values for N-7 against multiple YTH domains were robustly measured when altering the protein concentration or the order of reagents addition applied in the FP competition assay (Fig. S10, ESI†). In contrast, the negative control compound D-6 did not show competition against m6A RNA binding to all YTH-domain proteins (Fig. 3B). To further validate the measurements by the FP competition assay, we developed the RNA electrophoretic mobility shift assay (REMSA)45 to assess inhibitory activity of small molecule inhibitors for YTH-m6A RNA binding. We measured the inhibitory activity of N-7 against YTH domains of YTHDF1 and YTHDC1 using REMSA, which yielded an IC50 value of 45 ± 5 μM for YTHYTHDF1 and 30 ± 2 μM for YTHYTHDC1 (Fig. S11, ESI†). The REMSA results agree with those measured by the FP competition assay within error (Fig. 3B).Given that N-7 is a pan-inhibitor for YTH domains, we investigate its inhibition selectivity against other m6A RNA recognition proteins that do not contain a YTH domain. We chose to test FTO, one of the eraser proteins that recognize m6A RNA and catalyze the removal of the methylation of m6A in the presence of co-factors Fe2+ and α-ketoglutarate.46 We expressed and purified the FTO protein and confirmed binding between the purified FTO protein and FAM-m6A-RNA with the FP assay in the absence of catalytic co-factors (Fig. S12A–D, ESI†). We benchmarked the FP competition assay by measuring the inhibitory activity of a reported FTO inhibitor FB23,47 which shows IC50 of 0.6 ± 0.2 μM (Fig. S12E, ESI†). In contrast, N-7 did not show any detectable inhibition against FTO binding with m6A-RNA (Fig. S12F, ESI†). This suggests that N-7 shows selective binding to YTH domain proteins over FTO.
Together, these findings show that N-7 is a pan-inhibitor for the recognition between YTH domains and m6A RNA, and presents selective inhibition against YTH domains over a non-YTH m6A recognition protein FTO.
N-7 directly interacts with YTH-domain proteins
To probe the inhibitory mechanism, we assessed interactions between N-7 and FAM-m6A-RNA or the YTH domain proteins separately. On the one hand, we observed no apparent binding between N-7 and FAM-m6A-RNA by directly titrating N-7 into a constant concentration of FAM-m6A-RNA via the FP binding assay (Fig. 4A). On the other hand, we investigated the interaction between N-7 and YTH domain proteins via the thermal shift assay.48 As a positive control, we measured the thermal stabilization effect of the YTHYTHDF1 protein upon binding to m6A-RNA; incubation of 2 μM or 16 μM m6A-RNA significantly elevated the melting temperature of YTHYTHDF1 by 1.4 °C or 13.3 °C, respectively (Fig. S13, ESI†). Next, we characterized the effects of N-7 incubated with the five YTH domains. N-7 exhibited thermal stabilizations of all five YTH domain proteins; when 6 μM N-7 was incubated, the melting temperatures of the YTH domain proteins increased by 0.4–0.8 °C relative to the DMSO control (Fig. 4B and Fig. S14, ESI†). The stabilization effect became more prominent when an increasing amount of N-7 (50 μM) was added (Fig. 4B and Fig. S14, ESI†). In contrast, the negative control compound D-6 did not induce significant changes in the melting temperatures of the YTH domain proteins (Fig. S15, ESI†). | ||
| Fig. 4 N-7 directly interacts with YTH-domain proteins. (A) The FP binding data between N-7 and the FAM-m6A-RNA. Data are presented as mean ± SD with n = 3 biological replicates and no binding was observed. (B) The results of the thermal shift assay performed on YTHYTHDF1, YTHYTHDF2, YTHYTHDF3, YTHYTHDC1, or YTHYTHDC2 proteins in the presence of compound N-7 with varying concentrations. Data are presented as mean ± SD with n = 3 biological replicates. The two-tailed t-test was used to assess the statistical significance of the difference between two samples, with the p-value indicated as “ns” for p ≥ 0.05, “*” for p < 0.05, “**” for p < 0.01, or “***” for p < 0.001. | ||
The thermal shift assay results suggested that N-7 inhibited the YTH and m6A RNA recognition likely through directly interacting with the YTH-domain proteins, rather than the RNA. The magnitude of melting temperature elevation caused by N-7 is significantly lower than that caused by the m6A-RNA binding; this is consistent with less favorable binding energetics between N-7 and the YTH domains compared to the modified RNA. We reason that this change in energetics can result from weakened hydrogen bonding, van der Waals, charge–charge interactions, and/or a less favored hydrophobic effect for small molecule binding to the protein relative to the RNA.14
To gain further perspectives into potential structure–activity relationships (SAR), we performed an enrichment analysis of structure motifs in N-7, regarding the occurrence frequency of each motif in the overall library (320 compounds) versus that in the identified hits (13 compounds shown in Fig. S16, ESI†). Motif 1 (i.e. the purine) appears in 53% of compounds in the overall library and 100% of the screening hits with ∼2-fold enrichment. This suggests that purine is an important fragment for the inhibitor structure, compared to pyrimidine-derived structures within the library. Despite the importance of purine, we found no inhibitory activity for the 6-methylamino purine (Fig. S16A, ESI†). This suggests the purine structure alone is not sufficient for presenting potency. Motif 2 (i.e. purine with the [1-(hydroxymethyl)cyclobutyl]methanol group) occurs in 10% of the overall library, of which the occurrence gets significantly enriched by >5-fold within the screening hits, where 54% (7 out of 13) of the hit compounds contain the Motif 2 structure. This suggests that Motif 2 can be critical for the binding with YTH domains. Only one compound (i.e.N-7) contains Motif 3 (i.e. purine with 2,7-diazaspiro[4,4]nonan-8-one) in the overall library (Fig. S16C, ESI†). It is difficult to assess any enrichment of this motif. Among the 13 hit structures, Motif 3-equivalent positions showed greater variability than Motif 2, suggesting a certain tolerance of chemical diversity for the substitutes at the C6 of the purine (Fig. S3A, ESI†).
Lastly, we conducted molecular docking of N-7 onto the reported high-resolution structure of the YTH domain of YTHDF1 protein (PDBID: 4RCJ) to speculate the potential binding mode.14 The best docking pose revealed that N-7 occupies the m6A binding pocket; the [1-(hydroxymethyl)cyclobutyl]methanol group of N-7 adopts the same orientation as the 6-methyl group of m6A (Fig. S17, ESI†). The docking result suggests potential polar contacts between two hydroxyl groups of N-7 with the backbone NH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of C412, and the side chain of D401 (Fig. S17, ESI†). The suggested binding pose indicates the critical role of Motif 2 for the compound binding to the YTH domain. Motif 3 orients outwards relative to the binding pocket, which is consistent with the chemical diversity we observed among the hit structures (Fig. S3A, ESI†).
O groups of C412, and the side chain of D401 (Fig. S17, ESI†). The suggested binding pose indicates the critical role of Motif 2 for the compound binding to the YTH domain. Motif 3 orients outwards relative to the binding pocket, which is consistent with the chemical diversity we observed among the hit structures (Fig. S3A, ESI†).
The motif enrichment analysis of the screening results and molecular docking suggested Motif 2 can be a critical motif for binding to the YTH domain, whereas the spiro group of N-7 can be more tolerating of diverse chemical groups. Future efforts in resolving the atomic-resolution structures of the N-7/YTH domain protein complexes and synthesizing N-7 fragments and analogue compounds are critical for providing insights into the mechanism of inhibition and elucidating the structure–activity relationship.
Comparison of N-7 with reported YTH domain inhibitors
Next, we compared the inhibitory activity and selectivity of N-7 with four reported YTH domain inhibitors that are commercially available, including the natural product “SAC”,35 the “m6A nucleoside”,36,37 a pyrazolopyrimidine derivative compound “6”,36 and an indazole derivative compound “26”.38 These compounds were reported to inhibit YTH-m6A recognition against a single or subset of YTH domains in the literature, and the inhibitory activities (i.e. IC50) were measured using different assays including AlphaScreen35 and the homogeneous time-resolved fluorescence (HTRF) assay.36–38 As the IC50 values are difficult to compare when measured using different assay conditions, we systematically measured the inhibitory activities of four reported inhibitors with the same FP competition assay against five YTH domains (Table 1 and Fig. S18, ESI†).| Compound name | Structure | IC50 (μM) | ||||
|---|---|---|---|---|---|---|
| YTHYTHDF1 | YTHYTHDF2 | YTHYTHDF3 | YTHYTHDC1 | YTHYTHDC2 | ||
| a Reported IC50 value of SAC measured by the AlphaScreen assay in ref. 35. b Reported IC50 value of m6A nucleoside measured by the HTRF assay in ref. 36 and 37. c Reported IC50 value of compound 6 measured by the HTRF assay in ref. 36 and 37. d Reported IC50 value of compound 26 measured by the HTRF assay in ref. 38. e ND, not detected by FP competition assay. | ||||||
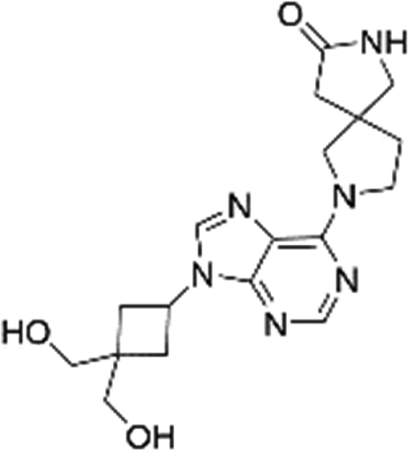
| 2-[9-[3,3-Bis(hydroxymethyl) cyclobutyl]purin-6-yl]-2,7-diazaspiro[4,4]nonan-8-one (N-7) |
|
39 ± 3 | 34 ± 5 | 35 ± 5 | 48 ± 11 | 30 ± 7 |
| (2R)-3-(3,4-Dihydroxyphenyl)-2-[(E)-3-[2-(3,4-dihydroxyphenyl)-7-hydroxy-1-benzofuran-4-yl]prop-2-enoyl]oxypropanoic acid (SAC)35 |
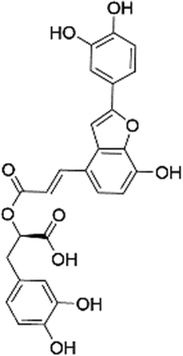
|
3.1 ± 2.5 | 6.0 ± 3.5 | 106 ± 41 | 29 ± 4 | 160 ± 42 |
| 1.4 ± 0.2a | 29.6 ± 3.7a | |||||
| (2R,3S,4R,5R)-2-(Hydroxymethyl)-5-[6-(methylamino)-9H-purin-9-yl]oxolane-2,3-diol (m6A nucleoside)36,37 |
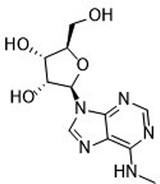
|
NDe | 295 ± 84 | ND | 2342 ± 896 | ND |
| 504b | 144b | |||||
| N,3-Dimethyl-2H-pyrazolo[4,3-d]pyrimidin-7-amine (6)36 |
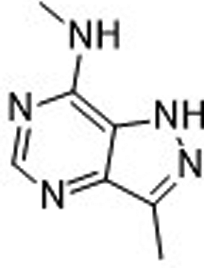
|
ND | ND | ND | 2000 ± 1452 | ND |
| 410c | >1000c | 39c | ||||
| N-Methyl-1H-indazole-3-carboxamide (26)38 |
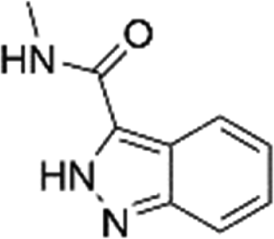
|
ND | ND | ND | 3980 ± 1376 | ND |
| 300d | 133d | 443d | 155d | |||
Based on our FP competition assay results, SAC exhibited inhibitory activity with an IC50 of 3.1 ± 2.5 μM against YTHYTHDF1, consistent with the reported IC50 value of 1.4 ± 0.2 μM within error measured by the AlphaScreen Assay (Table 1 and Fig. S18A, ESI†).35 Our data suggest a weak selectivity of SAC against YTHYTHDF2, which showed two times higher IC50 compared to YTHYTHDF1. Interestingly, we observed strong selectivity of SAC against YTHYTHDF3, YTHYTHDC1, and YTHYTHDC2 with IC50 values ranging from 29 to 160 μM (Table 1 and Fig. S18A, ESI†). Unlike N-7 being a pan-inhibitor, SAC presents selectivity among different YTH domains.35 This immediately suggests different binding modes of SAC with YTH domains from those of N-7; high-resolution structural and biochemical characterizations would be critical to elaborate the differences in molecular recognition.
The m6A nucleoside, compound 6, and compound 26 were identified and validated as binding fragments to YTHYTHDF2 and YTHYTHDC1 by high-throughput X-ray crystallography to aid structure-based inhibitor design for YTH domain proteins.36–38 These fragments show relatively weak inhibitory activities with the IC50 values reported within the sub-millimolar to millimolar range as measured using the HTRF assay38 (Table 1). Our measurements showed very weak inhibitory activities of the m6A nucleoside, 6 and 26 against all five YTH domains (IC50 > 2 mM or not detectable), except that the m6A nucleoside shows an IC50 of 295 ± 84 μM against YTHYTHDF2 (Fig. S18B, ESI†). Given the discrepancies in the IC50 values determined from the FP assay results and HTRF for compounds 6 and 26 against YTHYTHDC1, we performed orthogonal competition REMSA to confirm the minimal inhibitory activity for compounds 6 and 26 (Fig. S19, ESI†). While these fragments (6 and 26) can fit into the binding pocket in the crystal structures,36,38 the molecules alone do not appear to have high inhibitory activities, which suggests additional interactions are required to compete with m6A RNA binding.
In summary, among the compared compounds, SAC and N-7 are the most promising inhibitor structures. SAC contains more aromatic structures and presents a higher number of H-bond donors and acceptor groups than N-7, which can account for the selectivity among different YTH domains for SAC whereas N-7 is shown to be a pan-inhibitor. The N-7 structure may contain a robust motif that is important for binding to the highly similar pockets of all YTH domains.14 At the current stage, both SAC and N-7 compounds showed in vitro inhibitory activities in the micromolar range and will benefit from further structure optimization efforts for improving potency.
Conclusions
This current work reports the discovery of a new nucleoside analogue structure N-7 that functions as a pan-inhibitor for YTH domains recognition of m6A-RNA, with IC50 values in the range of 30–48 μM. N-7 exhibits a stabilizing effect of the YTH domains of human YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2 proteins as determined using a thermal shift assay, and no direct interaction was observed between N-7 and RNA. This suggests that the inhibitory mechanism primarily arose from the affinities between the compound and the YTH domain proteins. We envision that the development of new small molecule inhibitors will largely facilitate research into uncovering precise functions of m6A modification in physiology and diseases, and potentiate future therapeutic efforts by targeting the epitranscriptome.Experimental procedures
RNA oligonucleotides
RNA oligonucleotides including FAM-m6A-RNA, FAM-A-RNA, and m6A-RNA (Fig. 1C) were gifts from the He lab at the University of Chicago, which were originally produced in-house using an Expedite DNA synthesizer as previously reported.49 We dissolved oligonucleotides into water (Fisher Science, BP561-1) for usage in in vitro assays.Protein expression and purification
Genes of the YTH domain of YTHDF1 (amino acids 375–552), YTHDF2 (amino acids 383–553), YTHDF3 (amino acids 391–585), YTHDC1 (amino acids 345–509), and YTHDC2 (amino acids 1285–1424) were cloned into a pET28a vector with an N-terminal His-tag for protein over-expression in E. coli, respectively (Fig. S1 and S5–S8, ESI†). We followed previously reported methods14,17,44 to over-express and purify the YTH domain proteins from E. coli BL21(DE3) cells. A 5 mL overnight cell culture was used for inoculating 1 L of LB medium with 30 μg mL−1 kanamycin. The cells were cultured at 37 °C with shaking at 250 rpm until the OD600 reached 0.6–0.8, which typically took around 3 hours. Subsequently, 0.5 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added into the cell culture to induce over-expression. Cells were then incubated in the shaker at 37 °C for 3 hours and then harvested by centrifugation at 2500 rpm for 20 minutes at 4 °C.We prepared the lysis buffers for different YTH domain proteins based on the published protocols: 20 mM HEPES, pH 7.5, 200 mM NaCl, and 1 mM dithiothreitol (DTT) for YTHYTHDF1 and YTHYTHDF2 proteins50 and 100 mM Tris–HCl, pH 8.0, 500 mM NaCl and 1 mM DTT for YTHYTHDF3 protein51 and 20 mM Tris–HCl, pH 7.5, 400 mM NaCl and 1 mM DTT for YTHYTHDC1 and YTHYTHDC2 proteins.44,52 The cell pellets were resuspended and lysed in the corresponding lysis buffer by sonication for each protein. Clear cell lysates were obtained by taking the supernatant after centrifugation at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 minutes at 4 °C. We then added 1% (w/v) streptomycin sulfate (Acros Organics) to the supernatant to precipitate genomic DNA, performed centrifugation at 11000 rpm for 15 minutes at 4 °C, and recovered the supernatant afterward. The collected supernatant was subsequently incubated with pre-equilibrated His60 Ni Superflow Resin (Takara Bio) at 4 °C for 1 hour and then loaded onto an empty column; resins (bound with protein) were washed extensively using >20 mL of 10 mM imidazole solutions in the lysis buffer, and proteins were eluted under a gradient of imidazole concentrations (25 mM, 50 mM, 100 mM, 150 mM, 250 mM, and 500 mM) in the lysis buffer, and fractions of eluted proteins were collected into 2 mL tubes. All collected fractions were examined through 12% SDS-PAGE and those that contained predominantly the targeted protein were pooled together (Fig. S1 and S5–S8, ESI†), and concentrated using a concentrator (Sigma-Aldrich) down to around 3 mL. The concentrated protein was then desalted and exchanged into the storage buffer (25 mM HEPES, pH 7.5, 300 mM NaCl, 5% glycerol, 0.04% Triton X-100, and 1 mM DTT) using a PD-10 desalting column (GE Healthcare). The desalted proteins were concentrated to around 5 mg mL−1, aliquoted, flash-frozen by liquid nitrogen, and stored at −80 °C. Protein concentrations were measured via absorbance at 280 nm with 1 absorbance unit approximated to 1 mg mL−1 protein using a Nanodrop spectrophotometer (Thermo Scientific).
000 rpm for 15 minutes at 4 °C. We then added 1% (w/v) streptomycin sulfate (Acros Organics) to the supernatant to precipitate genomic DNA, performed centrifugation at 11000 rpm for 15 minutes at 4 °C, and recovered the supernatant afterward. The collected supernatant was subsequently incubated with pre-equilibrated His60 Ni Superflow Resin (Takara Bio) at 4 °C for 1 hour and then loaded onto an empty column; resins (bound with protein) were washed extensively using >20 mL of 10 mM imidazole solutions in the lysis buffer, and proteins were eluted under a gradient of imidazole concentrations (25 mM, 50 mM, 100 mM, 150 mM, 250 mM, and 500 mM) in the lysis buffer, and fractions of eluted proteins were collected into 2 mL tubes. All collected fractions were examined through 12% SDS-PAGE and those that contained predominantly the targeted protein were pooled together (Fig. S1 and S5–S8, ESI†), and concentrated using a concentrator (Sigma-Aldrich) down to around 3 mL. The concentrated protein was then desalted and exchanged into the storage buffer (25 mM HEPES, pH 7.5, 300 mM NaCl, 5% glycerol, 0.04% Triton X-100, and 1 mM DTT) using a PD-10 desalting column (GE Healthcare). The desalted proteins were concentrated to around 5 mg mL−1, aliquoted, flash-frozen by liquid nitrogen, and stored at −80 °C. Protein concentrations were measured via absorbance at 280 nm with 1 absorbance unit approximated to 1 mg mL−1 protein using a Nanodrop spectrophotometer (Thermo Scientific).
The FTO protein was overexpressed and purified from E. coli BL21 (DE3) cells following previously reported methods.53 Briefly, one liter of cell culture was grown at 37 °C for 3 hours in LB medium with 30 μg mL−1 kanamycin until the OD600 reached 0.6–0.8. Overexpression was induced by 0.5 mM IPTG and cells were incubated at 18 °C for 14–16 hours. Cells were lysed in 40 mL lysis buffer (20 mM Tris–HCl, 300 mM NaCl) by sonication, in the presence of a protease inhibitor and further purified with His60 Ni Superflow Resin. Proteins were eluted with 20 mM to 250 mM imidazole gradient in the context of the lysis buffer followed by the desalting column and finally kept in a storage buffer composed of 50 mM Tris–HCl pH 7.5, 150 mM KCl, 5% glycerol, and 0.04% Triton-X-100 at -80 °C (Fig. S12, ESI†).
MS analysis of intact proteins
Purified proteins were analyzed using an Agilent 1260 Infinity HPLC coupled with the 6230 ESI time-of-flight (ESI-TOF) mass spectrometer (MS). The protein samples were subjected onto a C8 column (100 × 4.5mm Phenomenex Aeris 3.6-μm Widepore XB-C8) and run through the column under an increasing gradient (5–95%) of non-polar buffer B in the aqueous buffer A (Buffer A: 95% water, 5% acetonitrile, 0.1% formic acid; Buffer B: 5% water, 95% acetonitrile, 0.1% formic acid). The eluted protein was injected into the MS and analyzed under the positive-ion mode. Total protein masses for the purified YTH domains and FTO were calculated through deconvolution using MagTran (Amgen).
FP binding assay for measuring binding affinities
Fluorescence polarization (FP) experiments were performed following the previously reported FP assay conditions for FAM-labeled RNA and YTH domains.50,54 Briefly, experiments were conducted in the binding buffer containing 25 mM Tris–HCl, pH 7.5, and 150 mM NaCl.50 The same binding buffer was used in this study unless otherwise specified. RNA samples were diluted to 50 nM from the stock solutions by the binding buffer. YTH domain proteins were thawed, buffer exchanged into the binding buffer at 4 °C using the 10 kDa-cutoff concentrators (Sigma-Aldrich), and adjusted to 2× the highest concentration used in the binding assay. 1 μL RNase inhibitor (SUPERaseIn, Invitrogen) was added. 40 μL protein solutions with various concentrations were prepared by serial dilution using the binding buffer. Subsequently, we added 40 μL diluted RNA into each 40 μL protein solution, mixed, and incubated at room temperature for 30 minutes. The final concentration of FAM-m6A-RNA or FAM-A-RNA was constant at 25 nM, and the YTH-domain protein concentrations varied from 10 nM to 24 μM. We performed three biological replicates for each binding measurement using the same protein purified from different batches. After incubation, samples were transferred to a black polypropylene 384-well round-bottom plate (Cellvis) and FP was measured on a SynergyTM Neo2 Multimode Microplate Reader at the wavelength of 485/20 nm for excitation and 528/20 nm for emission at 25 °C.We used the following equation to calculate the FP signal,43
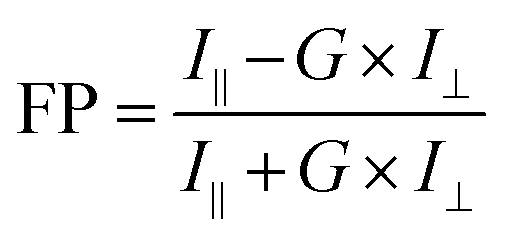
where D is the constant concentrations of FAM-m6A-RNA or FAM-A-RNA (D = 25 nM), B and C relate to the FP for free RNA and bound RNA, and Kd is the dissociation constant.
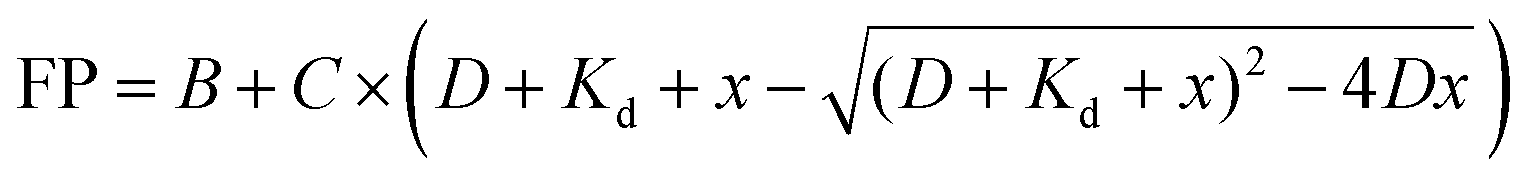
Binding between FTO and FAM-m6A-RNA was performed as described above in the 50 mM borate binding buffer (pH 7.5).
To evaluate the binding affinity between FAM-m6A-RNA and N-7, we prepared eight N-7 compound solutions in the same binding buffer with 5% DMSO with 0 to 400 μM concentrations via serial dilution. 20 μL of 50 nM of FAM-m6A-RNA was added to 20 μL compound solution. The final concentration for the FAM-m6A-RNA was 25 nM and that for N-7 ranged between 0 to 200 μM. Samples were incubated and FP was measured and analyzed as described above.
FP competition assays for dose–response measurements and screening
To set up the FP competition assay, we first used the non-fluorescent “m6A-RNA” (Fig. 1C) as a positive control inhibitor. We incubated 40 nM to 25 μM m6A-RNA with 2 μM YTHYTHDF1 and 25 nM FAM-m6A-RNA in the binding buffer with added RNase inhibitor at room temperature for 30 minutes. Samples were transferred onto the 384-well plate and measured on the plate reader as described above. We obtained the dose–response curves by plotting the FP values against concentrations of inhibitors. The inhibitory curves and IC50 values were obtained using the “dose–response inhibition” model in GraphPad Prism 6.0TM. The same assay condition was used for benchmarking the competition assay for other YTH domains (Fig. S9B, ESI†), except that 10 μM YTHYTHDC2 was used instead of 2 μM due to the weaker binding affinity (or increased Kd) for YTHYTHDC2 to m6A-RNA compared to other YTH domains (Fig. S9A, ESI†).To assess the performance of the FP competition assay for screening, we performed the assay with positive and negative control inhibitors 20 times each, where we used 20 μM non-fluorescent m6A-RNA and blank binding buffer as the positive and negative control, respectively. We calculated the Z′ factor using the following equation43 based on the measured FP of the positive and negative controls,
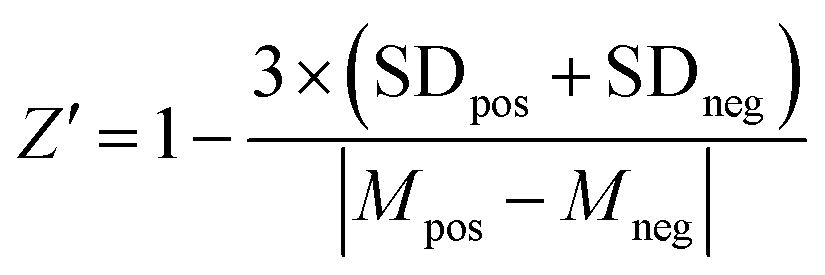
We purchased the Nucleoside Mimetics library from Enamine which contains 320 compounds dissolved in DMSO at 10 mM. All stock solutions of small molecules were prepared in 100% DMSO unless otherwise specified. To perform screening compounds, we first prepared RNA–protein solutions on the 384-well plate with 33.6 μL per well. We added 1.4 μL 1 mM compound into the RNA–protein solutions in each well, with the final concentrations of the YTHYTHDF1, FAM-m6A-RNA, and each compound at 2 μM, 25 nM, and 40 μM, respectively. Each compound was screened in technical duplicates (Fig. 2). Meanwhile, DMSO and the non-fluorescent m6A-RNA were used as the negative and positive controls for the inhibitory activity on the same plate. The plate was incubated at room temperature for 30 minutes and subjected to the plate reader for FP measurements.
To perform dose–response measurements, we ordered 40 mg of N-7 (Z2760966931) from Enamine. We examined the quality of purchased N-7 through LC-MS and 1H NMR spectroscopy (Fig. S4, ESI†). The dose–response FP competition assay was set up similarly to that described for the non-fluorescent m6A-RNA. Instead of the m6A-RNA, 1.5–200 μM N-7 were incubated with 2 μM YTH domain proteins and 25 nM FAM-m6A-RNA in the binding buffer.
As IC50 values may be subjected to changes under varying assay conditions, we performed several control conditions for the FP competition assay. Specifically, we adjusted the concentrations of the YTH domain proteins from 2 μM to the concentration around the Kd for each protein binding m6A-RNA (Fig. 1D and Fig. S10A, ESI†). Secondly, we tested the reported FP competition protocol which first incubated the compound with YTH domains, followed by adding the FAM-m6A-RNA35 (Fig. S10B, ESI†). Under these conditions, the measured IC50 values showed no significant changes (Fig. S10C, ESI†).
Reported inhibitors, compound 6 and compound 26, were ordered from Chemspace (CSMB00010870727 and CSSS00000159915). The m6A nucleoside was ordered from AmBeed. Salvianolic acid C was ordered from MedChemExpress (HY-N0319). 6-(methylamino)purine was purchased from Fisher Scientific. The inhibitory activities of these compounds were examined using the same dose–response FP competition assay as for N-7.
RNA electrophoretic mobility shift assay (REMSA)
Competitive REMSA was performed by incubating 20 nM YTHYTHDF1 or YTHYTHDC1 with various concentrations of N-7 (1–200 μM) and 25 nM FAM-m6A-RNA in a buffer containing 10 mM HEPES, 50 mM KCl, 1 mM EDTA, 0.05% Triton-X-100, 5% glycerol and RNase inhibitor, in a final volume of 20 μL. The reaction mixture was incubated at room temperature for 20 minutes, and then loaded onto an 8% polyacrylamide gel containing 0.2% glycerol. Electrophoresis was performed in 0.5× TBE buffer at 90 V and 4 °C for 60 minutes. FAM-m6A-RNA was detected using the ChemiDoc™ MP imaging system (BIO-RAD) with the fluorescence blotting module.Thermal shift assay
Thermal shift assays were set up according to the reported assay conditions using the SYPRO orange dye, which exhibited enhanced fluorescence in a more hydrophobic environment typically during protein unfolding.48 For the positive control, we incubated 2 μM or 16 μM m6A-RNA with 4 μM YTHYTHDF1 in the binding buffer with added RNase inhibitor and 5× SYPRO orange dye (Invitrogen) at room temperature for 15 minutes. After incubation, the thermal shift assay was performed in the QuantStudioTM 3 Real-Time PCR System (Thermo Fisher). Each sample was heated from 25 °C to 75 °C ramping by 0.2 °C s−1; meanwhile, fluorescence intensities were recorded every 0.3 °C with the excitation and emission wavelengths at 470 nm and 570 nm, respectively. Data were first normalized by setting the highest and lowest fluorescence intensities in each melting run to 100% and 0%, respectively. Data were then fitted to obtain the melting temperatures (Tm) using the Boltzmann Sigmoid equation48 within GraphPad Prism 6.0TM. As the YTHYTHDF1 protein specifically bound with m6A-RNA with Kd = 0.8 ± 0.2 μM through the FP binding assay (Fig. 1D), we expected the presence of m6A-RNA would promote the thermal stability of the protein. As expected, we observed that the addition of m6A-RNA showed significant stabilization of the YTHYTHDF1 (Fig. S13, ESI†).To assess whether N-7 interacted with the YTH domain proteins, we used the thermal shift assay to examine if there were any stabilization effects of the YTH domains by adding N-7. We incubated 6 μM or 50 μM N-7 with 4 μM YTHYTHDF1, YTHYTHDF2, YTHYTHDF3, YTHYTHDC1, or YTHYTHDC2 under the same conditions without the RNase inhibitor as above. As a negative control, we added an equal volume of DMSO rather than N-7 into each protein. The thermal shift assay measurements and analyses were performed in the same manner as described for the positive control. For each YTH domain protein, three biological replicates were performed to quantify the errors of the assay. The thermal stabilizations of the YTH domain proteins were analyzed by the change in the Tm (ΔTm) as the difference between the Tm measured on the protein with N-7 or DMSO, and the Tm of protein-only control (Fig. 4B and Fig. S14, ESI†). The interactions between D-6 and YTH domain proteins were assessed using the same method as for N-7 (Fig. S15, ESI†).
Molecular docking with AutoDock
The crystal structure of the YTHDF1 in complex with GGm6ACU (PDBID: 4RCJ) was chosen for molecular docking modeling.14 The structure was prepared for docking by removing water molecules and the bound RNA oligonucleotide GGm6ACU. The structure of N-7 was prepared for docking by generating conformers and optimizing geometry by AutoDock4.55 During molecular docking, the binding pocket of YTHDF1 was defined as 10 Å around the m6A binding site. The N-7 ligand was docked against the defined pocket of the YTHYTHDF1 protein via AutoDock. The resulting binding pose of N-7 with the most favorable docking score was reported and analyzed.Author contributions
H. Z. conceived the idea of the project and supervised the research. C.-H. W. performed all the experiments and data analysis with help from H. Z. H. Z. and C.-H. W. wrote the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
H. Z. and C.-H. W. have filed a provisional patent application for the inhibitors described in this manuscript.Acknowledgements
We acknowledge the financial supports provided by the National Institute of General Medical Sciences and Boston College. We thank the Office of the Vice Provost for Research at Boston College for providing the microplate reader used for the research. We thank Dr Qing Dai, Zhongyu Zou, and the He Lab at the University of Chicago for sharing the RNA oligonucleotides and plasmids for the YTH proteins used in this study. We thank Dr Thusitha Jayasundera, Lingchao Kong, and Xinyi Lyu for their assistance in collecting the LC-MS and NMR data for compound N-7. We acknowledge Prof. Jianmin Gao, Prof. Jia Niu, Prof. Eranthie Weerapana, and group members from the Zhou group for insightful discussions and advice. This work was supported by Boston College (the lab startup fund to H. Z.) and the National Institute of General Medical Sciences (R35 GM150789 to H. Z.).Notes and references
- R. P. Perry and D. E. Kelley, Cell, 1974, 1, 37–42 CrossRef CAS.
- R. Desrosiers, K. Friderici and F. Rottman, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3971–3975 CrossRef CAS PubMed.
- D. Dominissini, S. Moshitch-Moshkovitz, S. Schwartz, M. Salmon-Divon, L. Ungar, S. Osenberg, K. Cesarkas, J. Jacob-Hirsch, N. Amariglio, M. Kupiec, R. Sorek and G. Rechavi, Nature, 2012, 485, 201–206 CrossRef CAS PubMed.
- K. D. Meyer, Y. Saletore, P. Zumbo, O. Elemento, C. E. Mason and S. R. Jaffrey, Cell, 2012, 149, 1635–1646 CrossRef CAS PubMed.
- N. Liu, Q. Dai, G. Zheng, C. He, M. Parisien and T. Pan, Nature, 2015, 518, 560–564 CrossRef CAS PubMed.
- N. Liu, K. I. Zhou, M. Parisien, Q. Dai, L. Diatchenko and T. Pan, Nucleic Acids Res., 2017, 45, 6051–6063 CrossRef CAS PubMed.
- X. Wang, Z. Lu, A. Gomez, G. C. Hon, Y. Yue, D. Han, Y. Fu, M. Parisien, Q. Dai, G. Jia, B. Ren, T. Pan and C. He, Nature, 2014, 505, 117–120 CrossRef PubMed.
- I. A. Roundtree, G.-Z. Luo, Z. Zhang, X. Wang, T. Zhou, Y. Cui, J. Sha, X. Huang, L. Guerrero, P. Xie, E. He, B. Shen and C. He, eLife, 2017, 6, e31311 CrossRef PubMed.
- H. Shi, X. Wang, Z. Lu, B. S. Zhao, H. Ma, P. J. Hsu, C. Liu and C. He, Cell Res., 2017, 27, 315–328 CrossRef CAS PubMed.
- X. Wang, Boxuan S. Zhao, Ian A. Roundtree, Z. Lu, D. Han, H. Ma, X. Weng, K. Chen, H. Shi and C. He, Cell, 2015, 161, 1388–1399 CrossRef CAS PubMed.
- K. D. Meyer, D. P. Patil, J. Zhou, A. Zinoviev, M. A. Skabkin, O. Elemento, T. V. Pestova, S. B. Qian and S. R. Jaffrey, Cell, 2015, 163, 999–1010 CrossRef CAS PubMed.
- D. P. Patil, C.-K. Chen, B. F. Pickering, A. Chow, C. Jackson, M. Guttman and S. R. Jaffrey, Nature, 2016, 537, 369–373 CrossRef CAS PubMed.
- J. Liu, X. Dou, C. Chen, C. Chen, C. Liu, M. M. Xu, S. Zhao, B. Shen, Y. Gao, D. Han and C. He, Science, 2020, 367, 580–586 CrossRef CAS PubMed.
- C. Xu, K. Liu, H. Ahmed, P. Loppnau, M. Schapira and J. Min, J. Biol. Chem., 2015, 290, 24902–24913 CrossRef CAS PubMed.
- S. Zaccara, R. J. Ries and S. R. Jaffrey, Nat. Rev. Mol. Cell Biol., 2019, 20, 608–624 CrossRef CAS PubMed.
- M. N. Flamand, M. Tegowski and K. D. Meyer, Annu. Rev. Biochem., 2023, 92, 145–173 CrossRef CAS PubMed.
- T. Zhu, I. A. Roundtree, P. Wang, X. Wang, L. Wang, C. Sun, Y. Tian, J. Li, C. He and Y. Xu, Cell Res., 2014, 24, 1493–1496 CrossRef CAS PubMed.
- H. Du, Y. Zhao, J. He, Y. Zhang, H. Xi, M. Liu, J. Ma and L. Wu, Nat. Commun., 2016, 7, 12626 CrossRef CAS PubMed.
- O. H. Park, H. Ha, Y. Lee, S. H. Boo, D. H. Kwon, H. K. Song and Y. K. Kim, Mol. Cell, 2019, 74, 494–507 CrossRef CAS PubMed e498.
- S. H. Boo, H. Ha, Y. Lee, M.-K. Shin, S. Lee and Y. K. Kim, Cell Rep., 2022, 39, 110861 CrossRef CAS PubMed.
- W. Xiao, S. Adhikari, U. Dahal, Y.-S. Chen, Y.-J. Hao, B.-F. Sun, H.-Y. Sun, A. Li, X.-L. Ping, W.-Y. Lai, X. Wang, H.-L. Ma, C.-M. Huang, Y. Yang, N. Huang, G.-B. Jiang, H.-L. Wang, Q. Zhou, X.-J. Wang, Y.-L. Zhao and Y.-G. Yang, Mol. Cell, 2016, 61, 507–519 CrossRef CAS PubMed.
- Y. Gao, G. Pei, D. Li, R. Li, Y. Shao, Q. C. Zhang and P. Li, Cell Res., 2019, 29, 767–769 CrossRef CAS PubMed.
- R. J. Ries, S. Zaccara, P. Klein, A. Olarerin-George, S. Namkoong, B. F. Pickering, D. P. Patil, H. Kwak, J. H. Lee and S. R. Jaffrey, Nature, 2019, 571, 424–428 CrossRef CAS PubMed.
- J. Wang, L. Wang, J. Diao, Y. G. Shi, Y. Shi, H. Ma and H. Shen, Protein Cell, 2020, 11, 304–307 CrossRef PubMed.
- X. Dai, T. Wang, G. Gonzalez and Y. Wang, Anal. Chem., 2018, 90, 6380–6384 CrossRef CAS PubMed.
- Q. Zheng, H. Gan, F. Yang, Y. Yao, F. Hao, L. Hong and L. Jin, Cell Discovery, 2020, 6, 12 CrossRef CAS PubMed.
- C. B. Woodcock, J. R. Horton, J. Zhou, M. T. Bedford, R. M. Blumenthal, X. Zhang and X. Cheng, Nucleic Acids Res., 2020, 48, 10329–10341 CrossRef CAS PubMed.
- T. Lence, J. Akhtar, M. Bayer, K. Schmid, L. Spindler, C. H. Ho, N. Kreim, M. A. Andrade-Navarro, B. Poeck, M. Helm and J.-Y. Roignant, Nature, 2016, 540, 242–247 CrossRef CAS PubMed.
- H. Shi, X. Zhang, Y.-L. Weng, Z. Lu, Y. Liu, Z. Lu, J. Li, P. Hao, Y. Zhang, F. Zhang, Y. Wu, J. Y. Delgado, Y. Su, M. J. Patel, X. Cao, B. Shen, X. Huang, G.-l Ming, X. Zhuang, H. Song, C. He and T. Zhou, Nature, 2018, 563, 249–253 CrossRef CAS PubMed.
- J. Liu, B. T. Harada and C. He, Trends Cell Biol., 2019, 29, 487–499 CrossRef CAS PubMed.
- R. Shi, S. Ying, Y. Li, L. Zhu, X. Wang and H. Jin, Cell Death Dis., 2021, 12, 346 CrossRef CAS PubMed.
- N. S. Gokhale, A. B. R. McIntyre, M. J. McFadden, A. E. Roder, E. M. Kennedy, J. A. Gandara, S. E. Hopcraft, K. M. Quicke, C. Vazquez, J. Willer, O. R. Ilkayeva, B. A. Law, C. L. Holley, M. A. Garcia-Blanco, M. J. Evans, M. S. Suthar, S. S. Bradrick, C. E. Mason and S. M. Horner, Cell Host Microbe, 2016, 20, 654–665 CrossRef CAS PubMed.
- W. Zhang, T. Wu, Y. Zhang, W. Kang, C. Du, Q. You, X. Chen and Z. Jiang, Bioorg. Med. Chem., 2023, 90, 117373 CrossRef CAS PubMed.
- J. Liao, Y. Wei, J. Liang, J. Wen, X. Chen, B. Zhang and L. Chu, Cell Death Discovery, 2022, 8, 137 CrossRef CAS PubMed.
- Z. Zou, J. Wei, Y. Chen, Y. Kang, H. Shi, F. Yang, Z. Shi, S. Chen, Y. Zhou, C. Sepich-Poore, X. Zhuang, X. Zhou, H. Jiang, Z. Wen, P. Jin, C. Luo and C. He, Mol. Cell, 2023, 83, 4304–4317 CrossRef CAS PubMed.
- Y. Li, R. K. Bedi, F. Nai, V. von Roten, A. Dolbois, F. Zálešák, R. Nachawati, D. Huang and A. Caflisch, Eur. J. Med. Chem. Rep., 2022, 5, 100057 CAS.
- F. Nai, R. Nachawati, F. Zálešák, X. Wang, Y. Li and A. Caflisch, ACS Med. Chem. Lett., 2022, 13, 1500–1509 CrossRef CAS PubMed.
- R. K. Bedi, D. Huang, L. Wiedmer, Y. Li, A. Dolbois, J. A. Wojdyla, M. E. Sharpe, A. Caflisch and P. Sledz, ACS Chem. Biol., 2020, 15, 618–625 CrossRef CAS PubMed.
- M. Micaelli, A. Dalle Vedove, L. Cerofolini, J. Vigna, D. Sighel, S. Zaccara, I. Bonomo, G. Poulentzas, E. F. Rosatti, G. Cazzanelli, L. Alunno, R. Belli, D. Peroni, E. Dassi, S. Murakami, S. R. Jaffrey, M. Fragai, I. Mancini, G. Lolli, A. Quattrone and A. Provenzani, ACS Pharmacol. Transl. Sci., 2022, 5, 872–891 CrossRef CAS PubMed.
- Y.-G. Hong, Z. Yang, Y. Chen, T. Liu, Y. Zheng, C. Zhou, G.-C. Wu, Y. Chen, J. Xia, R. Wen, W. Liu, Y. Zhao, J. Chen, X. Gao and Z. Chen, Cancer Res., 2023, 83, 845–860 CrossRef CAS PubMed.
- L. Wang, X. Dou, S. Chen, X. Yu, X. Huang, L. Zhang, Y. Chen, J. Wang, K. Yang, J. Bugno, S. Pitroda, X. Ding, A. Piffko, W. Si, C. Chen, H. Jiang, B. Zhou, S. J. Chmura, C. Luo, H. L. Liang, C. He and R. R. Weichselbaum, Cancer Cell, 2023, 41, 1294–1308 CrossRef CAS PubMed.
- S. Yang, H. Zhang, Y. Li, F. Wang, G. Lin, T. Niu, H. Li, Y. Yi, H. Zhou, R. Yang, R. Yao, P. Zhou, Y. Li, M. Wu, M.-X. Chen, H. Xu, J. You, Y. Liao, C. Yang, A. Zhao, C. Chen, L. Li and Y. Wei, Discovery of a selective YTHDC1 inhibitor that targets acute myeloid leukemia, Research Square, 2023, preprint DOI:10.21203/rs.3.rs-2644364/v1.
- N. J. Moerke, Curr. Protoc. Chem. Biol., 2009, 1, 1–15 CrossRef PubMed.
- C. Xu, X. Wang, K. Liu, I. A. Roundtree, W. Tempel, Y. Li, Z. Lu, C. He and J. Min, Nat. Chem. Biol., 2014, 10, 927–929 CrossRef CAS PubMed.
- X. Dai, T. Wang, G. Gonzalez and Y. Wang, Anal. Chem., 2018, 90, 6380–6384 CrossRef CAS PubMed.
- G. Jia, Y. Fu, X. Zhao, Q. Dai, G. Zheng, Y. Yang, C. Yi, T. Lindahl, T. Pan, Y. G. Yang and C. He, Nat. Chem. Biol., 2011, 7, 885–887 CrossRef CAS PubMed.
- Y. Huang, R. Su, Y. Sheng, L. Dong, Z. Dong, H. Xu, T. Ni, Z. S. Zhang, T. Zhang, C. Li, L. Han, Z. Zhu, F. Lian, J. Wei, Q. Deng, Y. Wang, M. Wunderlich, Z. Gao, G. Pan, D. Zhong, H. Zhou, N. Zhang, J. Gan, H. Jiang, J. C. Mulloy, Z. Qian, J. Chen and C. G. Yang, Cancer Cell, 2019, 35, 677–691 CrossRef CAS PubMed e610.
- F. H. Niesen, H. Berglund and M. Vedadi, Nat. Protoc., 2007, 2, 2212–2221 CrossRef CAS PubMed.
- L. Hu, S. Liu, Y. Peng, R. Ge, R. Su, C. Senevirathne, B. T. Harada, Q. Dai, J. Wei, L. Zhang, Z. Hao, L. Luo, H. Wang, Y. Wang, M. Luo, M. Chen, J. Chen and C. He, Nat. Biotechnol., 2022, 40, 1210–1219 CrossRef CAS PubMed.
- B. Liu, D. K. Merriman, S. H. Choi, M. A. Schumacher, R. Plangger, C. Kreutz, S. M. Horner, K. D. Meyer and H. M. Al-Hashimi, Nat. Commun., 2018, 9, 2761 CrossRef PubMed.
- Y. Li, R. K. Bedi, E. V. Moroz-Omori and A. Caflisch, J. Chem. Inf. Model., 2020, 60, 5932–5935 CrossRef CAS PubMed.
- C. Ma, S. Liao and Z. Zhu, Biochem. Biophys. Res. Commun., 2019, 518, 678–684 CrossRef CAS PubMed.
- G. Jia, C. G. Yang, S. Yang, X. Jian, C. Yi, Z. Zhou and C. He, FEBS Lett., 2008, 582, 3313–3319 CrossRef CAS PubMed.
- H. Zhou, B. Sathyamoorthy, A. Stelling, Y. Xu, Y. Xue, Y. Z. Pigli, D. A. Case, P. A. Rice and H. M. Al-Hashimi, Biochemistry, 2019, 58, 1963–1974 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00105b |
| This journal is © The Royal Society of Chemistry 2024 |