
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bridge improvement work: maximising non-linear optical performance in polyoxometalate derivatives†
Claire F.
Jones
a,
Bethany R.
Hood
a,
Yovan
de Coene
 b,
Ivan
Lopez-Poves
a,
Benoît
Champagne
*c,
Koen
Clays
*b and
John
Fielden
*ad
b,
Ivan
Lopez-Poves
a,
Benoît
Champagne
*c,
Koen
Clays
*b and
John
Fielden
*ad
aSchool of Chemistry, University of East Anglia, Norwich, NR4 7TJ, UK
bDepartment of Chemistry, University of Leuven, Celestijnenlaan 200D, Leuven B-3001, Belgium
cUnit of Theoretical and Structural Physical Chemistry, Namur Institute of Structured Matter, University of Namur, Namur B-5000, Belgium
dDepartment of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK. E-mail: J.Fielden@Lancaster.ac.uk
First published on 11th January 2024
Abstract
New phenyl and stilbene-bridged polyoxometalate (POM) charge-transfer chromophores with diphenylamino donor groups produce, respectively, the highest intrinsic and absolute quadratic hyperpolarisabilities measured for such species. The β0,zzz obtained for the phenyl bridge – at 180 × 10−30 esu – is remarkable for a short conjugated system while changing to the stilbene (260 × 10−30 esu) produces a substantial increase in non-linearity for a minimal red-shift in the absorption profile. Together with TD-DFT calculations, the results show that maximising conjugation in the π-bridge is vital to high performance in such “POMophores”.
Polyoxometalates (POMs) are a diverse class of anionic molecular metal oxide clusters, which encompass a diverse range of properties.1 This range of properties can be expanded, and tuned through formation of hybrid POM-organic materials – for example by ion-pairing,2 more specific supramolecular interactions,3 and by covalent derivatisation.4 Due to enforced spatial proximity and (in some cases) through-bond electronic communication, covalent derivatisation gives the greatest potential for significantly adjusting the intrinsic properties of the POM such as redox potentials,4b and for emergence of physical properties not found independently in either subunit, for example long-range photo-induced charge separation.5
The strongest POM-organic electronic communication is seen in arylimido derivatives, where direct conjugation of the POM core and aromatic system occurs through the metal-nitrogen multiple bond, resulting in new electronic transitions and strongly shifted redox potentials.4e,6 We have exploited this unmatched electronic communication to construct molecular charge-transfer chromophores (POMophores) based on arylimido-Lindqvist ([Mo6O18NAr]2−) acceptor units attached to a number of organic donor groups and π-bridges.7 These materials have shown high 2nd order non-linear optical (NLO) coefficients, β, particularly considering their relatively high visible transparency and small π-systems. They can also have significant 3rd order properties,8 and recently demonstrated reversible, redox switched NLO responses.9 Thus, such POMophores have the potential to yield high-activity, switchable NLO materials while avoiding problems for efficiency and stability that result from reabsorption of second harmonic (SH) light.
However, there is still much to learn about basic structure–property relationships in POMophores. To date our work has predominantly featured dimethylamino donor groups, with phenyl or phenylacetylene bridges, yet a diphenylamino (–NPh2) donor gave the highest static hyperpolarizability (β0).7c Herein, we expand the family of –NPh2 donor POMophores showing a consistent performance advantage for –NPh2 over –NMe2, and suggested by prior calculations,10 introduce an alkene bridge for the first time obtaining the highest β0 yet for a POMophore.
Compounds 1 to 3 (Fig. 1 and 2 previously published7c) were obtained by synthesising the aniline precursor, then reacting with [NBu4]2[Mo6O19] using established coupling methods (Scheme S1, ESI†).4e,6b,7 Synthesis of 3 by Heck coupling on an iodo-substituted POM precursor was also investigated, an approach with little precedent,11 but isolation of significant quantities of the aniline derivative as well as 3 indicated that the imido bond poorly tolerates Heck conditions.
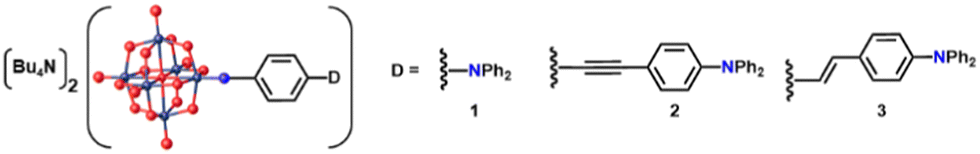 | ||
| Fig. 1 POMophores 1 to 3. Mo atoms are dark grey, O red and the imido-N blue. Compound 2 was previously published.7c | ||
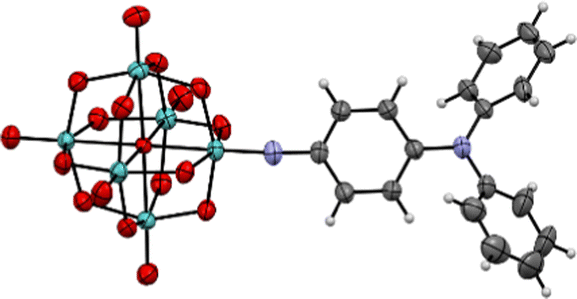 | ||
| Fig. 2 X-ray crystal structure of the molecular anion in compound 1. Disordered parts omitted for clarity, thermal ellipsoids are at the 30% probability level, C atoms are grey; Mo, green; O, red; N, blue. H atoms are white spheres of arbitrary radii. The crystal structure of 2 was previously published (CCDC 1837405†).7c | ||
The three compounds have been characterised by multiple techniques, including X-ray crystal structures for 1 (Fig. 2, Fig. S1 and Tables S1–S3, ESI†) and 2 (previously published).7c UV-vis spectra (Table 1; Fig. S2, ESI†) reveal a remarkable trend in λmax for the low energy intra-hybrid charge transfer (IHCT) bands of the three POMophores: a substantial blue shift of 20 nm (0.13 eV) from phenyl bridged 1 to diphenylacetylene bridged 2. In organic CT chromophores, including the –NO2 acceptor analogues of 1 to 3, extending conjugation invariably lowers CT transition energies,12 while in –NMe2 donor POMophores CT transitions are only very slightly higher in energy for the longer diphenylacetylene bridge.7b The strong blue shift for 2vs. 1 thus indicates a very strong influence for the donor group and bridge on the extent of charge transfer to the POM – whereby in 1 more involvement of the POM effectively lengthens the conjugation pathway and lowers the HOMO–LUMO energy gap. In compound 3, stronger planar conjugation resulting from the alkene rather than alkyne bridge restores the transition to slightly lower energy than in 1.
| λ max/nm (ε, 103 M−1 cm−1)a | E max/eVa | HWHM/nm | E max (calcd)/eVb (f) | E 1/2 vs. Fc0/+/V (ΔE/mV)c | |
|---|---|---|---|---|---|
| a Concentrations ca. 10−5 M in MeCN. b TD-DFT computed value (ωB97X-D/6-311G(d)/LanL2TZ). c Solutions ca. 10−3 M in analyte, 0.1 M in [NBu4][BF4] at a glassy carbon working electrode, scan rate 100 mV s−1. Ferrocene internal reference, ΔEp = 74 mV. | |||||
| 1 | 434 (34.3) | 2.86 | 33 | 3.22 (1.18) | −1.003 (76) |
| 2 | 414 (45.3) | 2.99 | 46 | 3.30 (2.43) | −0.949 (80) |
| 3 | 437 (43.5) | 2.84 | 46 | 3.17 (2.40) | −0.964 (72) |
For all three compounds, TD-DFT calculations (ωB97X-D/6-311G(d)/LanL2TZ, MeCN solvation by IEFPCM) reproduce experimental trends in the transition energies, and computed oscillator strengths closely follow the experimental trend in ε (Table 1 and Fig. S2 and S4, ESI†). Blue shifts of ca. 0.3 eV vs. experiment reflect the fact that these vertical excitation energies do not account for geometry relaxation of the excited state, or vibronic structure.13 The computed NTOs confirm that charge transfer is from the organic group to the POM, and qualitatively show greater participation of the POM in the acceptor (particle) orbitals for 1 than for 2 or 3 (Fig. 3). This is reflected in computed ground-to-excited state dipole moment changes (Δμ, Table S4, ESI†) that are almost identical in 1 to 2 and 3, and a charge transfer distance (dCT, Table S4, ESI†) only 20% shorter for 1, despite its much shorter conjugated bridge. Natural population analysis (NPA) of the ground states (Fig. S5, ESI†) is also consistent with more effective donor–acceptor communication in 1, through a higher calculated NPA negative charge on the {Mo6O18} unit and a less negative NPA charge on the –NPh2 donor than in either 2 or 3.
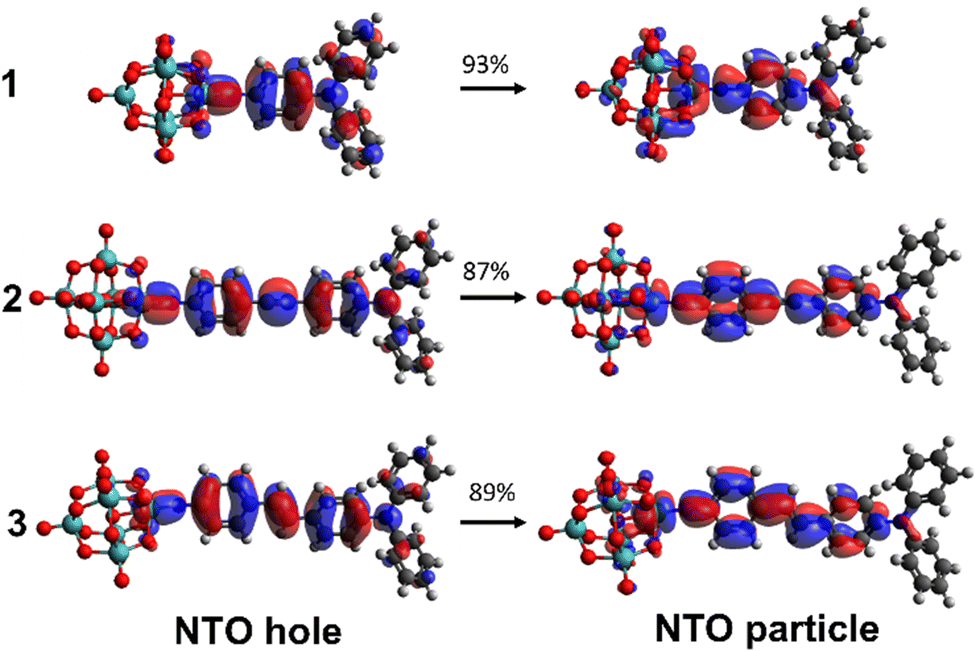 | ||
| Fig. 3 TD-DFT calculated (ωB97X-D/6-311G(d)/LanL2TZ) NTOs for the dominant transitions of the IHCT bands of 1 to 3. MeCN solvation is provided by IEFPCM. | ||
Electrochemical measurements (Table 1 and Fig. S3, ESI†) show [Mo6O18NR]2−/3− reductions for all three compounds that are highly reversible on the CV timescale, however, with no steric bulk around the imido group the reduced states are not stable enough for investigation of switched properties.9,14 The reduction potentials E1/2 trend more negative, in line with the trend in calculated NPA charges (Fig. S5, ESI†), as the strength of communication between –NPh2 donor and POM acceptor increases across the bridge from diphenylacetylene 2, to stilbene 3 and phenyl 1. The negative shift for 1, at ca. 55 mV vs.2, is rather smaller than reported for the analogous –NMe2 compound,7b as the availability of the N lone pair is reduced by the phenyl groups. The consequence of this is also seen in the X-ray crystal structure of 1, where contraction of ortho-to-meta-C–C bond distances (mean 1.381(7) Å) vs. meta-to-para (mean 1.396(8) Å) is much less significant than for –NMe2 (Table S3, ESI†), indicating a weaker contribution from quinoidal resonance forms.
Hyper-Rayleigh scattering (HRS) determined and TD-DFT computed β values for 1 to 3 are shown in Table 2, and a number of relevant comparisons with POM and –NO2 acceptors are presented in the ESI† (Table S5, ESI†). For 2 and 3, use of a 1064 nm fundamental gave good results, but for 1 a high level of two-photon fluorescence at 1064 nm necessitated use of a 1200 nm source. Subsequently 1200 nm was used to obtain data for all three compounds under the same conditions, and to extract non-resonant, static hyperpolarizabilities β0,zzz. The results show that all three compounds have high optical non-linearities: β0,zzz values are several times higher than reported for the technologically exploited DAS+ cation under non-resonant conditions and they are the three highest performing POMophores to date. The β0,zzz of 1, at 180 × 10−30 esu, is remarkably high for such a short π-bridge – as underlined by normalisation of β0 to N, the effective number of polarisable π-electrons in the bridge, to yield an extremely high intrinsic β (β0/N3/2), of 12.2. This more than doubles the highest intrinsic β previously obtained for a POMophore (the –NMe2 analogue of 1),7c while producing only a 10 nm red-shift in λmax and consequently pushing the performance well beyond the empirical β0/N3/2vs. λmax limits described by Kuzyk for planar organic chromophores.16 Similar percentage increases in smaller β values are seen in purely organic chromophores with short (i.e. phenyl) conjugated bridges on changing –NMe2/–NEt2 for –NPh2/NTol2 donor groups, and have been ascribed to extension of donor orbitals onto the aryl groups.12 Extending with the alkyne bridge in 2 slightly lowers β0, consistent with the blue shifted electronic transitions, while extending with an alkene to form 3 produces a ca. 50% increase in β0 (to 260 × 10−30 esu) vs. either of the other two compounds. This is with only a 3 nm red shift in λmaxvs. the phenyl bridge, and while the broader IHCT band and higher ε of 3 results in more absorption >450 nm, at the 600 nm SH wavelength it is quite minimal. Thus, while 1 has the highest intrinsic β of any POMophore to date, compound 3 gives the best absolute performance and retains an excellent transparency/non-linearity trade-off.
| β zzz,1064 | β zzz,1200 | β 0,zzz | β zzz,1064 (calcd)c | β zzz,1200 (calcd)c | β 0/N3/2d | |
|---|---|---|---|---|---|---|
| a β zzz calculated assuming a single dominant tensor component, measured using 1064 or 1200 nm fundamental laser beams. The quoted units (esu) can be converted into SI units (C3 m3 J−2) by dividing by a factor of 2.693 × 1020. b Non-resonant, static β estimated from βzzz,1200 using the two state model.15 c TD-DFT computed value (ωB97X-D/6-311G(d)/LanL2TZ) following B-convention for comparison with experiment. Values in parentheses account for differences of frequency dispersion between experiment and calculations. d N = Number of electrons in π-bridge:16 6 for 1 and 14 for 2 and 3. | ||||||
| /10−30 esu | ||||||
| 1 | — | 430 ± 30 | 180 ± 10 | 261 (287) | 199 (213) | 12.2 |
| 2 | 590 ± 20 | 376 ± 30 | 174 ± 10 | 373 (340) | 286 (267) | 3.32 |
| 3 | 1040 ± 20 | 640 ± 30 | 260 ± 10 | 454 (435) | 335 (328) | 4.96 |
TD-DFT computed unit sphere representations (Fig. 4) indicate that the β responses are dominated by a single tensor component directed along the molecular charge transfer axis, justifying the extraction of a single βzzz tensor from experiment. Dynamic β values, however, are underestimated vs experiment. In trend, they reproduce the relationship between phenyl-bridged 1 and stilbene 3 quite well, but over-estimate the non-linearity for 2 so that it is higher, rather than similar or lower than that of 1. This is likely to reflect the fact that β values are computed for an optimised, flat geometry where the dihedral angles (ϕ) between phenyl rings and alkene or alkyne bridge are close to 0°, lacking the effects of structural fluctuations.17 Yet, while the phenyl rings of 3 have a small rotational energy barrier of only ca. 5 kJ mol−1 between 0 and ± 30°, and a locally flat PES like that of trans-stilbene,18 there are substantial barriers to further rotation. For diphenylacetylenes such as 2 there is free rotation at room temperature with an energy barrier of only 2.5 kJ mol−1 across all values of ϕ.19 This means that less well conjugated rotamers contribute much more to the experimental measurements on 2 and lower β. Inspection of the computed NTOs (Fig. 2) shows that in 1, the donor orbitals extend strongly onto the –NPh2 rings: compared to –NMe2, this will result in a larger Δμ explaining the large (100%) increase in β (Table S5, ESI†). As the chromophores are extended, NTO holes extend less onto the –NPh2 rings and NTO particles extend less onto the POMs, so that Δμ and dCT only increase minimally with apparent conjugation length. A corresponding trend is seen in plots of computed ground-to-excited state electron density change (Fig. S6, ESI†). This reduces the performance advantage for the POM/-NPh2 combination compared to other POM-based and organic chromophores. Nonetheless, the β0,zzz for 3 is ca. 30% higher than its nitro analogue (202 × 10−30 esu, Table S5, ESI†) on the same set up, showing that the increased conjugation provided by the alkene-bridge enables the POM acceptor to clearly exceed the perfomance of –NO2 in an extended structure for the first time.
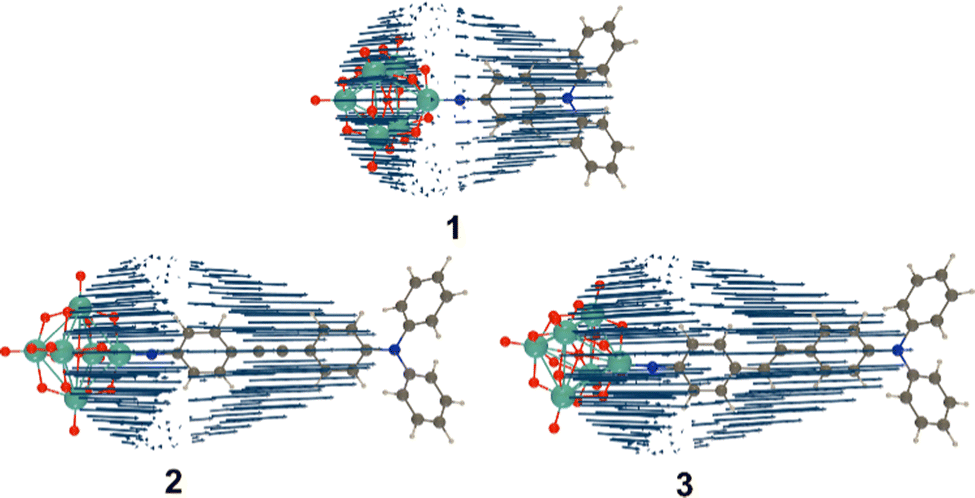 | ||
| Fig. 4 Unit sphere representations (USRs) of the β tensor (λ = 1200 nm) of compounds 1 to 3 calculated at the IEFPCM (solvent = acetonitrile) TDDFT/ωB97X-D/6-311G(d)/LanL2TZ level of theory. (USR factor of 0.0001). | ||
In summary, we have synthesised two new NLO-active POMophores with phenyl and stilbene bridges and diphenylamino donor groups, producing the highest intrinsic and absolute β0 values yet reported for such compounds. Together with the previously published diphenylacetylene-bridge analogue, these results show that a key for maximising the performance advantages of the imido-Lindqvist acceptor is to maximise the strength of conjugation across the organic bridge. Work to test other, stronger donor groups, and extended planar π-bridges, is under way.
In addition to the ESI† and deposited cif files, data can be obtained by contacting the corresponding author, and will be deposited at DOI: https://doi.org/10.17635/lancaster/researchdata/649.
We thank the EPSRC for support through grant EP/M00452X/1 to J. F., X-ray data were obtained in facilities established by EPSRC grant EP/S005854/1. BRH thanks the University of East Anglia for a studentship and Lancaster University for short-term postdoctoral support. C. F. J. and J. F. acknowledge funding from the Leverhulme Trust (RPG-2020-365) and thank Dr Joseph Wright of the University of East Anglia for assistance in re-synthesis of 3. YDC acknowledges the Fonds Wetenschappelijk Onderzoek (FWO) for junior postdoc (No. 1234222N). Calculations were performed on the UEA High Performance Cluster, Consortium des Equipements de Calcul Intensif (CECI, https://www.ceci-hpc.be) and the Technological Platform of High-Performance Computing, for which the authors acknowledge the financial support of the FNRS-FRFC, the Walloon Region, and University of Namur (Conventions No. 2.5020.11, U.G006.15, U.G018.19, U.G011.22, 1610468, and RW/GEQ2016).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) C. L. Hill, Chem. Rev., 1998, 98, 1 CrossRef CAS PubMed; (b) M. Hutin, M. H. Rosnes, D. L. Long and L. Cronin, Comprehensive Inorg. Chem. II, 2nd edn, 2013, vol. 2, pp. 241 Search PubMed.
- (a) P. Bolle, C. Menet, M. Puget, H. Serier-Brault, S. Katao, V. Guerchais, F. Boucher, T. Kawai, J. Boixel and R. Dessapt, J. Mater. Chem. C, 2021, 9, 13072 RSC; (b) Y. Gao, M. Choudhari, G. K. Such and C. Ritchie, Chem. Sci., 2022, 13, 2510 RSC.
- (a) M. Han, J. Hey, W. Kawamura, D. Stalke, M. Shionoya and G. H. Clever, Inorg. Chem., 2012, 51, 9574 CrossRef CAS PubMed; (b) Y. Wu, R. Shi, Y.-L. Wu, J. M. Holcroft, Z. Liu, M. Frasconi, M. R. Wasielewski, H. Li and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 4111 CrossRef CAS PubMed; (c) C. Falaise, M. A. Moussawi, S. Floquet, P. A. Abramov, M. N. Sokolov, M. Haouas and E. Cadot, J. Am. Chem. Soc., 2018, 140, 11198 CrossRef CAS PubMed; (d) B. Zhang, W. Guan, F. Yin, J. Wang, B. Li and L. Wu, Dalton Trans., 2018, 47, 1388 RSC.
- (a) A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382 RSC; (b) A. J. Kibler, N. Tsang, M. Winslow, S. P. Argent, H. W. Lam, D. Robinson and G. N. Newton, Inorg. Chem., 2023, 62, 3585 CrossRef CAS PubMed; (c) A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009 CrossRef CAS PubMed; (d) A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605 RSC; (e) J. Zhang, F. Xiao, J. Hao and Y. Wei, Dalton Trans., 2012, 41, 3599 RSC.
- (a) F. Odobel, M. Séverac, Y. Pellegrin, E. Blart, C. Fosse, C. Cannizzo, C. R. Meyer, K. J. Elliott and A. Harriman, Chem. – Eur. J., 2009, 15, 3130 CrossRef CAS PubMed; (b) K. J. Elliott, A. Harriman, L. Le Pleux, Y. Pellegrin, E. Blart, C. R. Mayer and F. Odobel, Phys. Chem. Chem. Phys., 2009, 11, 8767 RSC; (c) B. Matt, X. Xiang, A. L. Kaledin, N. Han, J. Moussa, H. Amouri, S. Alves, C. L. Hill, T. Lian, D. G. Musaev, G. Izzet and A. Proust, Chem. Sci., 2013, 4, 1737 RSC; (d) B. Matt, J. Fize, J. Moussa, H. Amouri, A. Pereira, V. Artero, G. Izzet and A. Proust, Energy Environ. Sci., 2013, 6, 1504 RSC.
- (a) J. B. Strong, G. P. A. Yap, R. Ostrander, L. M. Liable-Sands, A. L. Rheingold, R. Thouvenot, P. Gouzerh and E. A. Maatta, J. Am. Chem. Soc., 2000, 122, 639 CrossRef CAS; (b) Y. Wei, B. Xu, C. L. Barnes and Z. Peng, J. Am. Chem. Soc., 2001, 123, 4083 CrossRef CAS PubMed.
- (a) A. Al-Yasari, N. Van Steerteghem, H. El Moll, K. Clays and J. Fielden, Dalton Trans., 2016, 45, 2818 RSC; (b) A. Al-Yasari, N. Van Steerteghem, H. Kearns, H. El Moll, K. Faulds, J. A. Wright, B. S. Brunschwig, K. Clays and J. Fielden, Inorg. Chem., 2017, 17, 10181 CrossRef PubMed; (c) A. Al-Yasari, P. Spence, H. El Moll, N. Van Steerteghem, P. N. Horton, B. S. Brunschwig, K. Clays and J. Fielden, Dalton Trans., 2018, 47, 10415 RSC.
- A. Al-Yasari, H. El Moll, R. Purdy, K. B. Vincent, P. Spence, J.-P. Malval and J. Fielden, Phys. Chem. Chem. Phys., 2021, 23, 11807 RSC.
- B. R. Hood, Y. de Coene, A. V. Torre Do Vale Froes, C. F. Jones, P. Beaujean, V. Liégeois, F. MacMillan, B. Champagne, K. Clays and J. Fielden, Angew. Chem., Int. Ed., 2023, 62, e202215537 CrossRef CAS PubMed.
- E. Rtibi and B. Champagne, Symmetry, 2021, 13, 1636 CrossRef CAS.
- Y. Zhu, L. Wang, J. Hao, P. Yin, J. Zhang, Q. Li, L. Zhu and Y. Wei, Chem. – Eur. J., 2009, 15, 3076 CrossRef CAS PubMed.
- (a) C. R. Moylan, R. J. Tweig, V. Y. Lee, S. A. Swanson, K. M. Betterton and R. D. Miller, J. Am. Chem. Soc., 1993, 115, 12599 CrossRef CAS; (b) C. M. Whitaker, E. V. Patterson, K. L. Kott and R. J. McMahon, J. Am. Chem. Soc., 1996, 118, 9966 CrossRef CAS.
- F. Zutterman, V. Liégeois and B. Champagne, ChemPhotoChem, 2017, 1, 281 CrossRef CAS.
- B. R. Hood, PhD thesis, University of East Anglia, 2022.
- (a) J. L. Oudar and D. S. Chemla, J. Chem. Phys., 1977, 66, 2664 CrossRef CAS; (b) J. L. Oudar, J. Chem. Phys., 1977, 67, 446 CrossRef CAS.
- (a) K. Tripathy, J. Pérez Moreno, M. G. Kuzyk, B. J. Coe, K. Clays and A. M. Kelley, J. Chem. Phys., 2004, 121, 7932 CrossRef CAS PubMed; (b) K. Clays and B. J. Coe, Chem. Mater., 2003, 15, 642 CrossRef CAS.
- F. Castet, C. Tonnelé, L. Muccioli and B. Champagne, Acc. Chem. Res., 2022, 55, 3716 CrossRef CAS PubMed.
- M. M. Lin, D. Shorokhov and A. H. Zewail, J. Phys. Chem. A, 2009, 113, 4075 CrossRef CAS PubMed.
- (a) M. Wierzbicka, I. Bylí, C. Czaplewski and W. Wiczk, RSC Adv., 2015, 5, 29294 RSC; (b) K. Okuyama, T. Hasegawa, M. Ito and N. Mikami, J. Phys. Chem., 1984, 88, 1711 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and other experimental/computational details, CIF file. CCDC 2305545. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc05433k. |
| This journal is © The Royal Society of Chemistry 2024 |