
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffective and sustainable depolymerization of Nylon 66 – a transamidation for the complete recycling of polyamides†
Robin
Coeck
 and
Dirk E.
De Vos
*
and
Dirk E.
De Vos
*
Centre for Membrane Separations, Adsorption, Catalysis and Spectroscopy for Sustainable Solutions (cMACS), KU Leuven, 3001 Leuven, Belgium. E-mail: dirk.devos@kuleuven.be
First published on 11th January 2024
Abstract
The transamidation of polyamides with short primary amides is reported as an effective recycling technique. This novel depolymerization method is robust and only utilizes cheap and renewable reagents. The process requires a Nb2O5 catalyst, assisted by NH3, and operates at relatively mild reaction conditions (i.e. 200 °C and 3 bar NH3).
Polyamides (PAs) or nylon are an important class of nitrogen-containing polymers. With an annual production of more than 8 Mton, the PA market is mainly driven by the high demand for Nylon 6 (i.e. PA 6) and Nylon 66 (i.e. PA 66), with corresponding market shares of approximately 50% and 40%.1 Polyamides are known for their strength, durability, thermal and chemical stability. These excellent mechanical properties make them versatile and desirable materials for a variety of applications. PAs can be found in everyday objects such as clothing or are used for high-end applications such as 3D printing.2 The downside of PAs' durability is their high resilience towards degradation and depolymerization. This makes PAs notoriously difficult to recycle.
Over the past decades, many attempts were made to depolymerize PAs, e.g. via alcoholysis, ammonolysis, hydrolysis etc.3,4,6,8–10 In particular, the steam-assisted depolymerization of PAs proved to be very effective for lactam-based PAs such as Nylon 6. In supercritical liquid or with overheated steam molten Nylon 6 is depolymerized at temperatures of up to 420 °C.4 The generated caprolactam is immediately removed from the reaction mixture. Such a depolymerization was not successful for Nylon 66. During Nylon 66 hydrolysis, a reactive and unvolatile mixture of acids and amines is generated, which repolymerizes, thus leading to a Nylon 66 resin.5 In order to fully depolymerize Nylon 66, stoichiometric quantities of mineral acids or alkali are required, trapping either the α,ω-dicarboxylic acids or α,ω-diamines in their salt form.4d,6
To this date, the situation for Nylon 66 remains problematic. The lack of efficient recycling methods results in the predominant incineration of the material via waste-to-energy (WtE). While this method would be acceptable for plastics such as polyethylene (PE) or polypropylene (PP) this is not the case for nitrogen-containing plastics such as PA. Burning these polymers not only emits CO2, but also toxic NOx greenhouse gases (N2O has a global warming potential that is 265 times higher than that of CO2).7 The development of new and effective recycling methods is therefore essential to minimize potent greenhouse gas emission and to enable a circular economy.11
In the past few years, several new PA recycling methods were proposed that utilize hydrogen (H2) or ammonia (NH3) as the splitting agent. However, each of these new processes suffers from significant disadvantages. For instance, the teams of Milstein and Schaub proposed homogeneous hydrogenolysis catalysts.8 While both groups were able to successfully split polyamides into α,ω-diamines and diols, or, aminoalcohols, the performance of their catalysts for Nylon 66 is remarkably low. Additionally, these specialized catalysts are expensive and easily poisoned by additives or impurities that might be present in actual plastic (waste). In the work of Stuyck et al. Nylon 66 was depolymerized via a sustainable and green ammonolysis process.9 Reactions were performed with a homogeneous Lewis acid catalyst and in a simple glycol solvent, e.g. biosourced waste glycerol. Unfortunately, the ammonolysis of PAs is an equilibrium reaction. As a result, this method is unable to split Nylon 66 completely into monomeric compounds and a fairly reactive product mixture is obtained. To deal with this drawback, Coeck et al. coupled the ammonolysis to an irreversible reaction.10 This approach worked excellently for long-chain polyamides (LCPA) to generate α,ω-diamines, α,ω-dinitriles or amidonitriles. However, these new recycling methods have not yet been proven effective for Nylon 66.
In this work, we report a new and sustainable depolymerization method for PAs, optimized for Nylon 66 specifically. The depolymerization involves a transamidation reaction with a short monofunctional amide (see top Table 1). The trans-amidation of PAs is a fairly unknown reaction. However, during melting or extrusion (≥250 °C) some sporadic, uncatalyzed transamidations occur.12 In more recent years, spontaneous transamidation reactions at a high temperature allowed material scientists to generate “PA alloys”.13 However, such transamidations have never been performed catalytically or at a moderate temperature, nor was anyone ever able to effectively depolymerize PAs doing so.
|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Substrate | Catalyst | Solvent (R–CONH2) | P NH3 | Temp. (°C) | Time (h) | Product yieldsb | |||||
| ① | ② | ||||||||||
| a Amount of secondary amide bonds. b Leftover plastic not characterized. c S = N-butylbenzenesulfonamide, which was added to the mixture (0.022 g). d PP = polypropylene, which was added (0.25 g). e This value also corresponds to the adipamide yield, which is generated stoichiometrically. f Aside from the amides formed from Nylon 6, a yield of 5% caprolactam was obtained as well. | |||||||||||
| 1 | Nylon 66 | 0.5 Ma | — | — | Acetamide | 10 g | 3 bar | 200 | 5 | 1%e | 1% |
| 2 | Nylon 66 | 0.5 Ma | Nb2O5 | 0.1 g | Acetamide | 10 g | — | 200 | 5 | 4%e | 4% |
| 3 | Nylon 66 | 0.5 Ma | Nb2O5 | 0.1 g | Acetamide | 10 g | 1 bar | 200 | 5 | 7%e | 7% |
| 4 | Nylon 66 | 0.5 Ma | Nb2O5 | 0.1 g | Acetamide | 10 g | 3 bar | 200 | 5 | 12%e | 13% |
| 5 | Nylon 66 | 0.5 Ma | Nb2O5 | 0.1 g | Acetamide | 10 g | 5 bar | 200 | 5 | 9%e | 8% |
| 6 | Nylon 66 | 0.5 Ma | Nb2O5 | 0.2 g | Acetamide | 10 g | 3 bar | 200 | 16 | 65%e | 23% |
| 7 | Nylon 66 | 0.5 Ma | Nb2O5 | 0.2 g | Acetamide | 10 g | 3 bar | 225 | 16 | 94%e | 5% |
| 8 | Nylon 66 + Sc | 0.5 Ma | Nb2O5 | 0.2 g | Acetamide | 10 g | 3 bar | 200 | 16 | 69%e | 20% |
| 9 | Nylon 66 + PPd | 0.5 Ma | Nb2O5 | 0.2 g | Acetamide | 10 g | 3 bar | 200 | 16 | 69%e | 20% |
| 10 | Nylon 6 | 0.1 Ma | Nb2O5 | 0.2 g | Acetamide | 10 g | 3 bar | 200 | 16 | 23%f | 35%f |
| 11 | Nylon 66 | 0.1 Ma | Nb2O5 | 0.2 g | Acetamide | 10 g | 3 bar | 200 | 72 | 98%e | 2% |
| 12 | Nylon 66 | 0.5 Ma | Nb2O5 | 0.2 g | Acetamide | 10 g | 3 bar | 200 | 72 | 93%e | 7% |
| 13 | Nylon 66 | 1.25 Ma | Nb2O5 | 0.5 g | Acetamide | 10 g | 3 bar | 200 | 72 | 89%e | 9% |
| 14 | Nylon 66 | 0.5 Ma | Nb2O5 | 0.2 g | Propionamide | 10 g | 3 bar | 200 | 72 | 92%e | 8% |

To start, acetamide was chosen as the most ideal primary amide reagent because it can be cheaply biosourced (e.g. from acetic acid) and is readily biodegradable.14 In a blank reaction at 200 °C barely 1% of monomeric compounds was obtained (Table 1, entry 1), proving the need for a catalyst. Upon addition of ortho-Nb2O5, the yield of N,N′-hexamethylene bis(acetamide) (1) increased substantially (entries 2–5). This comes to no surprise; we previously reported the ability of Nb2O5 to activate secondary amide bonds.10 Ammonia (NH3) was also introduced into the system, to assist in breaking down the polymer by ammonolysis. Under the applied conditions, virtually no amine products were detected. Thus, NH3 merely acts as a co-catalyst. An optimum in NH3 pressure was observed at 3 bar, with a yield of 12% N,N′-hexamethylene bis(acetamide) and 13% of the dimer (i.e. N1-(6-acetamidohexyl)adipamide (2)). Upon further addition of NH3, the global catalytic performance decreases, most likely due to a poisoning effect of the high NH3 concentration on the Nb2O5 surface.
Next, the reaction time was prolonged from 5 to 16 hours (entry 6). A spectacular increase in monomer yield (i.e. from 12% to 65% (1)) illustrates that the polymer is chopped up at random, gradually breaking it down into smaller pieces. By slightly increasing the reaction temperature to 225 °C (entry 7), Nylon 66 was completely depolymerized into monomers (94% (1)) and dimers (5% (2)). Note that for every molecule of N,N′-hexamethylene bis(acetamide), a molecule of adipamide is generated as well. In addition to our desired products, we also detected very low quantities of pyrimidines and triazines, resulting from the trimerization of acetonitrile (i.e. dehydrated acetamide).15 This side reaction is caused by the Nb2O5 catalyst, which is known for its ability to (partially) dehydrate primary amides to nitriles.10,16 In order to maximize the solvent's reusability, we did not further increase the reaction temperature.
The system was also proven to be very robust (entries 8–10). The addition of other plastics (e.g. polypropylene, entry 9) or sulfur-containing plasticizers (e.g. N-butylbenzenesulfonamide, entry 8) did not hinder the reaction. In particular, the catalytic system's resilience towards plasticizers is a significant advance over current recycling techniques. Such sulfur-containing molecules typically poison metal-based catalysts almost immediately.8,10 In addition, we succeeded in breaking down Nylon 6 as well (entry 10). Therefore, our innovative depolymerization system has the potential to be a valuable approach for carpet recycling, where Nylon 66/Nylon 6/PP are commonly mixed and/or copolymerized.2,3a
Next, reactions were performed with a long reaction time of 72 h and with a variable concentration of PA (entries 11–13). These experiments allow to identify the position of the thermodynamic equilibrium under each set of reaction conditions. In general, one could consider a single polymer string of Nylon 66 as a series of 1,6-diaminohexane (HMDA) molecules trapped in secondary amide bonds. It appears that the thermodynamic stability of the amide bond in HMDA-adipyl is similar to that of HMDA-acetyl. In other words, the results depicted in entries 11–13 nearly perfectly match the situation in which each amino group of a molecule HMDA does not differentiate between a condensation reaction with adipamide or acetamide (i.e. R1–NH2 + NH2–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–R2 to R1–NH–C(O)–R2). For example, in entry 12, the reaction contains 169 mmol carbonyl groups from acetamide and 5 mmol from adipamide (ratio of 97
O)–R2 to R1–NH–C(O)–R2). For example, in entry 12, the reaction contains 169 mmol carbonyl groups from acetamide and 5 mmol from adipamide (ratio of 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). If no differentiation occurs, one would expect a N,N′-hexamethylene bis(acetamide) yield of 94% (= 0.97 × 0.97), which matches the experimental value of 93% very well. It is worth noting that even at a high polymer concentration, i.e. 12.5 wt% Nylon 66 (entry 13), the plastic can still be depolymerized completely into mainly monomeric compounds (i.e. 89% yield) with the dimer as the secondary side product (9% yield). This is the consequence of the large molar excess of the small molecule acetamide versus adipamide (i.e. 169 mmol acetamide versus 6.25 mmol adipamide).
:3). If no differentiation occurs, one would expect a N,N′-hexamethylene bis(acetamide) yield of 94% (= 0.97 × 0.97), which matches the experimental value of 93% very well. It is worth noting that even at a high polymer concentration, i.e. 12.5 wt% Nylon 66 (entry 13), the plastic can still be depolymerized completely into mainly monomeric compounds (i.e. 89% yield) with the dimer as the secondary side product (9% yield). This is the consequence of the large molar excess of the small molecule acetamide versus adipamide (i.e. 169 mmol acetamide versus 6.25 mmol adipamide).
Finally, as an alternative to acetamide, a transamidation reaction was performed with propionamide, which poses fewer health risks for mammals (e.g. humans; entry 14).14 Although the final monomer yield is slightly lower than with acetamide, a final yield of 92% N,N′-hexamethylene bis-(propionamide) was obtained with 8% of the dimer (i.e. N1-(6-propionamido-hexyl)adipamide) as the only side product.
Based on prior knowledge and our experimental results, we propose a reaction mechanism for the ammonia assisted transamidation of PAs (Scheme 1).10 Initially, the carbonyl group of the secondary amide (i.e. PA) needs the Lewis acid site (L) on the catalytic surface to be reactive (a). The positive effect of NH3 on the reaction rate is in agreement with the next step, i.e. a nucleophilic attack of NH3 on the surface-adsorbed amide to form a primary amide and a free amine group (b)(c).
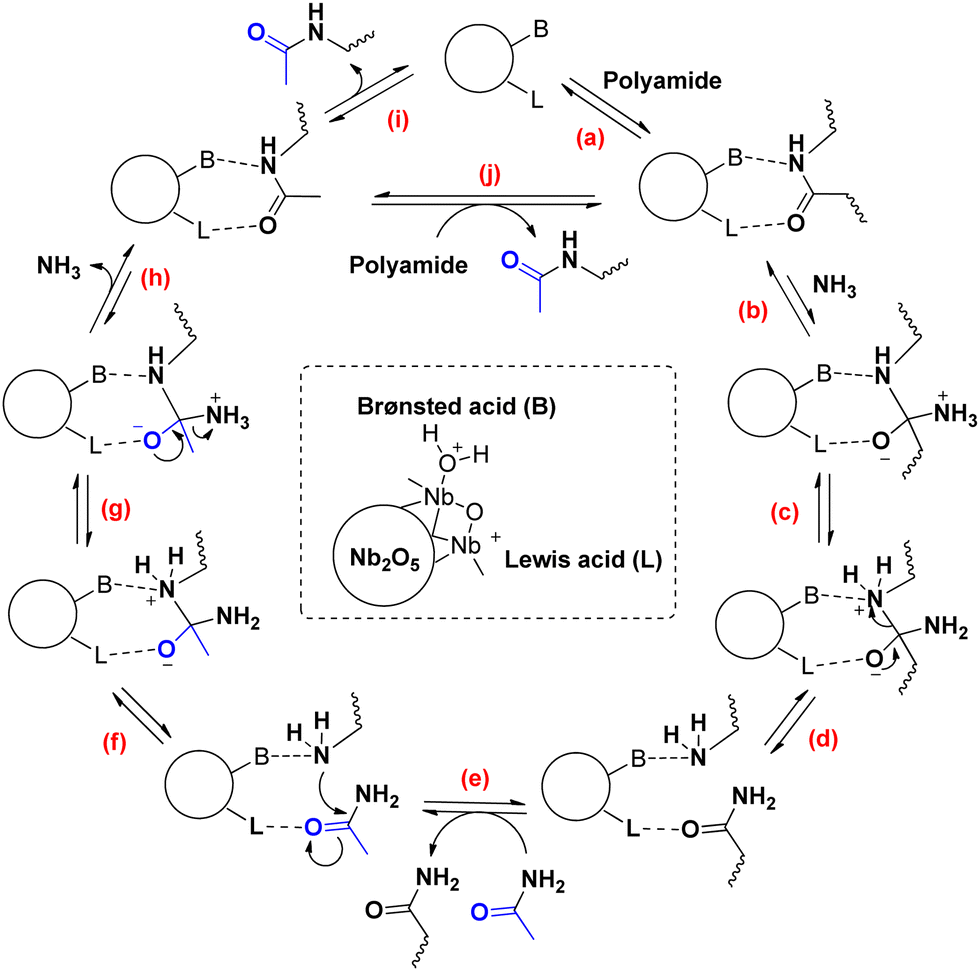 | ||
| Scheme 1 Proposed reaction mechanism for the ammonia assisted transamidation of polyamides with acetamide on a Nb2O5 catalyst. | ||
During this reaction, a neighboring (Brønsted) acid group facilitates the leaving of the amine group (d). If the surface-adsorbed primary amide end-group is displaced by an acetamide molecule from the solvent (e), the ammonolysis reaction can be reversed. First, the free amine chain end attacks the now activated carbonyl group of acetamide (f). The resulting intermediate releases a molecule of NH3, yielding the final transamidation product (g)(h). After desorption of the product, a new secondary amide molecule can coordinate to the catalytic surface (i)(j). All elementary steps in this mechanism are reversible. Nevertheless, the PA is depolymerized completely as a consequence of the large molar excess of the small molecule acetamide versus adipamide.
In summary, we have developed a very robust recycling method that only uses renewable and/or low-cost materials and reagents. Nylon 66 could be depolymerized into easily processable monomers at relatively high concentrations, i.e. 12.5 wt% of Nylon 66 (in acetamide). After a transamidation reaction with acetamide, two distinct monomers are obtained: N,N′-hexamethylene bis(acetamide) and adipamide (Scheme 2). These monomers differ greatly in chemical properties, facilitating an easy separation. Adipamide's limited solubility in most organic solvents allows a separation via crystallization. In addition, the boiling point of these monomers is very different, allowing classical isolation methods such as distillation.
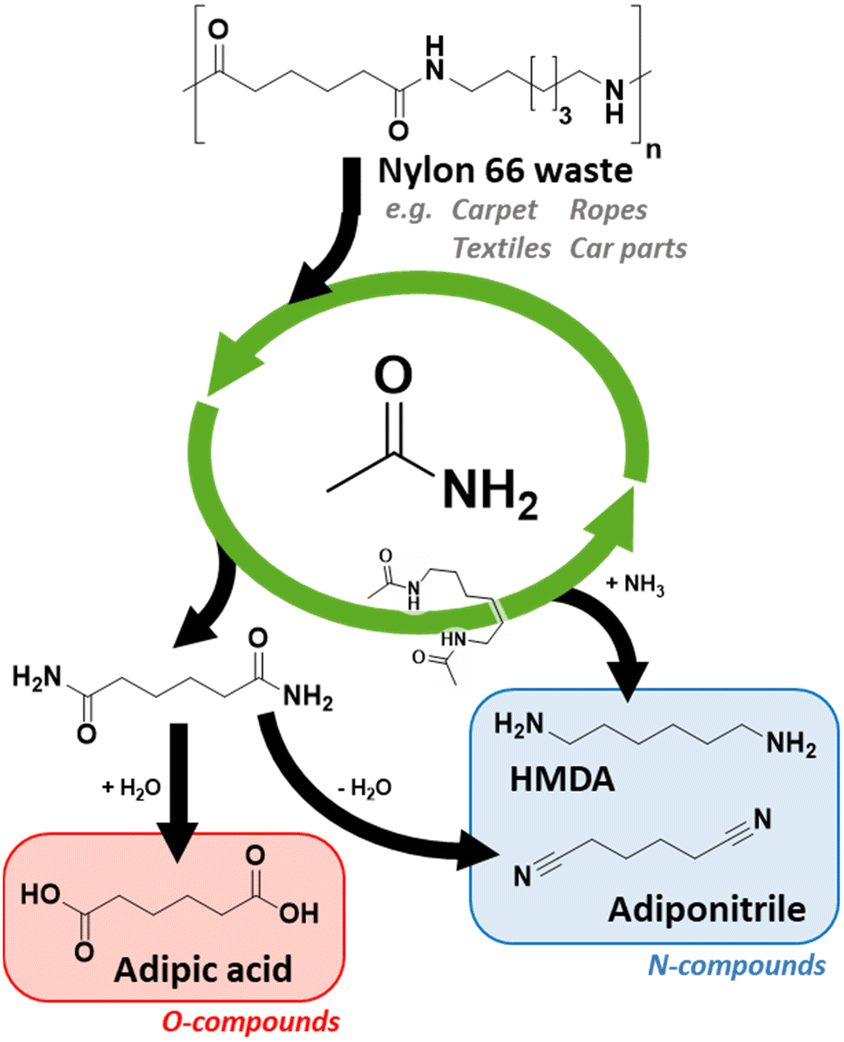 | ||
| Scheme 2 Complete recycling of Nylon 66 waste based on a transamidation depolymerization technique. | ||
After isolation, N,N′-hexamethylene bis(acetamide) can be reconverted to HMDA via, e.g., ammonolytic reactive distillation.10a In doing so, acetamide is liberated, closing the loop for this primary amide molecule. Alternatively, one can utilize the homogeneous amide hydrogenation catalysts developed by the teams of Milstein and Schaub.8
The resulting overall reaction for such a process would be as follows:
Nylon 66 (repeating unit) + 4 H2 + 2 acetamide → HMDA + 2 ethanol [+ adipamide].
Although in this case acetamide could not be recycled, the overall reaction could still be economically feasible.
Considering adipamide, this molecule is a well-known intermediate in the PA industry. One of the first (industrial) synthesis methods of adiponitrile was via the dehydration of adipamide/ammonium adipate (under a stream of NH3).17 If adiponitrile is the desired end product, a (complete) separation of N,N′-hexamethylene bis(acetamide) and adipamide might be superfluous. As a proof of concept, we performed a niobia catalyzed transfer-dehydration with acetonitrile as the solvent and sacrificial water scavenger. Although severe solvent degradation was observed, we obtained a 65% adiponitrile yield without major degradation of the N,N′-hexamethylene bis(acetamide) monomer (Fig. 1).
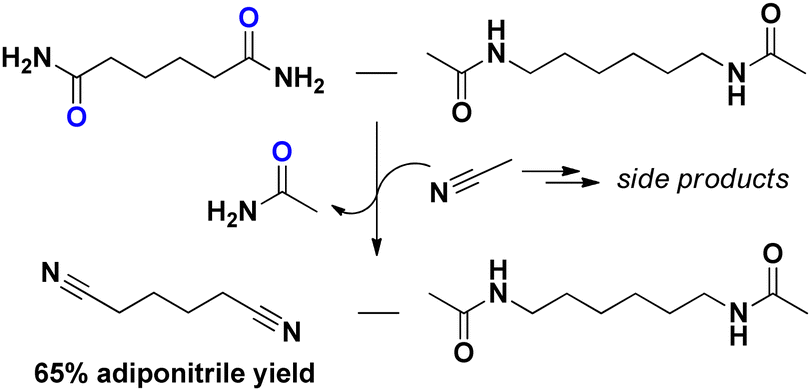 | ||
| Fig. 1 Transfer dehydration of a 1–1 molar mixture of N,N′-hexamethylene bis(acetamide) and adipamide to acetamide. Reaction conditions: Adipamide (1 mmol), N,N′-hexamethylene bis(acetamide) (1 mmol), 200 °C, Nb2O5 (0.2 g), acetonitrile (10 mL), 16 hours. | ||
Alternatively, adipamide could be hydrolyzed with water to the original adipic acid. For this reaction, Nb2O5 would be an excellent catalyst as well.18 Hence, the developed transamidation process could potentially serve as the foundation for the closed-loop recycling of Nylon 66.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Statista. Market volume of polyamide industry worldwide from 2017 to 2018 with a forecast until 2025, https://www.statista.com/; (b) Grand View Research. Global Nylon Market Size, Share & Trends Report, 2030, https://www.grandviewresearch.com/.
- B. Herzog, M. I. Kohan, S. A. Mestemacher, R. U. Pagilagan, K. Redmond and R. Sarbandi, Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2020, vol. 14, pp. 1–47 Search PubMed.
- (a) C. Mihut, D. K. Captain, F. Gadala-Maria and M. D. Amiridis, Polym. Eng. Sci., 2001, 41, 1457–1470 CrossRef CAS; (b) A. Kamimura, K. Ikeda, S. Suzuki, K. Kato, Y. Akinari, T. Sugimoto, K. Kashiwagi, K. Kaiso, H. Matsumoto and M. Yoshimoto, ChemSusChem, 2014, 7, 2473–2477 CrossRef CAS PubMed.
- (a) K. Kaiso, T. Sugimoto, K. Kashiwagi and A. Kamimura, Chem. Lett., 2011, 40, 370–371 CrossRef CAS; (b) K. Kaiso and T. Sugimoto, European Pat., EP1975156A, 2008 Search PubMed; (c) S. Sifniades, A. B. Levy and J. A. J. Hendrix, US Pat., US5932724, 1999 Search PubMed; (d) E. F. Moran, World Pat., WO1994008942A, 1994 Search PubMed.
- Carpet America Recovery Effort, CARE 2016 Annual Report, https://carpetrecovery.org/.
- (a) M. G. Peter, US Pat., US3069465A, 1962 Search PubMed; (b) D. D. Davis and E. D. Wilhoit, World Pat., WO9700846A, 1997 Search PubMed.
- S. L. Konopa, J. A. Mulholland, M. J. Realff and P. M. Lemieux, J. Air Waste Manage. Assoc., 2008, 58, 1070–1076 CrossRef CAS PubMed.
- (a) A. Kumar, N. von Wolff, M. Rauch, Y.-Q. Zou, G. Shmul, Y. Ben-David, G. Leitus, L. Avram and D. Milstein, J. Am. Chem. Soc., 2020, 142, 14267–14275 CrossRef CAS PubMed; (b) W. Zhou, P. Neumann, M. Al Batal, F. Rominger, A. S. K. Hashmi and T. Schaub, ChemSusChem, 2021, 14, 4176–4180 CrossRef CAS PubMed.
- W. Stuyck, K. Janssens, M. Denayer, F. De Schouwer, R. Coeck, K. V. Bernaerts, J. Vekeman, F. De Proft and D. E. De Vos, Green Chem., 2022, 24, 6923–6930 RSC.
- (a) R. Coeck, A. De Bruyne, T. Borremans, W. Stuyck and D. E. De Vos, ACS Sustain. Chem. Eng., 2022, 10, 3048–3056 CrossRef CAS; (b) R. Coeck, M. Houbrechts and D. E. De Vos, Chem. Sci., 2023, 14, 7944–7955 RSC.
- (a) J. H. Clark, T. J. Farmer, L. Herrero-Davila and J. Sherwood, Green Chem., 2016, 18, 3914–3934 RSC; (b) W. R. Stahel, Nature, 2016, 531, 435–438 CrossRef CAS PubMed; (c) T. Keijer, V. Bakker and J. C. Slootweg, Nat. Chem., 2019, 11, 190–195 CrossRef CAS PubMed; (d) J.-P. Lange, Green Chem., 2002, 4, 546–550 RSC.
- J. G. Ramos and M. A. Lozano, An. R. Soc. Esp. Fis. Quim., Ser. B, 1960, 56B, 833–840 CAS.
- (a) L. Wang, X. Dong, Y. Gao, M. Huang, C. C. Han, S. Zhu and D. Wang, Polymer, 2015, 59, 16–25 CrossRef CAS; (b) K. L. L. Eersels, G. Groeninckx, Y. Mengerink and S. van der Waals, Macromolecules, 1996, 29, 6744–6749 CrossRef CAS.
- G. L. Kennedy and R. D. Short, CRC Crit. Rev. Toxicol., 1986, 17, 129–182 CrossRef CAS PubMed.
- (a) X. Tao, T. Liu, H. Tao, R. Liu and Y. Qian, J. Mol. Catal. A: Chem., 2003, 201, 155–160 CrossRef CAS; (b) J. H. Forsberg, V. T. Spaziano, S. P. Klump and K. M. Sanders, J. Heterocycl. Chem., 1988, 25, 767–770 CrossRef CAS.
- R. Coeck, J. Meeprasert, G. Li, T. Altantzis, S. Bals, E. A. Pidko and D. E. De Vos, ACS Catal., 2021, 11, 7672–7684 CrossRef CAS.
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, Wiley, Weinheim, Germany, 2003, vol. 11, pp. 239–266 Search PubMed.
- S. M. A. H. Siddiki, M. N. Rashed, A. S. Touchy, M. A. R. Jamil, Y. Jing, T. Toyao, Z. Maeno and K. Shimizu, Catal. Sci. Technol., 2021, 11, 1949–1960 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc05462d |
| This journal is © The Royal Society of Chemistry 2024 |