
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorophosphoniums as Lewis acids in organometallic catalysis: application to the carbonylation of β-lactones†‡
Marie-Hélène
Pietraru
 ,
Louise
Ponsard
,
Nicolas
Lentz
,
Pierre
Thuéry
,
Emmanuel
Nicolas
* and
Thibault
Cantat
*
,
Louise
Ponsard
,
Nicolas
Lentz
,
Pierre
Thuéry
,
Emmanuel
Nicolas
* and
Thibault
Cantat
*
Université Paris-Saclay, CEA, CNRS, NIMBE, Gif-sur-Yvette 91191, France. E-mail: emmanuel.nicolas@cea.fr; thibault.cantat@cea.fr
First published on 4th January 2024
Abstract
We describe the synthesis and characterisation of four organic Lewis acids based on fluorophosphoniums, with tetracarbonyl cobaltate as the counter-anion: [R3PF]+[Co(CO)4]− (with R = o-Tol, Cy, iPr, and tBu). Their catalytic activity was investigated for the carbonylation of β-lactones to succinic anhydrides. In the presence of [tBu3PF]+[Co(CO)4]− IV (3 mol%), 90% of succinic anhydride was afforded from β-propiolactone after 16 h at 80 °C, at a very mild pressure of 2 bar of carbon monoxide. Our study sets the first example of the use of a main-group cation as a Lewis acidic partner in the cobalt-catalyzed carbonylation of β-lactones.
Despite the description of syntheses and characterizations of some fluorophosphoniums since the 1960s,1 their application as Lewis acidic catalysts only emerged recently. Since 2012,2 the group of Stephan has reported an extensive array of fluorophosphoniums, which have contributed to the expansion of metal-free catalytic processes, for reactions such as hydrosilylation of ketones,3 imines, nitriles, and olefins,4 isomerization of olefins,4b hydrodefluorination of fluoroalkanes,4d,e,5 dehydrocoupling of silanes with amines, alcohols,4d,e,5a acids, and thiols,6 transfer hydrogenation of alkenes,6 hydrodeoxygenation of ketones,4c–e,5a deoxygenation of phosphine oxides,7 Friedel–Crafts dimerization,4d,e,5a Diels–Alder reaction, or Nazarov cyclization.8
Encouraged by these results, we envisioned extending the use of Lewis acidic fluorophosphoniums to organometallic catalysis, and transition metal-catalyzed carbonylation was chosen as a relevant application. Indeed, ring-expanding carbonylation of heterocycles has been reported using a combination of a Lewis acid and low-valent cobalt carbonyl species. The carbonylation of epoxides was first investigated by Alper et al. in 2001,9 using various sources of cobalt carbonyl complexes as catalyst, assisted by boronic Lewis acids ([Co2(CO)8]/PPNCl or [PPN]+[Co(CO)4]−, assisted by BF3·Et2O or B(C6F5)3, PPN = bis(triphenylphosphine)iminium). The reaction was improved in the following years by the group of Coates with porphyrin or salphen-based tetradentate chromium or aluminum Lewis acids to obtain unprecedented activity in the carbonylation of epoxides to lactones (Scheme 1, Cat. 1),10 lactones to anhydrides (Scheme 1, Cat. 2),11 or the direct double carbonylation of epoxides to anhydrides (Scheme 1, Cat. 3).12 An aluminum phthalocyanine complex was also reported as a suitable Lewis acid for the carbonylation of epoxides.13 Recently, the groups of Yoon14 and of Dincă and Román-Leshkov15 reported supported heterogeneous catalysts based on these tetradentate chromium or aluminum Lewis acids.
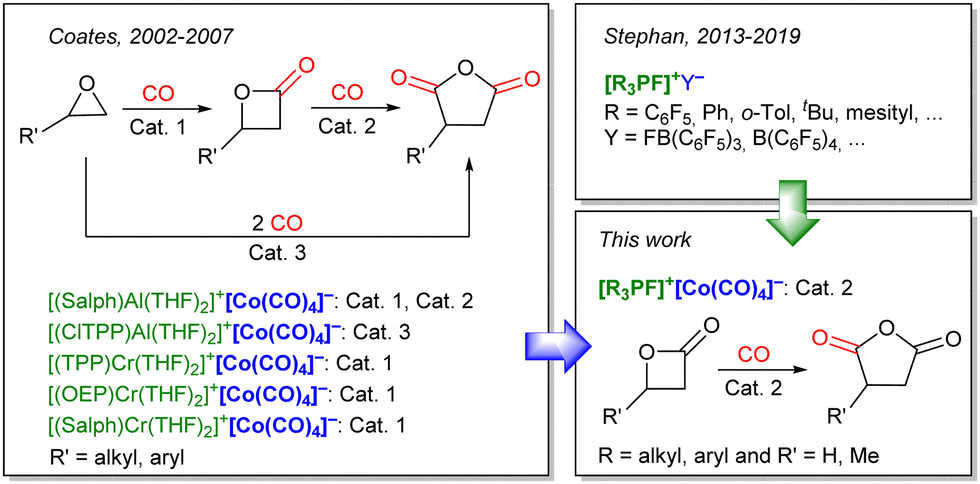 | ||
| Scheme 1 Left: Selected examples of [Lewis acid]+[Co(CO)4]− ion pairs reported by the group of Coates, used as homogeneous catalysts for the carbonylation of epoxides and β-lactones. Salph: N,N′-bis(3,5-di-tert-butylsalicylidene)phenylenediamine; OEP: octaethylporphyrinato; TPP: 5,10,15,20-tetraphenylporphyrin. Top right: Selected examples of ion pairs including a fluorophosphonium cation, reported by the group of Stephan. Bottom right: This work, where novel [Lewis acid]+[Co(CO)4]− ion pairs including a fluorophosphonium cation, are engaged in the carbonylation of β-lactones to succinic anhydrides. | ||
In these reactions, the role of the Lewis acidic cation is decisive for the activity of the catalytic system (Scheme 2):12 the cation should be Lewis acidic enough to bind to the lactone and accelerate its ring-opening through bimolecular nucleophilic substitution by [Co(CO)4]− (step 1). Furthermore, following the carbonylation (step 2), the cation should facilitate the eventual closure of the ring to liberate the product (step 3). Achieving optimal reaction outcomes requires a delicate balance and fine-tuning of the cation's Lewis acidity: while a more potent Lewis acid could accelerate the rate of step 1, it might also lead to the retention and entrapment of intermediates, and the ring-closure step towards product formation (step 3).
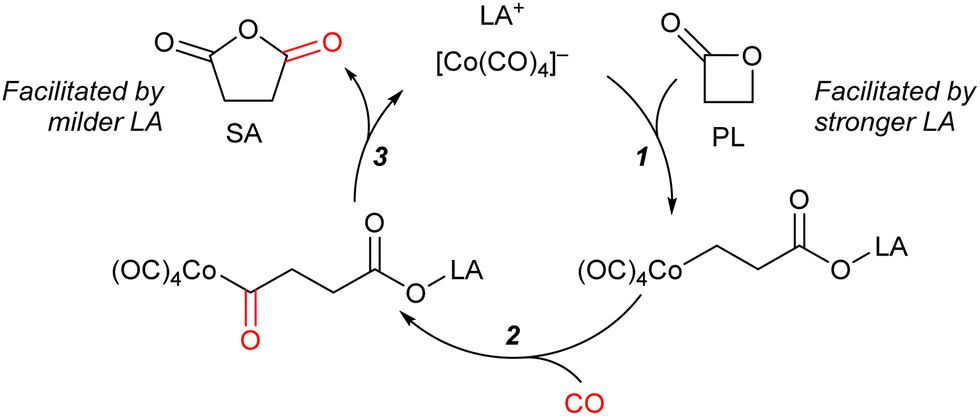 | ||
| Scheme 2 Mechanistic model proposed by Coates for the carbonylation of PL to SA. | ||
Since the low-lying P–F σ* orbital of fluorophosphoniums provides them high Lewis acidity,16 and since additional tuning of the Lewis acidity can be brought through the choice of stronger or milder electron-withdrawing substituents around the phosphorus atom, these cations can be envisaged as suitable promoters for the carbonylation of heterocycles catalyzed by [Co(CO)4]−.
Therefore, we envisioned ion pairs of the formula [R3PF]+[Co(CO)4]− as possible well-defined catalysts for the carbonylation of heterocycles. Although using main-group instead of metal-based Lewis acids seemed advantageous, uncertainty remained regarding their compatibility with the conditions, reactants, and substrates in carbonylations. This work aimed thus at evaluating the performances of [R3PF]+ species as Lewis acidic partners in a dual organometallic catalytic process.
Herein we report the syntheses and characterizations of four fluorophosphoniums associated with a tetracarbonyl cobaltate anion, which were then engaged in the carbonylation of β-propiolactone (PL) to succinic anhydride (SA), and of β-butyrolactone (BL) to methyl succinic anhydride (MeSA).
The starting difluorophosphines were synthesized according to the literature procedure from the corresponding phosphine and XeF2,17 and, without purification, were reacted with an in situ generated triethylsilyltetracarbonylcobalt18 in toluene to give the ion pairs I–IV as yellow, green, or blue powders in good to high yields after purification and isolation (Scheme 3). Et3SiF is formed as a by-product.
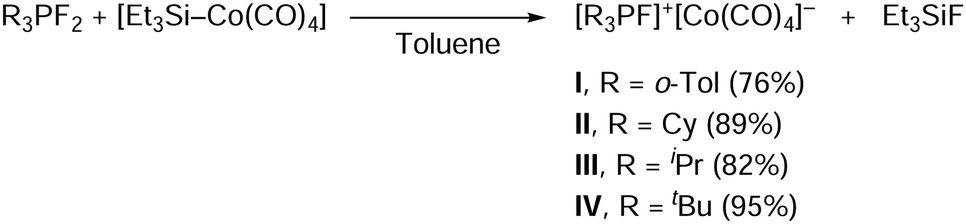 | ||
| Scheme 3 Synthetic pathway towards fluorophosphonium tetracarbonylcobaltates I–IV. | ||
Syntheses were attempted with other aryl substituents around the phosphorus atom: whereas the ion pair [(o-Tol)3PF]+[Co(CO)4]−I could be easily obtained, the syntheses of [Ph3PF]+[Co(CO)4]− and [Mes3PF]+[Co(CO)4]− were however unsuccessful. The resulting products either lacked stability (for R = Ph) or did not form (for R = Mes) (Mes: mesityl; see ESI,‡ Section S3.1).
The 19F and 31P NMR spectra of complexes I–IV exhibited characteristic doublets corresponding to the phosphorous–fluorine coupling (J ≈ 1000 Hz), with chemical shifts similar to the fluorophosphoniums reported by the groups of Schmutzler19 or Stephan17 with Br−, PhPF5−, FB(C6F5)3− or B(C6F5)4− as counter-anions (Table 1).
| Entry | Catalyst | J P–F (Hz) | δ(31P) (ppm) | δ(19F) (ppm) |
|---|---|---|---|---|
| a From ref. 17. b From ref. 19. | ||||
| 1 | [o-Tol3PF]+[Co(CO)4]− (I) | 993 | 103.2 | −125.4 |
| 2a | [o-Tol3PF]+[FB(C6F5)3]− | 994 | 104.3 | −125.5 |
| 3a | [o-Tol3PF]+[B(C6F5)4]− | 993 | 103.2 | −125.5 |
| 4 | [Cy3PF]+[Co(CO)4]− (II) | 993 | 133.0 | −171.1 |
| 5 | [iPr3PF]+[Co(CO)4]− (III) | 997 | 146.4 | −169.4 |
| 6b | [iPr3PF]+Br− | 966 | 145.0 | −167.0 |
| 7b | [iPr3PF]+[PhPF5]− | 962 | 147.0 | −167.0 |
| 8 | [tBu3PF]+[Co(CO)4]− (IV) | 1019 | 150.6 | −171.4 |
| 9a | [tBu3PF]+[FB(C6F5)3]− | 1019 | 148.5 | −171.6 |
| 10a | [tBu3PF]+[B(C6F5)4]− | 1019 | 147.5 | −171.6 |
As described by the group of Stephan,17 the more electron-withdrawing the substituents on the fluorophosphonium, the lower the 31P NMR chemical shifts, and the higher the 19F NMR chemical shifts: the aryl-substituted salt I showcases a 31P NMR chemical shift around 100 ppm and a 19F NMR chemical shift around −125 ppm, while for alkyl-substituted II, III, and IV, δ(31P) ≈ 140 ppm, and δ(19F) ≈ −170 ppm.
Crystals of I, II, and IV were obtained by diffusion of pentane in a DME solution (Fig. 1). The cobalt and phosphorus atoms adopt tetrahedral geometries in the molecular structure of I, II, and IV. While complexes I and IV are devoid of symmetry, II presents with mirror symmetry. The P–F bond lengths are all similar, around 1.555 Å, suggesting that the bulkiness of the substituents does not have much influence on the length of the P–F bond in our fluorophosphoniums; on the contrary, the group of Stephan17 reported that the P–F bond length in [o-Tol3PF][FB(C6F5)3] was 1.5543(3) Å (vs. 1.549(1) Å in I), which was significantly lower than 1.628(2) Å in [tBu3PF][FB(C6F5)3] (vs. 1.5563(9) Å in IV). The sums of the C–P–C angles are respectively 337.1, 341.6, and 344.6° in I, II, and IV, consistent with the increasing bulkiness of the substituents on the phosphorus atom. The anion and cation are well separated, with Co–F distances of 3.5881(8), 4.3906(10), and 5.5795(8) Å in I, II, and IV, respectively, much larger than the sum of the ionic radii of cobalt and fluorine (ca. 1.9 Å).20
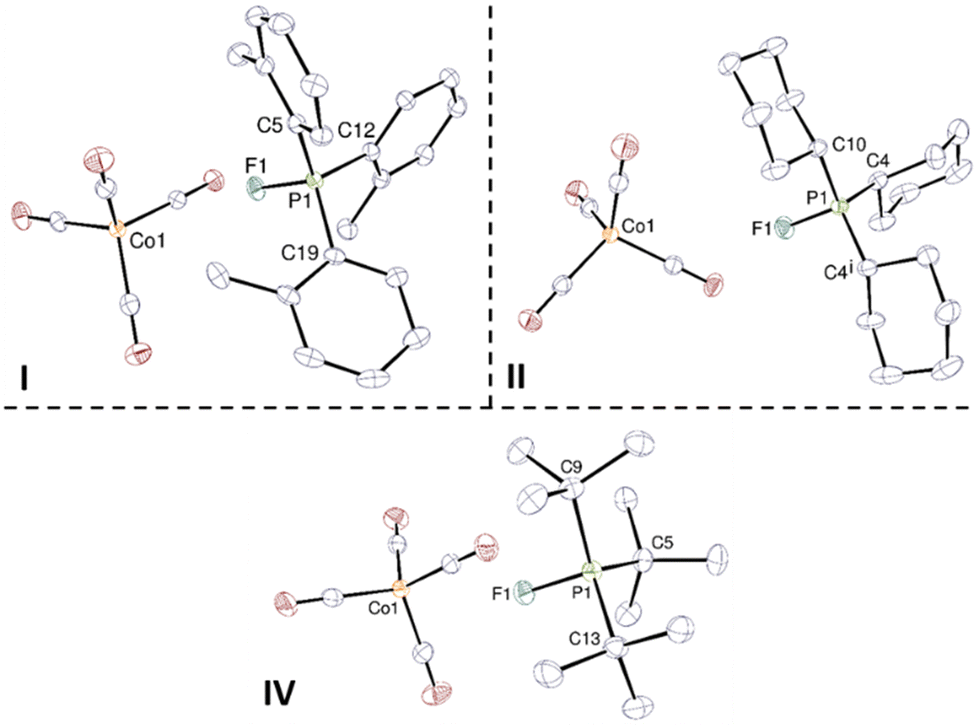 | ||
| Fig. 1 Crystal structures of I, II and IV. Displacement ellipsoids are shown at the 50% probability level and hydrogen atoms are omitted. Symmetry code for II: i = x, 3/2 − y, z. Selected bond distances (Å) and bond angles (°). For I: P1–F1 1.5486(8); C5–P1–C12 110.68(6); C5–P1–C19 114.14(6); C12–P1–C19 112.27(6). For II: P1–F1 1.5547(10); C4–P1–C4i 118.57(7); C4–P1–C10 111.51(4). For IV: P1–F1 1.5563(9); C5–P1–C9 114.35(7); C9–P1–C13 114.97(6); C13–P1–C5 115.27(7). | ||
Fluorophosphonium tetracarbonylcobaltates I–IV were engaged in the carbonylation of PL to SA (Table 2). Under 50 bar of CO, in toluene, after 16 h of reaction, whereas only 58% of SA was obtained in presence of aryl-substituted I (Table 2, entry 1), alkyl-substituted II, III, and IV, which contain a cation with a milder Lewis acidity, afforded higher yields of SA, respectively 75, 70, and 75% (Table 2, entries 2–4). This result is in line with the balanced Lewis acidity required to avoid trapping of intermediate carboxylate-LA adducts (see Scheme 2).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | [LA]+[Co(CO)4]− | P CO (bar) | T (°C) | PL conversion (%) | SA yield (%) | Selectivity (%) |
| a Conditions: PL (2 mol L−1), mesitylene (internal standard, 10 mol%), and [LA]+ [Co(CO)4]− (3 mol%) in toluene (1 mL), heated for 16 h under CO pressure. SA yields and PL conversions measured by GC-MS analysis. | ||||||
| 1 | I | 50 | 80 | 74 | 58 | 78 |
| 2 | II | 50 | 80 | 87 | 75 | 86 |
| 3 | III | 50 | 80 | 88 | 70 | 80 |
| 4 | IV | 50 | 80 | 85 | 75 | 88 |
| 5 | IV | 50 | 100 | 95 | 74 | 78 |
| 6 | IV | 20 | 80 | 96 | 78 | 81 |
| 7 | IV | 10 | 80 | 99 | 91 | 92 |
| 8 | IV | 2 | 80 | 96 | 90 | 94 |
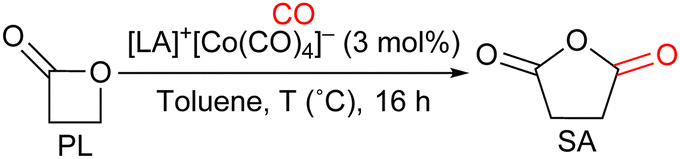
Further optimization was performed with complex IV: a similar yield of SA was obtained when the reaction was run at 100 °C instead of 80 °C, while the conversion of PL increased from 85 to 95%, suggesting that a higher temperature reduced the selectivity (Table 2, entry 5). Lowering the CO pressure contributed to a better catalytic activity: 78% of SA was obtained at 20 bar, 91% at 10 bar, and 90% at 2 bar, with almost complete conversion of PL in each case (Table 2, entries 6–8). Compared to the system proposed by the group of Coates, which yielded 98% of SA from the carbonylation of PL under 14 bar of CO in the presence of [(salph)Al(THF)2]+[Co(CO)4]− (0.3 mol%) after 24 h at 24 °C, our catalytic system afforded SA in high yields as well, with only low to moderate pressure of CO; however, the metallic-based LA of Coates appears to induce more activity, and at room temperature, since our process required the application of a higher catalytic loading and a higher temperature to achieve similar activity in a comparable duration.
The same reaction conditions were applied to the more challenging carbonylation of BL to MeSA in presence of IV (Table 3).
|
|
|||||
|---|---|---|---|---|---|
| Entry | T (°C) | Duration (h) | BL conversion (%) | MeSA yield (%) | Selectivity (%) |
| a Conditions: BL (2 mol L−1) and mesitylene (internal standard, 10 mol%) with IV (3 mol%) in toluene (1 mL), heated for the indicated duration under CO (10 bar). MeSA yields and BL conversions measured by GC-MS analysis. b 9% of CA was obtained as a side-product. c 49% of CA was obtained as a side-product. | |||||
| 1 | 80 | 16 | 3 | 4 | >99 |
| 2 | 100 | 16 | 33 | 33 | >99 |
| 3 | 100 | 48 | 92 | 72 | 78 |
| 4 | 120 | 16 | 74 | 62 | 84 |
| 5b | 120 | 52 | 89 | 64 | 72 |
| 6c | 140 | 16 | 86 | 28 | 33 |
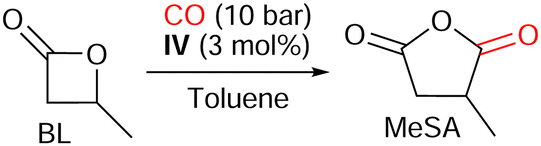
The presence of the methyl group hinders the electrophilic site of the lactone and causes the rate-determining ring-opening of BL by nucleophilic attack of [Co(CO)4]− to be more difficult (see ESI,‡ Section S2.2),12 and indeed, only traces of MeSA were obtained after 16 h at 80 °C, under 10 bar of CO (Table 3, entry 1). Providing more energy to the system thanks to elevated reaction temperatures unlocked the activity of the catalytic system: 33% of MeSA was afforded after 16 h at 100 °C, 62% at 120 °C, but only 28% at 140 °C (Table 3, entries 2, 4, and 6). Nonetheless, higher temperatures induced a reduction of the selectivity as well: CO2 and H2 were detected in the gas phase after carbonylation runs at 100, 120, or 140 °C (see ESI,‡ Section S3.2), while isomerization of BL to crotonic acid was observed at 140 °C, and even at 120 °C after a prolonged heating (Table 3, entries 5, and 6). This suggests that a compromise should be found in the choice of temperature between activation and selectivity. The 48-hour-long reaction at 100 °C enabled the almost complete conversion of BL and yielded 72% of MeSA (Table 3, entry 3).
In summary, we described the synthesis and characterization of four fluorophosphonium tetracarbonylcobaltates (I–IV). To highlight the potential of fluorophosphoniums as organic Lewis acids in a transition metal-catalyzed reaction, the catalytic activities of I–IV were evaluated in the carbonylation of PL to SA. Alkyl-substituted fluorophosphoniums were the best candidates, and further optimization demonstrated that the carbonylation of PL and BL to respectively SA and MeSA could be achieved in good to high yields in presence of IV at low pressure.
For financial support of this work, we acknowledge CEA, CNRS, University Paris-Saclay, the CHARMMMAT Laboratory of Excellence, and the European Research Council (ERC Consolidator grant agreement no. 818260). N. L. and L. P. were supported by a fellowship from the European Union Horizon 2020 research and innovation program under grant agreement no. 768919 (Carbon4PUR project). We thank Thierry Bernard (CEA) for help in the conception and realization of the autoclaves, and Antonin Homassel for his help in performing the stoichiometric reaction between BL and complex IV.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. F. Nixon and R. Schmutzler, Spectrocs. Acta, 1964, 20, 1835–1842 CrossRef CAS; (b) R. Schmutzler, J. Am. Chem. Soc., 1964, 86, 4500–4502 CrossRef CAS; (c) R. Baumgärtner, W. Sawodny and J. Goubeau, Z. Anorg. Allg. Chem., 1964, 333, 171–180 CrossRef.
- L. J. Hounjet, C. B. Caputo and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 4714–4717 CrossRef CAS PubMed.
- L. Süsse, J. H. W. LaFortune, D. W. Stephan and M. Oestreich, Organometallics, 2019, 38, 712–721 CrossRef.
- (a) M. Perez, Z. W. Qu, C. B. Caputo, V. Podgorny, L. J. Hounjet, A. Hansen, R. Dobrovetsky, S. Grimme and D. W. Stephan, Chem. – Eur. J., 2015, 21, 6491–6500 CrossRef CAS PubMed; (b) M. Perez, L. J. Hounjet, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 18308–18310 CrossRef CAS PubMed; (c) M. H. Holthausen, M. Mehta and D. W. Stephan, Angew. Chem., Int. Ed., 2014, 126, 6656–6659 CrossRef; (d) M. H. Holthausen, R. R. Hiranandani and D. W. Stephan, Chem. Sci., 2015, 6, 2016–2021 RSC; (e) J. M. Bayne, M. H. Holthausen and D. W. Stephan, Dalton Trans., 2016, 45, 5949–5957 RSC.
- (a) I. Mallov and D. W. Stephan, Dalton Trans., 2016, 45, 5568–5574 RSC; (b) C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374–1377 CrossRef CAS PubMed; (c) M. H. Holthausen, M. Mehta and D. W. Stephan, Angew. Chem., Int. Ed., 2014, 53, 6538–6541 CrossRef CAS PubMed.
- M. Perez, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10917–10921 CrossRef CAS PubMed.
- M. Mehta, I. Garcia de la Arada, M. Perez, D. Porwal, M. Oestreich and D. W. Stephan, Organometallics, 2016, 35, 1030–1035 CrossRef CAS.
- M. Vogler, L. Süsse, J. H. W. LaFortune, D. W. Stephan and M. Oestreich, Organometallics, 2018, 37, 3303–3313 CrossRef CAS.
- J. T. Lee, P. J. Thomas and H. Alper, J. Org. Chem., 2001, 66, 5424–5426 CrossRef CAS PubMed.
- (a) Y. D. Getzler, V. Mahadevan, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1174–1175 CrossRef CAS PubMed; (b) J. A. Schmidt, V. Mahadevan, Y. D. Getzler and G. W. Coates, Org. Lett., 2004, 6, 373–376 CrossRef CAS PubMed; (c) J. A. Schmidt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 11426–11435 CrossRef CAS PubMed; (d) J. W. Kramer, E. B. Lobkovsky and G. W. Coates, Org. Lett., 2006, 8, 3709–3712 CrossRef CAS PubMed; (e) T. L. Church, Y. D. Getzler and G. W. Coates, J. Am. Chem. Soc., 2006, 128, 10125–10133 CrossRef CAS PubMed; (f) V. Mahadevan, Y. D. Getzler and G. W. Coates, Angew. Chem., Int. Ed., 2002, 114, 2905–2908 CrossRef.
- Y. D. Getzler, V. Kundnani, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 6842–6843 CrossRef CAS PubMed.
- J. M. Rowley, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 4948–4960 CrossRef CAS PubMed.
- J. Jiang, S. Rajendiran and S. Yoon, Asian J. Org. Chem., 2019, 8, 151–154 CrossRef CAS.
- (a) J. Jiang and S. Yoon, J. Mater. Chem. A, 2019, 7, 6120–6125 RSC; (b) V. Ganesan and S. Yoon, ACS Appl. Mater. Interfaces, 2019, 11, 18609–18616 CrossRef CAS PubMed; (c) V. Ganesan and S. Yoon, Inorg. Chem., 2020, 59, 2881–2889 CrossRef CAS PubMed.
- (a) H. D. Park, M. Dinca and Y. Roman-Leshkov, ACS Cent. Sci., 2017, 3, 444–448 CrossRef CAS PubMed; (b) H. D. Park, M. Dinca and Y. Roman-Leshkov, J. Am. Chem. Soc., 2018, 140, 10669–10672 CrossRef CAS PubMed.
- A. R. Jupp, T. C. Johnstone and D. W. Stephan, Dalton Trans., 2018, 47, 7029–7035 RSC.
- C. B. Caputo, D. Winkelhaus, R. Dobrovetsky, L. J. Hounjet and D. W. Stephan, Dalton Trans., 2015, 44, 12256–12264 RSC.
- A. J. Chalk and J. F. Harrod, J. Am. Chem. Soc., 1967, 89, 1640–1647 CrossRef CAS.
- R. Bartsch, O. Stelzer and R. Schmutzler, Z. Naturforsch. B, 1981, 36, 1349–1355 CrossRef.
- R. D. Shannon, Acta Cryst. A, 1976, 32, 751–767 CrossRef.
Footnotes |
| † This article was posted as a preprint on ChemRxiv. DOI: 10.26434/chemrxiv-2023-d3r4g. |
| ‡ Electronic supplementary information (ESI) available: Experimental details and data. CCDC 2286472–2286474. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc04282k |
| This journal is © The Royal Society of Chemistry 2024 |