
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEarly-stage biosynthesis of phenalinolactone diterpenoids involves sequential prenylation, epoxidation, and cyclization†
Tyler A.
Alsup
 a,
Zining
Li
a,
Caitlin A.
McCadden
a,
Annika
Jagels
ab,
Diana P.
Łomowska-Keehner
a,
Erin M.
Marshall
ab,
Liao-Bin
Dong
c,
Sandra
Loesgen
ab and
Jeffrey D.
Rudolf
*a
a,
Zining
Li
a,
Caitlin A.
McCadden
a,
Annika
Jagels
ab,
Diana P.
Łomowska-Keehner
a,
Erin M.
Marshall
ab,
Liao-Bin
Dong
c,
Sandra
Loesgen
ab and
Jeffrey D.
Rudolf
*a
aDepartment of Chemistry, University of Florida, Gainesville, Florida, USA. E-mail: jrudolf@chem.ufl.edu
bWhitney Laboratory for Marine Bioscience, University of Florida, St. Augustine, FL, USA
cState Key Laboratory of Natural Medicines, School of Traditional Chinese Pharmacy, China Pharmaceutical University, Nanjing 211198, Jiangsu, China
First published on 5th August 2024
Abstract
The chemical logic associated with assembly of many bacterial terpenoids remains poorly understood. We focused our efforts on the early-stage biosynthesis of the phenalinolactone diterpenoids, demonstrating that the anti/anti/syn-perhydrophenanthrene core is constructed by sequential prenylation, epoxidation, and cyclization. The functions and timing of PlaT1–PlaT3 were assigned by comprehensive heterologous reconstitution. We illustrated that the UbiA prenyltransferase PlaT3 acts on geranylgeranyl diphosphate (GGPP) in the first step of phenalinolactone biosynthesis, prior to epoxidation by the flavin-dependent monooxygenase PlaT1 and cyclization by the type II terpene cyclase PlaT2. Finally, we isolated eight new-to-nature terpenoids, expanding the scope of the bacterial terpenome. The biosynthetic strategy employed in the assembly of the phenalinolactone core, with cyclization occurring after prenylation, is rare in bacteria and resembles fungal meroterpenoid biosynthesis. The findings presented here set the stage for future discovery, engineering, and enzymology efforts in bacterial meroterpenoids.
Introduction
Terpenoids are the largest family of natural products (NPs) with over 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 known members wielding a vast array of biological activities.1,2 Their activities against bacteria, fungi, viruses, and cancer cell lines have driven natural products discovery efforts to identify novel terpenoids for applications in human health. Bacteria are commonly regarded as a trivial source of terpenoids; <2% of all known terpenoids are of bacterial origin.2 However, there is a vast diversity of bacterial terpenoids and meroterpenoids and advances in genome sequencing and bioinformatics continue to reveal the immense biosynthetic potential encoded within bacterial genomes.3–5 While terpenoid discoveries in bacteria are rapidly advancing in the genomic era, substantial knowledge gaps remain in how many of these complex small molecules are biosynthesized.
000 known members wielding a vast array of biological activities.1,2 Their activities against bacteria, fungi, viruses, and cancer cell lines have driven natural products discovery efforts to identify novel terpenoids for applications in human health. Bacteria are commonly regarded as a trivial source of terpenoids; <2% of all known terpenoids are of bacterial origin.2 However, there is a vast diversity of bacterial terpenoids and meroterpenoids and advances in genome sequencing and bioinformatics continue to reveal the immense biosynthetic potential encoded within bacterial genomes.3–5 While terpenoid discoveries in bacteria are rapidly advancing in the genomic era, substantial knowledge gaps remain in how many of these complex small molecules are biosynthesized.
The phenalinolactones, diterpenoid glycosides isolated from Streptomyces sp. Tü 6071,6 are highly functionalized meroterpenoids with moderate antibacterial activities (Fig. 1A). The phenalinolactones bear an anti/anti/syn-perhydrophenanthrene core and several peripheral decorations including γ-butyrolactone, L-amicetose and 5-methylpyrrole-2-carboxylic acid moieties.2,6 Extensive work has been carried out to elucidate many of the late-stage tailoring and functionalization steps;7–11 however, the biosynthesis of the diterpenoid scaffold, i.e., the perhydrophenanthrene core, remains elusive. Bioinformatic analysis of the pla BGC (Fig. 1B) led to an initial biosynthetic proposal (Fig. 1C). Geranylgeranyl diphosphate (GGPP), formed by the polyprenyl synthase PlaT4, may first be epoxidized at the terminal alkene by the putative flavin-dependent monooxygenase, PlaT1, to yield 14,15-epoxy-GGPP.7 The oxidosqualene cyclase-like (Fig. S1, ESI†) type II terpene cyclase (TC),12,13 PlaT2, was then proposed to catalyze cyclization via epoxide ring opening of epoxy-GGPP to afford the tricyclic core. The diphosphate moiety at C-15 of the cyclized product is presumed to be retained for the UbiA-like prenyltransferase, PlaT3, to subsequently add a three carbon α-keto acid;7 the origin of this moiety is still unknown.7,8 Inactivation of plaT1, plaT2, and plaT3 in the native producer abolished production of the phenalinolactones and no intermediates were detected, supporting that these genes play crucial roles in formation of the diterpenoid core.7 Since this initial proposal, similar hypotheses were made for the structurally related brasilicardins,14 tiancilactones,15 and atolypenes16 (Fig. 1A); each of these BGCs have homologs of PlaT1–PlaT4. The tetraterpenoid longestin (KS-505a) is also proposed to undergo the same three reactions, although a different timing was suggested.17,18 There is currently no direct evidence for the functions of PlaT1–PlaT4, or their homologs, the timing in which they act, or their respective substrates and products.
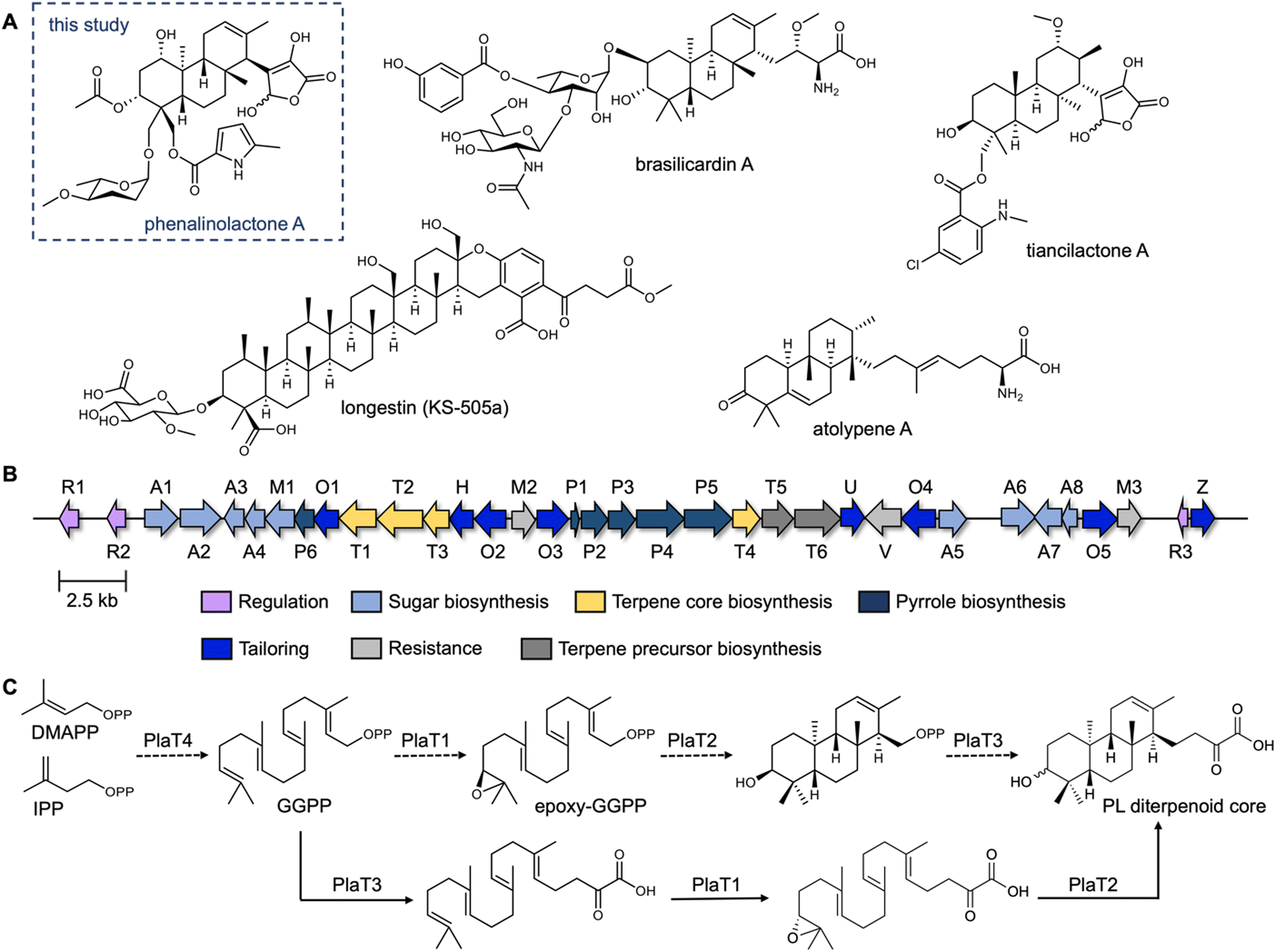 | ||
| Fig. 1 The phenalinolactone family of bacterial terpenoids and their proposed early-stage biosynthesis. (A) Phenalinolactone A and related terpenoids from bacteria.6,14–16,19 (B) Phenalinolactone biosynthetic gene cluster (pla) from Streptomyces sp. Tü 6071. The roles, either confirmed or putative, of each gene are annotated according to the legend. The four core genes studied here, PlaT1–PlaT4, which are conserved throughout this family of terpenoids, are colored in yellow. (C) Previously proposed biosynthetic scheme suggesting the timing of PlaT1–PlaT4 (ref. 7 dashed lines) and biosynthetic scheme proposed in this study (solid lines). | ||
Here, we report a revised proposal for the biosynthesis of the phenalinolactone diterpenoid core. Using heterologous expression, we (i) confirmed that the assembly of the diterpenoid core requires the four key enzymes PlaT1–PlaT4, (ii) demonstrated that prenylation by PlaT3 and epoxidation by PlaT1 occur prior to cyclization by PlaT2, and (iii) isolated eight novel phenalinolactone congeners. These findings support that each member of this family of bacterial terpenoids, as well as hundreds of their related BGCs,15 likely follow a similar biosynthetic strategy, but with distinct enzymes and modifications to achieve chemical diversity. This study also lays the foundation for investigating the mechanistic details of each of these enzymes.
Results and discussion
Formation of the diterpenoid core requires four enzymes
We first set out to determine if PlaT1–PlaT4 were required and sufficient for assembly of the diterpene scaffold. The four-gene cassette was reconstituted in the heterologous hosts Streptomyces albus J1074 and Streptomyces venezuelae under the control of the constitutive sp44 promoter (Fig. S2, ESI†).20 LC-MS analysis of the extracts revealed the production of two new metabolites 1 (m/z 374, [M − H]−) and 2 (m/z 376, [M − H]−) in S. albus::plaT4321; only 1 was produced in S. venezuelae::plaT4321 (Fig. 2A and B). Large-scale (18 L) fermentation of S. albus::plaT4321 was carried out to isolate the metabolites for structural characterization. High-resolution ESIMS (HRESIMS) of 1 gave an [M + H]+ ion at m/z 376.2852, consistent with a molecular formula of C23H38NO3 (calc. [M + H]+ at 376.2846); 2 had a molecular formula of C23H40NO3 based on an observed [M + H]+ ion at m/z 378.3010 (calc. [M + H]+ at 378.3003) (Fig. S3, ESI†). The structure of 1 was determined, by extensive 1D and 2D NMR spectroscopy (Table S4 and Fig. S4–S9, ESI†), to be a C23 tricyclic terpenoid bearing an amino acid tail and a ketone at C-3 (Fig. 2C). The 13C NMR data of 2 were almost identical to that of 1, with the exception of the presence of a signal at 79.70 ppm instead of the ketone signal at 221.07 ppm (Table S5 and Fig. S10–S15, ESI†), supporting that 2 was the C-3 hydroxy analog of 1. Using an 800 MHz NMR, the C-3 hydroxyl of 2 was determined to be in an equatorial position based on the splitting pattern and coupling constants of its axial H-3 (dd, J = 11.8, 4.7 Hz) counterpart, which was also observed in the known biosynthetic intermediate PL CD6.7 Although there are several overlapping signals in the 1H NMR spectrum complicating the 2D NOESY analysis, several key correlations seen in 1, including H-5/H-9, H-9/H3-22, H3-22/H2-15, H3-21/H-1eq, H3-21/H3-19, H3-21/H-6ax, and H3-20/H-3ax support the assigned anti/anti/syn core of phenalinolactone A (Fig. 2D and Fig. S9, S15, ESI†); several of these NOESY correlations also match those reported for phenalinolactone A.6 Overall, the structure of 2, including its R-configured hydroxyl at C-3, is consistent with that of PL CD6 (Fig. 2C). A previous biosynthetic proposal suggested that a 3-keto analog of 2, PL CD7, was an immediate precursor to PL CD6 and that cyclization by PlaT2 may result in an S-configured C-3 hydroxyl that is epimerized through successive oxidation and reduction at C-3;7 however, our result supports that the R-configured hydroxyl is the direct product of cyclization. Although oxidation of the hydroxyl of 2 to the ketone of 1 could be carried out by a host oxidative enzyme,7 there is no direct evidence in our work to support it. Limited amounts of 1 and 2, as well as inconclusive DFT-based configurational assignment of the stereocenter at C-17, made the determination of the absolute configurations of 1 and 2 impossible. We suspect, however, that C-17 may be S configured in line with that of the brasilicardins and atolypenes.14,16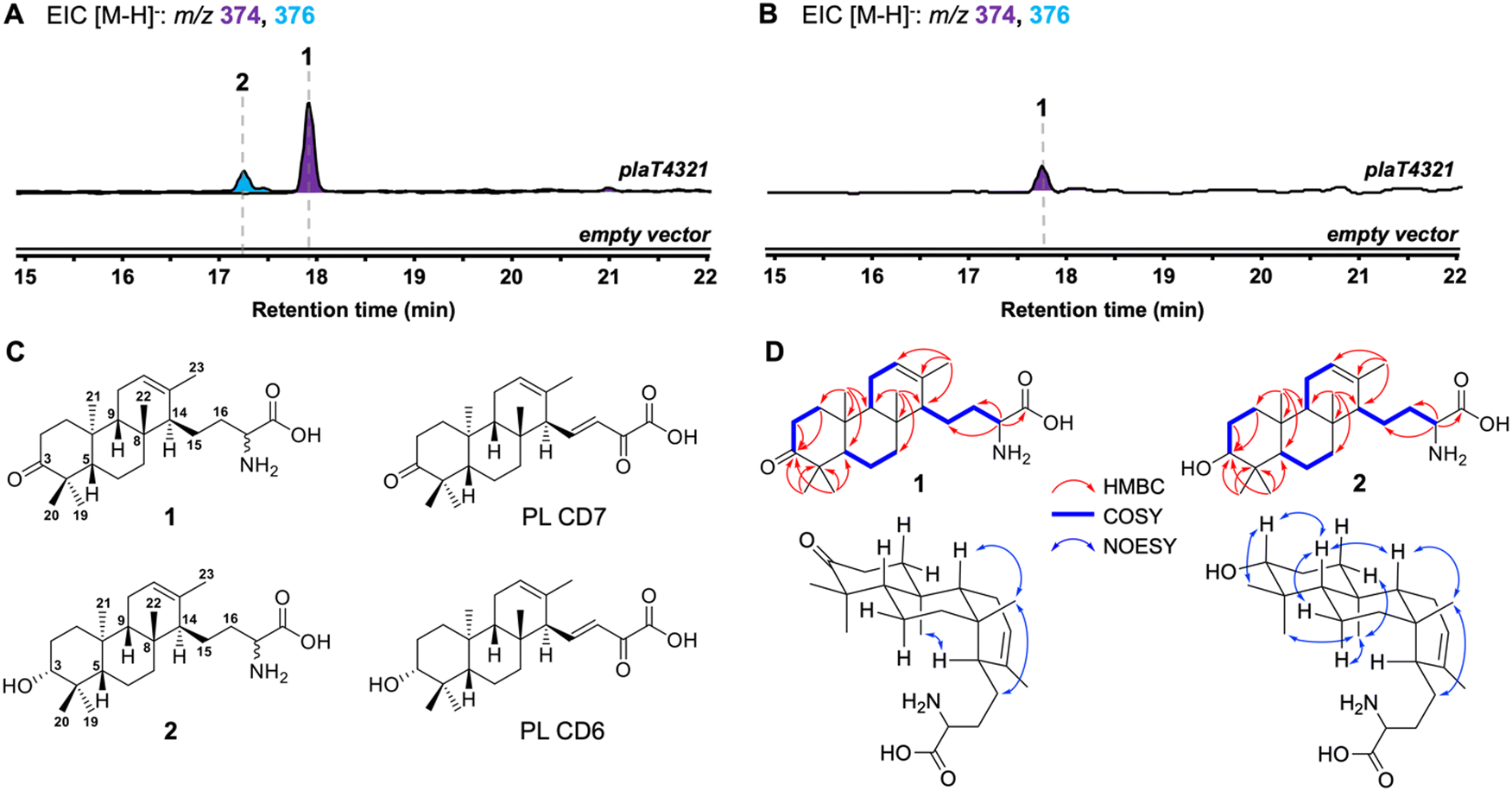 | ||
| Fig. 2 Reconstitution of the first four steps in phenalinolactone biosynthesis. (A) and (B) LCMS-EIC analysis of extracts obtained from S. albus (A) and S. venezuelae (B) hosts. (C) Cyclized diterpenoids (1 and 2) isolated in this study and previously isolated phenalinolactones PL CD6 and PL CD7 (ref. 7) (D) Key HMBC, COSY, and NOESY correlations of 1 and 2. As several 2D NOESY correlations were obscured by overlapping signals, particularly in 1, only relevant and high confidence correlations are shown here. | ||
The tricyclic scaffolds of 1 and 2 are akin to those of the phenalinolactone intermediates PL CD6 and PL CD7, albeit with two key differences. Most notably, the amino acid moiety on 1 and 2 is different than the α-keto acid tail seen in PL CD6 and PL CD7 (Fig. 2C). The presence of the amino acid moiety was surprising given that no phenalinolactones nor their known on-pathway intermediates, e.g., PL CD6, possess this functional group. The tetronic acid moiety in the phenalinolactones is built from an α-keto acid tail (Fig. 1).7 The acyclic tail of 1 and 2, however, resembles the immunosuppressive brasilicardins and the sesterterpenoid atolypenes (Fig. 1A), whose BGCs both harbor aminotransferases that are proposed to convert the α-keto acid into the α-amino acid.16,21,22 As no genes encoding an aminotransferase were included in the plaT1–plaT4 construct, we surmised that the amino acid tails of 1 and 2 arise from an endogenous aminotransferase in the heterologous hosts. However, we cannot rule out that the prenyl acceptor in the UbiA reaction is an amino acid. Isolated products 1 and 2 also lack the double bond at C-15/C-16, thus none of the four PlaT1–PlaT4 enzymes are responsible for this oxidation. Furthermore, no related diterpenoids with this olefin were detected in either heterologous host indicating that they lack the proper machinery to perform this oxidation.
Epoxidation and prenylation precede cyclization
To determine the timing of the three key steps, epoxidation, prenylation, and cyclization, in the biosynthesis of the terpene core of phenalinolactone, we reconstituted combinations of plaT1–plaT4 in S. albus and S. venezuelae (Fig. S16, ESI†). As anticipated, removing plaT2 (i.e., plaT431), the gene encoding the terpene cyclase, abolished production of 1, 2, or any other tricyclic terpenoids in S. albus::plaT431 and S. venezuelae::plaT431 (Fig. 3A and B). Instead, a number of new metabolites, 3–10, with m/z ([M − H]−) values ranging between 339 and 394 were detected in both strains. Metabolites 3–10 were observed in the extracts of S. venezuelae::plaT431 while only 8–10 were seen in S. albus::plaT431 (Fig. 3A and B). A large-scale fermentation (18 L) of S. venezuelae::plaT431 was carried out to isolate 3–7 for structural elucidation. Compounds 3–5 were determined to have 23, 22, and 20 carbons, respectively, based on HRESIMS and 13C NMR analyses (Tables S6–S8 and Fig. S17–S32, ESI†). The 13C NMR spectra for 3–5 also revealed two signals between 70 and 80 ppm suggesting the presence of a pair of hydroxyl groups in each of the molecules. For 5, key HMBC correlations of H3-18 with C-10, C-11, and C-12, along with H-10 with C-9 and C-11 confirmed oxidation of the C-10/C-11 olefin; similar HMBC correlations were also observed for 3 and 4 (Fig. 3C). HRESIMS and comprehensive NMR supported 3 to be an α-amino acid as observed in the tails of the cyclized terpenoids (1 and 2), whereas 13C NMR resonances at 177.36 and 178.19 ppm, respectively, established 4 and 5 as carboxylic acids. Similarly, the molecular formulas of 6 (C20H36O5) and 7 (C22H38O5) were deduced by HRESIMS (Fig. S33, ESI†). The 13C NMR spectra of 6 and 7 (Tables S9 and S10 and Fig. S34 and S45, ESI†) supported the presence of 20 and 22 carbons, respectively, with each showing four signals (70–90 ppm) for oxygenated carbons and a single signal consistent with the carboxylic acid seen in 4 and 5. A distinct spin system was observed in the 1H-1H COSY spectra of 6 (H2-12/H2-13/H-14) and 7 (H2-14/H2-15/H-16) and taken together with corroborative HMBC correlations, the presence of a tetrahydrofuran ring was determined (Fig. 3C). The relative configurations of the three stereocenters at the tetrahydrofuran ends of 6 and 7 were established based on NOESY correlations, chemical shifts, and comparison with similar compounds. In 6, there is a strong NOESY correlation between H-10 and H-14, supporting that H-14 is on the opposite face of CH3-18 (Fig. S39, ESI†). In addition, the downfield chemical shifts of C-10, C-12, and C-14, in comparison with previously isolated sesquiterpenoids,23,24 indicate a relative configuration of 10R*, 11R*, 14R*; the configuration of C-3 remains undetermined. The chemical shifts and NOESY correlations in 7 are essentially identical, also supporting its relative configuration of 12R*, 13R*, 16R*.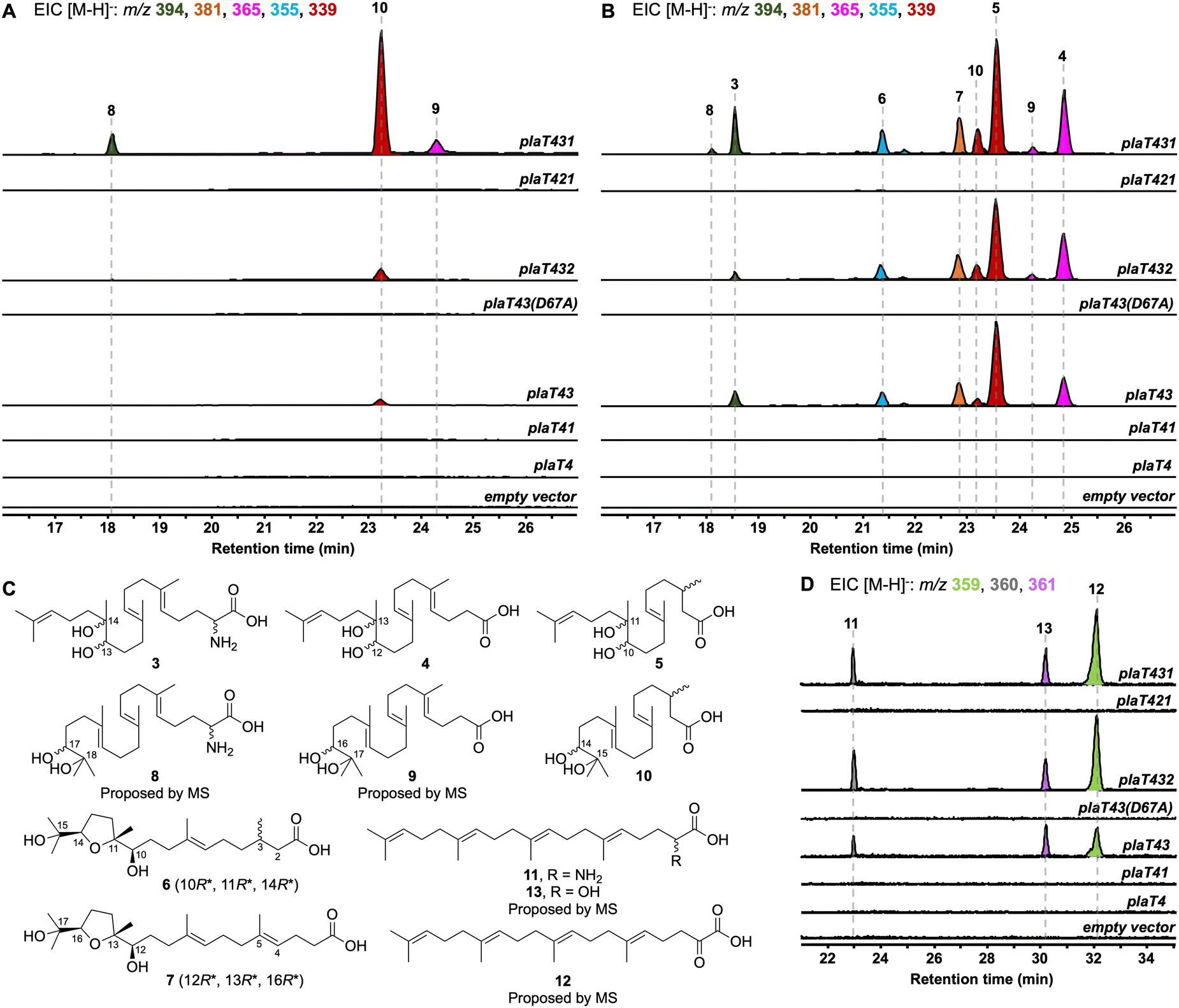 | ||
| Fig. 3 Reconstitution of various combinations of genes involved in biosynthesis of the phenalinolactone diterpenoid core. LCMS-EIC analysis of extracts obtained from S. albus (A and D) and S. venezuelae (B) hosts. (C) Isolated and structurally determined terpenoid metabolites. The relative configurations of 6 and 7 are given. | ||
Compounds 8–10 had m/z values identical to 3–5 but with slightly different retention times (Fig. 3A). A large-scale fermentation (18 L) of S. albus::plaT431 was conducted to isolate 10, which gave the chemical formula of C20H36O4 by HRESIMS (Fig. S46, ESI†). The 1H and 13C NMR spectra of 10 were similar to that of 5, with a doublet methyl signal (δH 0.96 ppm) for C-20 and 13C resonances at 73.34 ppm, 78.48 ppm, and 177.36 ppm, consistent with the diol and carboxylic acid functionalities in 5 (Table S11 and Fig. S47–S51, ESI†). Analysis of the 2D NMR data of 10 confirmed the position of the diol at C-14/C-15, with key 1H–13C HMBC correlations of H3-19 with C-14 and C-15 and H3-20 with C-14 and C-15. Although we did not isolate 8 and 9, their structures were proposed to be the C23 and C22 equivalents of 3 and 4, respectively, based on HRESIMS (Fig. S46, ESI†) and comparisons of their retention times and relative yields to those of 3–5 (Fig. 3). The presence of diols at C-10/C-11 of 5 (and the corresponding positions in 3 and 4) and at C-14/C-15 in 10 suggests epoxidation of either an internal or terminal alkene of the prenyl tail. The terminal position is required for the biosynthesis of 1 and 2via cyclization by PlaT2. The presence of C23 (3 and 8), C22 (4, 7, and 9), and C20 (5, 6, and 10) oxidized metabolites hints towards oxidative degradation of the carboxylic acid end of the metabolites, similar to what was previously seen for bacterial isoprenoid and fatty acid degradation (Fig. S52, ESI†).25,26
Prenylation by PlaT3 is the first step in phenalinolactone biosynthesis
We next set out to determine the order of epoxidation and prenylation by PlaT1 and PlaT3, respectively. LC-MS analysis of our pla constructs revealed that plaT3 is required for production of 3–10. Hosts expressing plaT43, plaT432, or plaT431 produced several diterpenoids, whereas hosts expressing plaT4, plaT41, or plaT421, i.e., those without plaT3, did not produce any detectable diterpenoids (Fig. 3A and B). The presence of several oxidized terpenoids seen in hosts expressing plaT43 (or plaT432), when no oxidase was included, complicated our study of the timing of epoxidation and prenylation. Oxidation was seen at two different positions, the middle alkene (3–5), terminal alkene (8–10), or both (6 and 7; Fig. S53, ESI†), particularly in S. venezuelae and even in the absence of plaT1. We suspected that an endogenous enzyme(s) in the host Streptomyces was oxidizing the prenylated C23 intermediate. This idea was supported by the presence of 3–7 only in S. venezuelae, which must have an endogenous enzyme that oxidizes the internal alkene but is absent in S. albus. However, we could not rule out these oxidations were occurring prior to prenylation. Thus, we targeted an Asp residue for mutagenesis that was previously shown to be essential for catalyzing prenylation in characterized UbiA family prenyltransferases.27,28 We generated the construct plaT43(D67A), which rendered it inactive, to ensure that PlaT3 was not acting as an oxidase and to support that its prenylation activity was required to produce 3–10. Gratifyingly, the expression of plaT43(D67A) in both hosts did not yield any of the metabolites 3–10 (Fig. 3A and B).Further examination of the LC-MS data from S. albus hosts harboring plaT3 uncovered three additional metabolites, 11–13, with m/z ([M − H]−) values of 359, 361, and 360, respectively (Fig. 3D). Unfortunately, very low yields of these metabolites in large-scale fermentation prevented isolation and structure elucidation by NMR. However, HRESIMS gave an [M − H]− value of m/z 360.2907 (calc. [M − H]− at 360.2908) for 11, indicating a molecular formula of C23H39NO2 (Fig. S54, ESI†). Consistent with the production of 3–10, metabolites 11–13 were not produced in any constructs lacking plaT3 or the inactive mutant plaT43(D67A) (Fig. 3D). The presence of 11–13 further supports that prenylation occurs prior to both oxidation and cyclization, implicating that PlaT3 acts on GGPP in the first step of phenalinolactone biosynthesis (Fig. 4). Since the active sites of functionally characterized UbiA prenyltransferases accommodate the diphosphate of a prenyl donor and a small, often aromatic, prenyl acceptor, it is logical that PlaT3 catalyzes the condensation of GGPP and a three-carbon α-keto acid prenyl acceptor.27–29
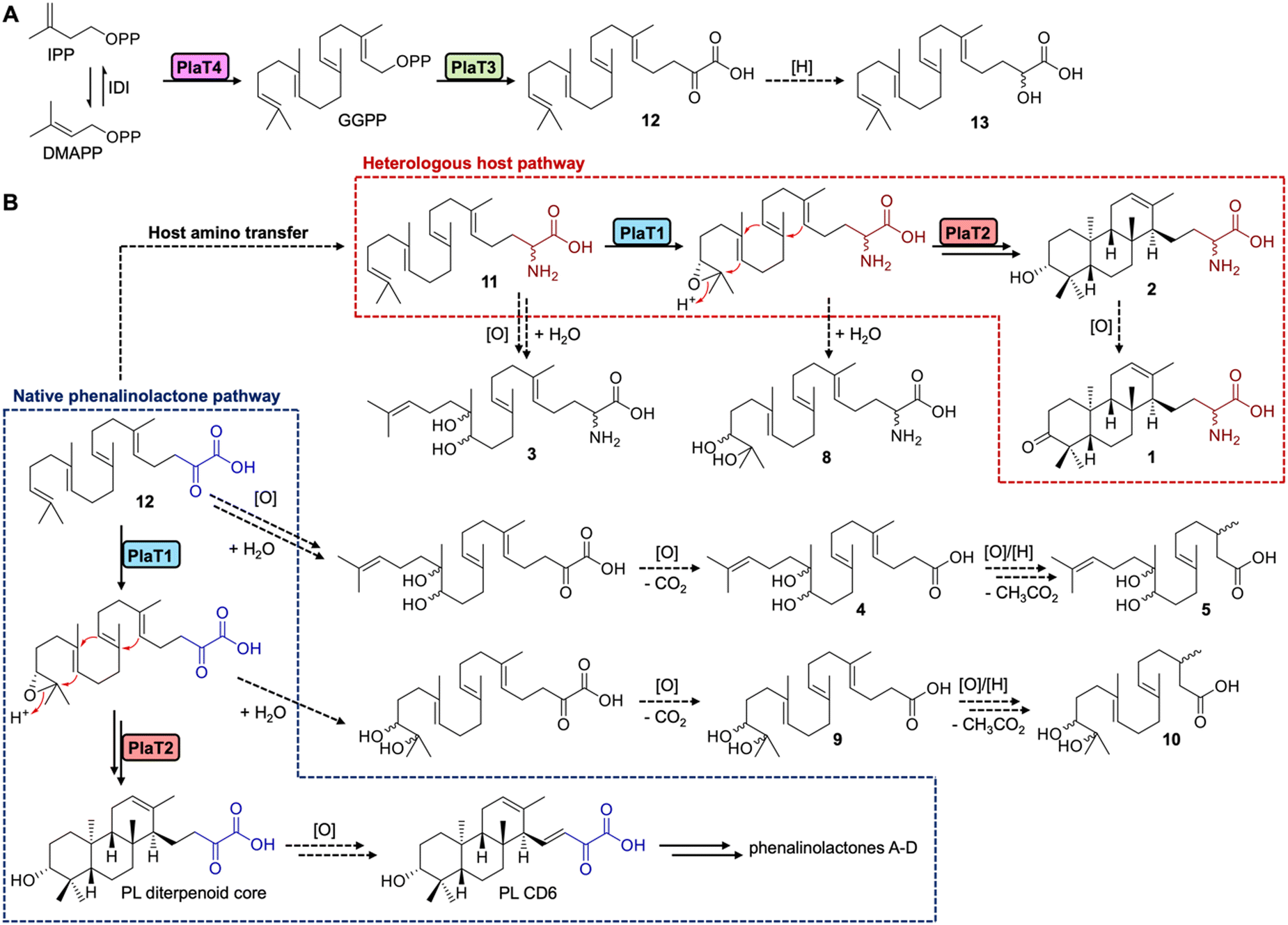 | ||
| Fig. 4 Proposed pathway for the biosynthesis of the phenalinolactone diterpenoid core. (A) Proposed biosynthesis of prenylated intermediates 12 and 13 from IPP and DMAPP. (B) Proposed biosynthetic scheme from the common intermediate 12 for amino acid and keto acid pathways. Dashed arrows labeled only with [H] or [O] denote off-pathway enzymatic reduction or oxidation steps in the heterologous hosts, or undetermined enzymatic steps in the native producer (towards PL CD6). In the heterologous hosts amino transfer may also occur after oxidation (3 and 8) or cyclization (1 and 2). | ||
Overall, an updated proposal for the early-stage biosynthesis of the phenalinolactones involves sequential prenylation by PlaT3, epoxidation by PlaT1, and cyclization by PlaT2 (Fig. 4). In addition, the direct formation of the R-configured hydroxyl at C-3 in 2 by the combined efforts of PlaT1–PlaT4 supports two new insights into phenalinolactone biosynthesis. First, PlaT1 installs a 17R,18-epoxide that PlaT2 then opens to yield the 3R-hydroxylated diterpene core. This stereoselective epoxidation is the same as that proposed in brasilicardin, but results in the opposite stereo configuration found in tiancilactone, longestin, and a proposed intermediate in atolypene biosynthesis,30 suggesting homologous epoxidases in these pathways differentially control biosynthesis and perhaps cyclization. Second, there is no requirement of successive oxidation and reduction to isomerize the C-3 hydroxyl group, as previously proposed.7 The cyclized perhydrophenanthrene core can simply be oxidized to PL CD6 for downstream processing to the phenalinolactones (Fig. 4).
Conclusions
Bacteria represent a treasure trove of biosynthetic potential. While genomic data has driven a reinvigorated effort in the discovery of novel natural products, many biosynthetic pathways have yet to be fully elucidated. By employing heterologous expression, we uncovered new insights into early-stage biosynthesis of the phenalinolactone diterpenoids, demonstrating that assembly of the anti/anti/syn-perhydrophenanthrene core is accomplished via sequential prenylation, epoxidation, and cyclization. These findings are in stark contrast to the previously proposed pathway, where PlaT1 was thought to epoxidize GGPP prior to prenylation by PlaT3. Epoxidation of prenyl diphosphates has not been previously observed in terpenoid pathways; epoxidation typically occurs on substrates lacking the diphosphate group (e.g., squalene or prenylated meroterpenoids). In fact, modification of prenyl diphosphates is rare, with only methylation and hydroxylation known.31,32 Structurally and mechanistically, PlaT2 is akin to oxidosqualene cyclases (OSCs) involved in steroid biosynthesis. Both OSC and PlaT2 accommodate an epoxy substrate lacking the diphosphate head group and initiate cyclization via protonation of the epoxide ring to yield terpenoid scaffolds with C-3 hydroxy functionalities. Interestingly, early-stage biosynthesis of the phenalinolactones follows a similar biosynthetic logic as often described in fungal meroterpenoid pathways (Fig. S55, ESI†), where prenylation and epoxidation occur prior to cyclization. Overall, we propose that the phenalinolactones and related family members (i.e. brasilicardins, tiancilactones, and atolypenes) likely follow a shared biosynthetic rationale, although definitive evidence via in vitro data is still required to confirm that possibility. In parallel to this study, an analogous biosynthetic investigation was performed on the atolypene sesterterpenoids, indicating that prenylation and epoxidation also precede cyclization in ato biosynthesis.30 However, the timing of prenylation and epoxidation in the atolypenes may be different. No products were seen in the construct containing the ato polyprenyl synthase (AtoC) and UbiA prenyltransferase (AtoD) and a hydrolyzed derivative of 14,15-epoxygeranylgeraniol was isolated from a construct without AtoD.30 In conclusion, our findings underline Nature's highly evolved strategies for terpenoid scaffold assembly, providing new insights into terpene biochemistry and establishing the foundation for future discovery, engineering, and enzymology efforts. We envision that a thorough biosynthetic understanding of this family of bacterial terpenoids will help to guide genome mining of novel natural products and facilitate engineering of enzymes and pathways for the production of bioactive metabolites.Experimental procedures
All experimental procedures are described in the ESI.†Author contributions
Conceptualization, J. D. R.; methodology, T. A. A. and J. D. R.; investigation, T. A. A., Z. L., C. A. M., A. J., D. Ł. K., and E. M.; data analysis, T. A. A., Z. L., C. A. M., A. J., D. Ł. K., E. M., L.-B. D., S. L., and J. D. R.; writing – original draft, T. A. A. and J. D. R.; writing – review & editing, T. A. A., Z. L., C. A. M., A. J., D. Ł. K., E. M., L.-B. D., S. L., and J. D. R.; visualization, T. A. A. and J. D. R.; supervision, J. D. R.; funding acquisition, S. L. and J. D. R.Data availability
The data supporting this article have been included as part of the ESI.† The raw NMR data of 1–7 and 10 were deposited in the Natural Product Magnetic Resonance Database (np-mrd.org) under accession numbers NP0333388–NP0333396. Plasmids generated in this study will be made available upon request.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded in part by NIH Grants R00 GM124461 and R35 GM142574 (to J. D. R.) and NSF Grant CHE-2020110 (to S. L.). T. A. A. was supported in part by an NIH Chemistry-Biology Interface Research Training Program Grant T32 GM136583. We acknowledge the University of Florida Mass Spectrometry Research and Education Center (MSREC), which is supported by the NIH (S10 OD021758-01A1). We acknowledge both the University of Florida Center for Nuclear Magnetic Resonance Spectroscopy and the University of Florida McKnight Brain Institute at the National High Magnetic Field Laboratory's Advanced Magnetic Resonance Imaging and Spectroscopy (AMRIS) Facility, which is supported by the US NSF Cooperative Agreement No. DMR-1644779 and the State of Florida, for NMR support. We would like to give special thanks to Drs James Rocca and Ion Ghiviriga for their incredible NMR support and Dr Donovon Adpressa for DFT calculations. Finally, we thank Prof. Dr Axel Zeeck for generously gifting Streptomyces sp. Tü 6071.References
- Dictionary of Natural Products, https://dnp.chemnetbase.com (accessed Mar 19, 2024).
- J. D. Rudolf, T. A. Alsup, B. Xu and Z. Li, Nat. Prod. Rep., 2021, 38, 905–980 RSC.
- D. E. Cane and H. Ikeda, Acc. Chem. Res., 2012, 45, 463–472 CrossRef CAS PubMed.
- M. Nett, H. Ikeda and B. S. Moore, Nat. Prod. Rep., 2009, 26, 1362–1384 RSC.
- Y. Yamada, T. Kuzuyama, M. Komatsu, K. Shin-ya, S. Omura, D. E. Cane and H. Ikeda, Proc. Natl. Acad. Sci. U. S. A., 2014, 112, 857–862 CrossRef PubMed.
- K. Gebhardt, S. W. Meyer, J. Schinko, G. Bringmann, A. Zeeck and H.-P. Fiedler, J. Antibiot., 2011, 64, 229–232 CrossRef CAS PubMed.
- C. Dürr, H.-J. Schnell, A. Luzhetskyy, R. Murillo, M. Weber, K. Welzel, A. Vente and A. Bechthold, Chem. Biol., 2006, 13, 365–377 CrossRef PubMed.
- M. Daum, H. J. Schnell, S. Herrmann, A. Günther, R. Murillo, R. Müller, P. Bisel, M. Müller and A. Bechthold, ChemBioChem, 2010, 11, 1383–1391 CrossRef CAS PubMed.
- T. Binz, S. C. Wenzel, H.-J. Schnell, A. Bechthold and R. Müller, ChemBioChem, 2008, 9, 447–454 CrossRef CAS PubMed.
- C. Kiske, A. Erxleben, X. Lucas, L. Willmann, D. Klementz, S. Günther, W. Romer and B. Kammerer, Rapid Commun. Mass Spectrom., 2014, 28, 1459–1467 CrossRef CAS PubMed.
- M. Myronovskyi, E. Welle, V. Fedorenko and A. Luzhetskyy, Appl. Environ. Microbiol., 2011, 77, 5370–5383 CrossRef CAS PubMed.
- D. W. Christianson, Chem. Rev., 2017, 117, 11570–11648 CrossRef CAS PubMed.
- X. Pan, J. D. Rudolf and L.-B. Dong, Nat. Prod. Rep., 2024, 41, 402–433 RSC.
- H. Shigemori, H. Komaki, K. Yazawa, Y. Mikami, A. Nemoto, Y. Tanaka, T. Sasaki, Y. In, T. Ishida and J. Kobayashi, J. Org. Chem., 1998, 63, 6900–6904 CrossRef CAS PubMed.
- L.-B. Dong, J. D. Rudolf, M.-R. Deng, X. Yan and B. Shen, ChemBioChem, 2018, 19, 1727 CrossRef CAS PubMed.
- S.-W. Kim, W. Lu, M. K. Ahmadi, D. Montiel, M. A. Ternei and S. F. Brady, ACS Synth. Biol., 2019, 8, 109–118 CrossRef CAS PubMed.
- S. Nakanishi, K. Osawa, Y. Saito, I. Kawamoto, K. Kuroda and H. Kase, J. Antibiot., 1992, 45, 341–347 CrossRef CAS PubMed.
- T. Ozaki, S. S. Shinde, L. Gao, R. Okuizumi, C. Liu, Y. Ogasawara, X. Lei, T. Dairi, A. Minami and H. Oikawa, Angew. Chem., Int. Ed., 2018, 57, 6629–6632 CrossRef CAS PubMed.
- F. Wei, W. Li, R. Song and Y. Shen, Nat. Prod. Commun., 2018, 13, 1433–1436 Search PubMed.
- S. J. Moore, H.-E. Lai, S.-M. Chee, M. Toh, S. Coode, K. Chengan, P. Capel, C. Corre, E. L. C. de los Santos and P. S. Freemont, ACS Synth. Biol., 2021, 10, 402–411 CrossRef CAS PubMed.
- A. Botas, M. Eitel, P. N. Schwarz, A. Buchmann, P. Costales, L. E. Núñez, J. Cortés, F. Morís, M. Krawiec, M. Wolański, B. Gust, M. Rodriguez, W.-N. Fischer, B. Jandeleit, J. Zakrzewska-Czerwińska, W. Wohlleben, E. Stegmann, P. Koch, C. Méndez and H. Gross, Angew. Chem., Int. Ed., 2021, 60, 13536–13541 CrossRef CAS PubMed.
- P. N. Schwarz, A. Buchmann, L. Roller, A. Kulik, H. Gross, W. Wohlleben and E. Stegmann, Biotechnol. J., 2018, 13, 1700527 CrossRef PubMed.
- L. Li, R. Liu, L. Han, Y. Jiang, J. Liu, Y. Li, C. Yuan and X. Huang, Magn. Reson. Chem., 2016, 54, 606–609 CrossRef CAS PubMed.
- T. Inui, Y. Wang, D. Nikolic, D. C. Smith, S. G. Franzblau and G. F. Pauli, J. Nat. Prod., 2010, 73, 563–567 CrossRef CAS PubMed.
- W. Seubert, J. Bacteriol., 1960, 79, 426–434 CrossRef CAS PubMed.
- K. Rose and A. Steinbüchei, Appl. Environ. Microbiol., 2005, 71, 2803–2812 CrossRef CAS PubMed.
- S. Ren, N. A. W. de Kok, Y. Gu, W. Yan, Q. Sun, Y. Chen, J. He, L. Tian, R. L. H. Andringa, X. Zhu, M. Tang, S. Qi, H. Xu, H. Ren, X. Fu, A. J. Minnaard, S. Yang, W. Zhang, W. Li, Y. Wei, A. J. M. Driessen and W. Cheng, Cell Rep., 2020, 33, 108294 CrossRef CAS PubMed.
- H. Huang, E. J. Levin, S. Liu, Y. Bai, S. W. Lockless and M. Zhou, PLoS Biol., 2014, 12, 1001911 CrossRef PubMed.
- K. Zhang, G. Zhang, X. Hou, C. Ma, J. Liu, Q. Che, T. Zhu and D. Li, Org. Lett., 2022, 24, 2025–2029 CrossRef CAS PubMed.
- Z. Wang, T. A. Alsup, X. Pan, L. Li, J. Tian, Z. Yang, X. Lin, H.-M. Xu, J. D. Rudolf and L.-B. Dong, ChemRxiv, 2024 DOI:10.26434/chemrxiv-2024-0fz4z.
- C. Ignea, M. H. Raadam, A. Koutsaviti, Y. Zhao, Y.-T. Duan, M. Harizani, K. Miettinen, P. Georgantea, M. Rosenfeldt, S. E. Viejo-Ledesma, M. A. Petersen, W. L. P. Bredie, D. Staerk, V. Roussis, E. Ioannou and S. C. Kampranis, Nat. Commun., 2022, 13, 5188 CrossRef CAS PubMed.
- H. Tsutsumi, Y. Katsuyama, M. Izumikawa, M. Takagi, M. Fujie, N. Satoh, K. Shin-ya, Y. Ohnishi, J. Am. Chem. Soc., 2018, 140, 6631–6639 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00138a |
| This journal is © The Royal Society of Chemistry 2024 |