
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aluminyl derived ethene functionalization with heteroallenes, leading to an intramolecular ligand rearrangement†
Andrea
O’Reilly
 a,
Michael G.
Gardiner
b,
Claire L.
McMullin
*c,
J. Robin
Fulton
*a and
Martyn P.
Coles
*a
a,
Michael G.
Gardiner
b,
Claire L.
McMullin
*c,
J. Robin
Fulton
*a and
Martyn P.
Coles
*a
aSchool of Chemical and Physical Sciences, Victoria University of Wellington, PO Box 600, Wellington 6012, New Zealand. E-mail: martyn.coles@vuw.ac.nz; j.robin.fulton@vuw.ac.nz
bResearch School of Chemistry, The Australian National University, Canberra, ACT 2601, Australia
cDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: cm2025@bath.ac.uk
First published on 22nd December 2023
Abstract
The aluminacyclopropane K[Al(NON)(η-C2H4)] ([NON]2− = [O(SiMe2NDipp)2]2−, Dipp = 2,6-iPr2C6H3) reacts with CO2 and iPrN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CNiPr to afford ring-expanded products of C–C bond formation. The latter system undergoes a 1,3-silyl retro-Brook rearrangement of the NON-group, to afford the [NNO]2− ligand ([NNO]2− = [N(Dipp)SiMe2N(Dipp)SiMe2O]2−). The mechanism of transformation was examined by density functional theory (DFT).
CNiPr to afford ring-expanded products of C–C bond formation. The latter system undergoes a 1,3-silyl retro-Brook rearrangement of the NON-group, to afford the [NNO]2− ligand ([NNO]2− = [N(Dipp)SiMe2N(Dipp)SiMe2O]2−). The mechanism of transformation was examined by density functional theory (DFT).
A defining goal in applied inorganic chemistry is the effective conversion of low-cost chemical feedstocks into value-added chemicals. Much of this research is focused on the combination of small organic molecules in the coordination sphere of an s- or p-block element to generate more complex molecules, relying on activation and subsequent bond forming/breaking processes of the substrates. This area has traditionally been dominated by transition-metals, although select main group metal complexes are emerging as viable alternatives,1 with justifications for this research often citing the cost advantage of using earth abundant elements in these transformations.
The ready availability and economic accessibility of aluminium has led to the chemistry of this element being prominent in this field, with a fertile area involving low-valent Al(I) and Al(II) species.2 In 2018, a new class of anionic Al(I) complex containing nucleophilic aluminyl anions was introduced.3 These compounds have already demonstrated many examples of bond-breaking and bond-forming reactions initiated by the electron rich Al(I) centre, with examples of C–C bond formation featuring prominently in this research.
Acrylic acid (propenoic acid) is an important commodity chemical with a global market volume of approximately 8 million metric tonnes in 2022.4 Processes used in the production of acrylates include the hydroxy-carbonylation of alkynes (Reppe chemistry)5 and the oxidation of propene (Fig. 1).6 An attractive alternative (that also helps to mitigate problems associated with greenhouse gases) involves the direct combination of CO2 with alkenes.7 A key step in this process is the C–C bond formation between the two substrates and, although endergonic under normal experimental conditions,8 a series of systems based on d-block metals Ti,9 Zr,10 V,11 Mo and W,12 Fe,13 Ru,14 Rh,15 Ni,16 and Pd17 promote this reaction. However, examples of CO2/alkene coupling at main group metal centres are limited to matrix isolation experiments involving condensates of Mg atoms with C2H4/CO2.18
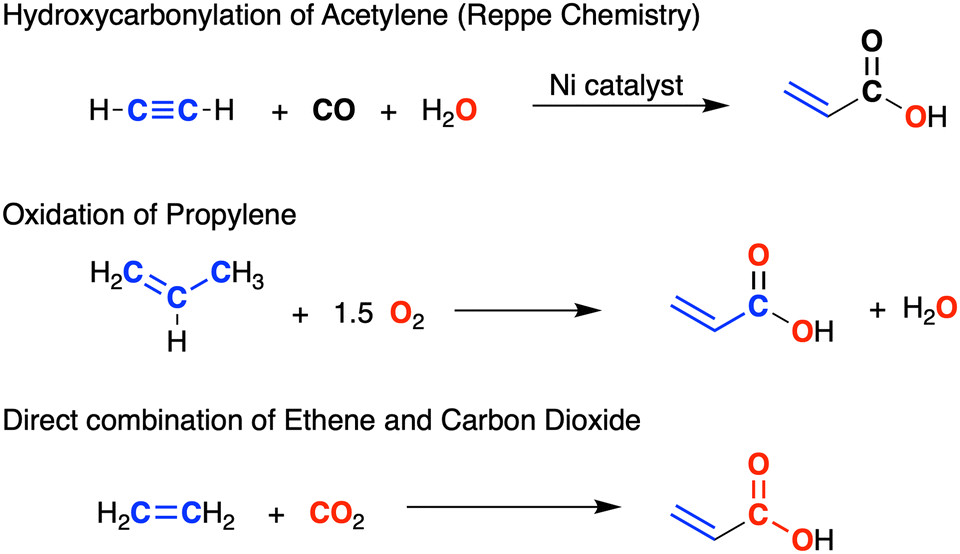 | ||
| Fig. 1 Methods for the production of acrylic acid. | ||
Previous work has shown that the Al(I) compound Al(BDI) (BDI = [HC(CMeNDipp)2]2−) reacts reversibly with alkenes to generate the corresponding aluminacyclopropane species.19 Although the products contain “activated” alkene bonds, reactivity is limited to the reversible insertion of CO into an Al–C bond of the aluminacyclopropane derived from norbornene.20 This area has been extended to anionic aluminacyclopropane analogues derived from potassium aluminyl compounds,21 and we have shown that the potassium aluminyl K[Al(NON)]22 reacts with ethene to form K[Al(NON)(η-C2H4)] (I).23 Reaction of I with CO under ambient conditions afforded the isolable CO insertion product, which underwent a thermal rearrangement to afford the propan-1,3-diyl-κ2C-1-one containing product. In this study we demonstrate C–C bond forming reactions between an aluminacyclopropane and heteroallenes. In addition, we have studied an unusual ligand rearrangement of the normally innocent NON-ligand.
The reaction of aluminacyclopropane I with 13CO2 proceeds under ambient conditions to form a new product, 1 (Scheme 1). 1H NMR spectroscopy of 1 shows a non-symmetrical product in which the high field singlet in I (−1.40 ppm) has been split into two multiplets centred at 1.79 and 0.17 ppm, consistent with insertion of CO2 into a single Al–C bond. 13C{1H} NMR spectroscopy shows a low field peak at 188.8 ppm for the labelled carbon of the metallacycle, which couples to the CH2 methylene peaks (1JCC = 49 Hz, 2JCC = 6 Hz). IR spectroscopy shows a strong absorption at 1578 cm−1, assigned to an exocyclic CO group. Unfortunately, quenching the reaction with HCl afforded a complex mixture of unidentified products.
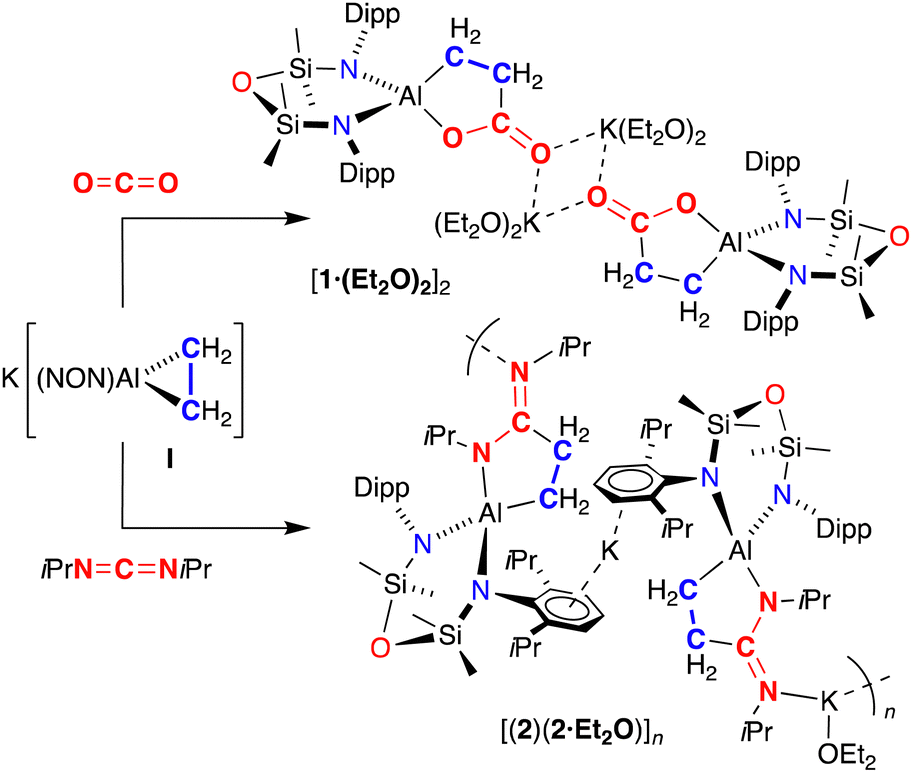 | ||
| Scheme 1 Synthesis of 1 and 2. | ||
Crystals of 1 were isolated from Et2O as the dimeric species, [{K(Et2O)2}(Al{NON}){κ2C,O-CH2CH2C(O)O}]2 [1·(Et2O)2]2 in which C–C bond formation between CO2 and a carbon of the aluminacyclopropane has occurred (Fig. 2). Each aluminium is spirocyclic, defined by the chelated NON-ligand and a 3-oxido-3-oxopropan-1-ide ligand. The core of the dimer consists of a K2O2 motif in which the solvated potassium atoms engage in μ-K⋯O interactions with a neighbouring unit. The endocyclic C–C distances (range: 1.509(5) Å to 1.545(4) Å) indicate single bonds within the metallacycle. However, the endocyclic (1.292(5) Å and 1.302(5) Å) and exocyclic (1.233(4) Å and 1.233(5) Å) C–O distances indicate a degree of delocalization when compared to the expected values for single (1.350 Å) and double (1.201 Å) bonds.24
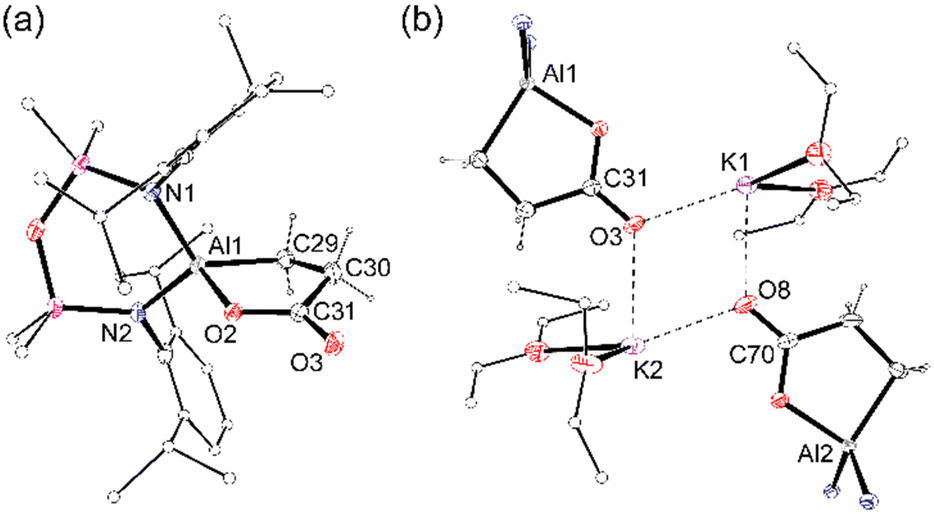 | ||
| Fig. 2 (a) Displacement ellipsoid plot (30% probability, H-atoms except CH2CH2 of metallacycle omitted, C-atoms except at key positions represented as spheres) of one of the [Al(NON){κ2C,O–CH2CH2C(O)O}]− anions from [1·(Et2O)2]2. Selected bond lengths (Å) and angles (°) {corresponding value from second anion}: Al1–O2 1.8514(19) {1.847(2)}, Al1–C29 1.980(4) {1.984(4)}, C29–C30 1.545(4) {1.517(5)}, C30–C31 1.513(5) {1.509(5)}, C31–O2 1.292(5) {1.302(5)}, C31–O3 1.233(4) {1.233(5)}; C29–Al1–O2 89.77(12) {90.10(13)}, O2–C31–O3 123.1(3) {121.9(3)}, C30–C31–O2 116.2(3) {116.3(3)}, C30–C31–O3 120.7(4) {121.8(4)}. (b) Dimeric core of [1·(Et2O)2]2. | ||
The reaction of I with diisopropylcarbodiimide (iPrNCNiPr) was also performed, affording colourless crystals 2 (Scheme 1). NMR spectroscopic data of 2 resemble those for 1, with a similar reduction in molecular symmetry evident from new methylene peaks at 2.44 and 0.36 ppm, and a low field resonance in the 13C{1H} NMR spectrum at 174.2 ppm for the NCN quaternary carbon. A distinct absorption at 1604 cm−1 in the IR spectrum is assigned to a ν(CN) stretch.
Crystals of 2 suitable for an X-ray diffraction study were grown from the slow evaporation of a toluene/Et2O solution (Fig. 3). The extended structure shows a 1-D polymer consisting of repeating dimeric [(2·Et2O)(2)] units. Each dimer is formed by K⋯π(arene) interactions involving the Dipp-substituents of neighbouring units, supplemented by close contacts with hydrogen atoms of the metallacyclic CH2 groups. The dimers are linked through solvated K(Et2O) units that bond to the exocyclic nitrogen atoms of imine groups. As anticipated, the carbodiimide substrate has inserted into one of the aluminacyclopropane Al–C bonds to afford isopropyl(3-(isopropylimino)propan-1-id-3-yl)amide ligands. The C–N bonds in the AlC3N rings (1.370(2) Å and 1.373(2) Å) are longer than the exocyclic C–N bonds (1.305(2) Å and 1.307(2) Å).
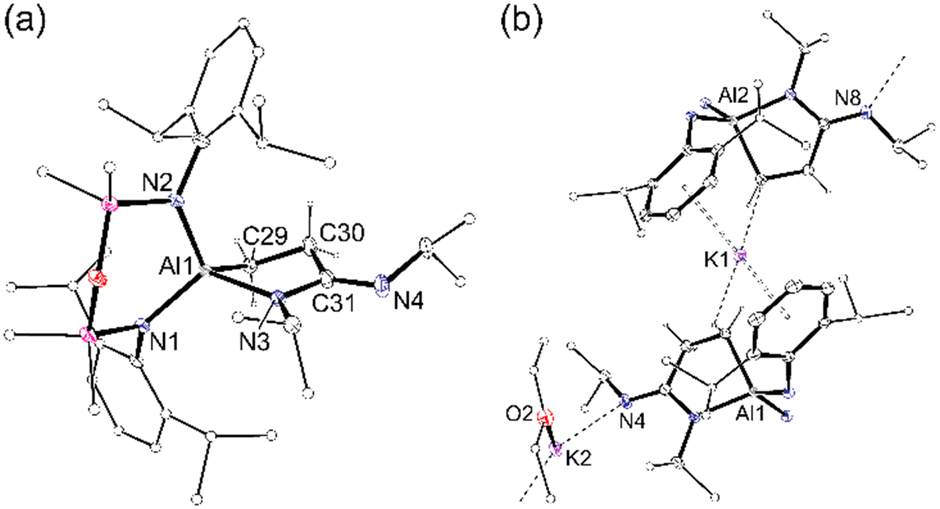 | ||
| Fig. 3 (a) Displacement ellipsoid plot (30% probability, H-atoms except CH2CH2 of metallacycle omitted, C-atoms except at key positions represented as spheres) of one of the [Al(NON){κ2C,N-CH2CH2C(NiPr)NiPr}]− anions from [(2·Et2O)(2)]. Selected bond lengths (Å) and angles (°) {corresponding value from second anion}: Al1–N3 1.9300(15) {1.9333(15)}, Al1–C29 1.9740(18) {1.9667(18)}, C29–C30 1.533(2) {1.531(2)}, C30–C31 1.534(2) {1.531(2)}, C31–N3 1.370(2) {1.373(2)}, C31–N4 1.305(2) {1.307(2)}; N1–Al–N2 109.91(6) {109.75(6)}, C29–Al1–N3 86.64(7) {88.02(7)}, N3–C31–N4 123.04(17) {122.90(16)}, C30–C31–N3 113.15(15) {113.49(14)}, C30–C31–N4 123.75(17) {123.56(16)}. (b) Dimeric monomer unit of [(2·Et2O)(2)]n. | ||
On standing for several days at room temperature, a sample of non-solvated K[Al(NON){CH2CH2C(NiPr)NiPr}] (2) transformed to a new species 3, the 1H NMR of which shows a further reduction in symmetry. Monitoring a sample in benzene showed that this process is complete after 2 days at 80 °C (Fig. S13, ESI†). No such conversion was noted for 1 (100 °C, 5 days). The 1H NMR spectrum of 3 shows that each proton of the AlC3N metallacycle is in a different environment, with multiplets observed at 2.52, 1.99, 0.12 and −0.46 ppm (1H). Furthermore, the resonances formally associated with the NON-ligand also reflect a reduction in symmetry compared with 2, with four septets for the NON-Dipp substituents, and non-equivalent SiMe2 groups (0.73, 0.36, 0.07 and 0.01 ppm, 3H). These data suggest de-symmetrisation of the NON-ligand, while the retention of a ν(CN) stretch at 1608 cm−1 indicate that the integrity of the AlC3N metallacycle is retained.
The slow evaporation of a benzene solution of 3 afforded crystals suitable for X-ray diffraction (Fig. 4). The structure of 3 showed that while the isopropyl(3-(isopropylimino)propan-1-id-3-yl)amide moiety did indeed remain intact at the Al centre, the NON-ligand had undergone a 1,3-silyl retro-Brook rearrangement to afford the corresponding [NNO]2− ligand, [N(Dipp)SiMe2N(Dipp)SiMe2O]2− (Scheme 2). This phenomenon has been studied in the context of [NON]2− dianions, where Li2[NONR] was observed to undergo intramolecular rearrangements in THF, with τ1/2 that vary from 5.7 × 101 s to 1.5 × 108 s, depending on the R-substituent.25 However, to the best of our knowledge this process has not been noted for a chelated NON-ligand, in which the N,N′-diamido coordination mode is normally considered stable.
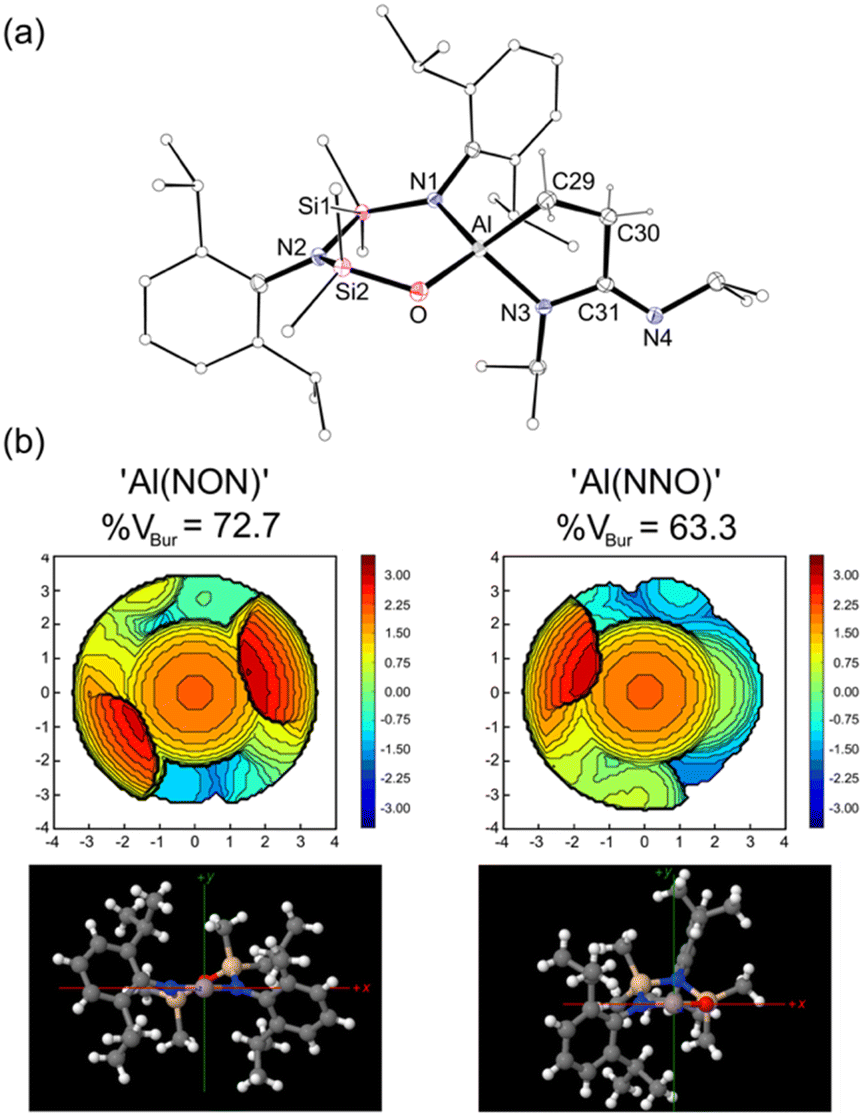 | ||
| Fig. 4 (a) Displacement ellipsoid plot (‘−x, 1 − y, 1 − z, 30% probability, H-atoms except CH2CH2 of metallacycle omitted, C-atoms except at key positions represented as spheres) of the [Al(NNO){κ2C,N-CH2CH2C(NiPr)NiPr}]− anion from [3]2. Selected bond lengths (Å) and angles (°): Al–O 1.7694(10), Al–N1 1.8789(11), Al–C29 1.9739(14), Al–N3 1.9041(11), C29–C30 1.538(2), C30–C31 1.5285(18), C31–N3 1.3722(17), C31–N4 1.3019(18), O–Al–N1 104.83(5), Al–O–Si2 122.75(6), C29–Al–N3 88.45(5), N3–C31–N4 122.99(12), C30–C31–N3 112.99(11), C30–C31–N4 123.99(12). (b) Topographic steric maps of ‘Al(NON)’ and ‘Al(NNO)’ fragments (3.5 Å radius around Al, Bondi radii scaled by 1.17). | ||
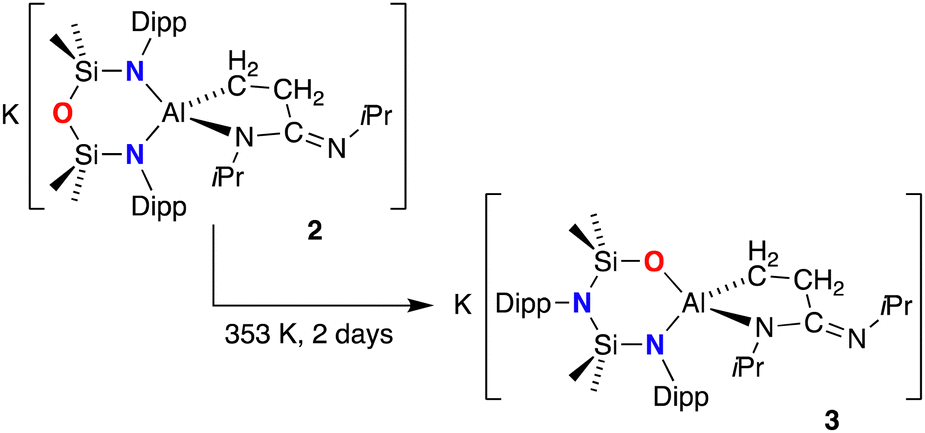 | ||
| Scheme 2 Conversion of (non-solvated) 2 to 3. | ||
The asymmetric unit of 3 consists of [K(C6H6)][Al(NNO){κ2C,N-CH2CH2C(NiPr)NiPr}] located on an inversion centre, forming a dimeric unit [3]2 linked by contacts between potassium and the exocyclic Nimine of the partner complex (K⋯N4′ = 2.7498(11) Å, Fig. S16, ESI†). The benzene-solvated potassium is bonded to the oxygen of the [NNO]2− ligand, with a K–O distance of 2.6643(9) Å. The bond lengths and angles within the [CH2CH2C(NiPr)NiPr]2− group are essentially the same as those noted in 2. However, switching from an N,N′- to an N,O-bonding mode for the [NNO]2− ligand reduces the bite angle from 109.91(6)° {109.75(6)°} to 104.83(5)°. Furthermore, the relocation of one of the Dipp-substituents to the rear of the ligand significantly reduces the steric demand of the ligand at the aluminium centre. This was confirmed quantitatively using % volume buried measurements (%VBur),26 performed on the ‘Al(NON)’ and ‘Al(NNO)’ fragments (Fig. 4b), which generate values of 72.7% for [NON]2− and 63.3% for [NNO]2−.
The mechanism for the rearrangement of 2 to 3 was examined using DFT (Fig. 5. See ESI† for full computational details). The starting point for the calculations was the non-solvated monomer K[Al(NON){CH2CH2C(NiPr)NiPr}] (2DFT), in which the potassium is η6-bound to a Dipp substituent with additional K⋯H support from a methylene hydrogen atom. The first transition-state TS(2DFT-A)‡ was located at 15.8 kcal mol−1 and involves Al–N2 dissociation, leading to the four-membered aluminacycle A where the formally NON-ligand is coordinated through the remaining nitrogen atom (N1) and the backbone oxygen. Although the oxygen-atom is commonly involved in tridentate bonding of the NON-ligand, examples of exclusively κN,O-bonded species are restricted to potassium27 and magnesium.28 From A, after rotation about O–Si2, bond formation between the dissociated N2 nitrogen atom and Si1 occurs in transition state TS(A–B)‡, which is rate determining with a barrier of 15.6 kcal mol−1. This leads to a second intermediate B in which Si1–O bond cleavage has occurred, and an NNO-coordination mode has been achieved. The reaction pathway from B to the computed monomer K[Al(NNO){CH2CH2C(NiPr)NiPr}] 3DFT, with an energy of −14.6 kcal mol−1, has also been identified (Fig. S18, ESI†). It plots the migration of the K-atom from the N1-Dipp group to oxygen, whilst maintaining contacts to the ‘CN2’ component of the [{CH2CH2C(NiPr)NiPr}]-ligand.
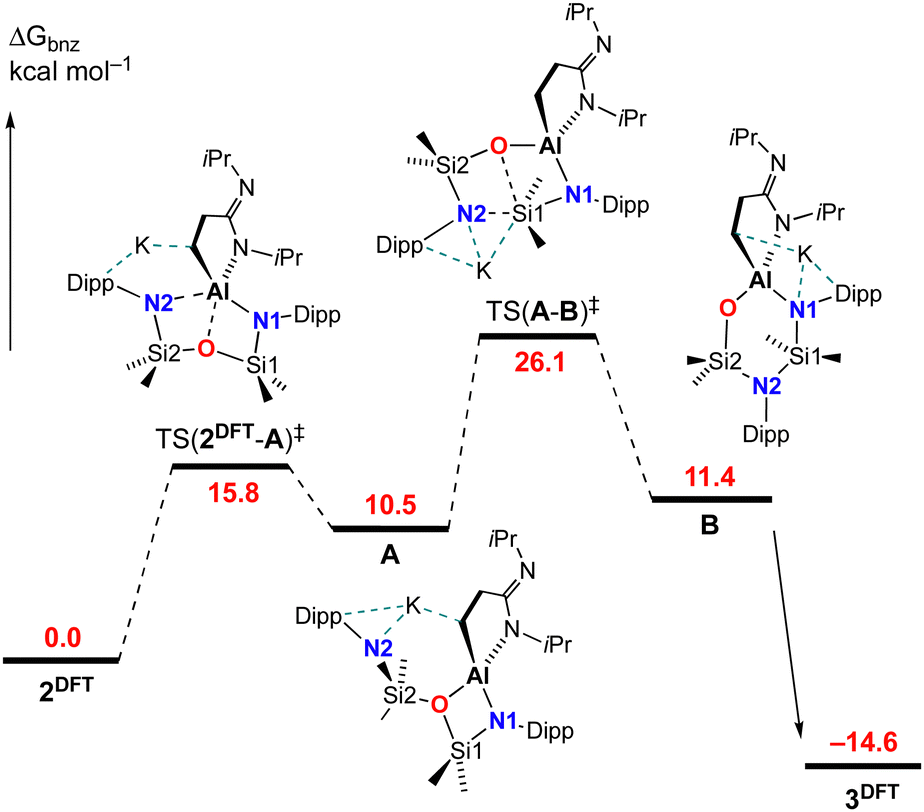 | ||
| Fig. 5 Computed free energy profile (BP86-D3BJ(PCMC6H6)/BS2//BP86/BS1 in kcal mol−1) for 1,3-silyl retro-Brook rearrangement converting K[Al(NON){CH2CH2C(NiPr)NiPr}] (2DFT) to K[Al(NNO){CH2CH2C(NiPr)NiPr}] (3DFT). | ||
In summary we have shown that the aluminacyclopropane anion [Al(NON)(η-C2H4)]− reacts with CO2 and iPrNCNiPr to form cyclo-AlC3X (X = O or N{R}) products via C–C bond formation. Upon heating an intramolecular retro-Brook rearrangement of the chelating NON-ligand occurs at aluminium. DFT calculations identified a key intermediate that involves a bimetallic Al/K species where the chelating NON-diamide has adopted a ring-contracted N,O-coordination. The results of equivalent insertion/NON → NNO rearrangements during the reaction of I with other unsaturated substrates will be reported in a subsequent manuscript.
Conflicts of interest
There are no conflicts to declare.References
- (a) C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS; (b) P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- K. Hobson, C. J. Carmalt and C. Bakewell, Chem. Sci., 2020, 11, 6942–6956 RSC.
- (a) M. P. Coles and M. J. Evans, Chem. Commun., 2023, 59, 503–519 RSC; (b) J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702–1713 CrossRef CAS PubMed.
- https://www.statista.com/statistics/1245262/acrylic-acid-market-volume-worldwide/. Retrieved Nov. 08, 2023.
- A. Brennführer, H. Neumann and M. Beller, ChemCatChem, 2009, 1, 28–41 CrossRef.
- M. M. Lin, Appl. Catal., A, 2001, 207, 1–16 CrossRef CAS.
- (a) T. Schaub, A. S. K. Hashmi and R. A. Paciello, J. Org. Chem., 2019, 84, 4604–4614 CrossRef CAS PubMed; (b) X. Wang, H. Wang and Y. Sun, Chemistry, 2017, 3, 211–228 CrossRef CAS; (c) M. Hollering, B. Dutta and F. E. Kühn, Coord. Chem. Rev., 2016, 309, 51–67 CrossRef CAS; (d) M. Limbach, Adv. Organomet. Chem., 2015, 63, 175–202 CrossRef CAS.
- P. N. Plessow, A. Schäfer, M. Limbach and P. Hofmann, Organometallics, 2014, 33, 3657–3668 CrossRef CAS.
- S. A. Cohen and J. E. Bercaw, Organometallics, 1985, 4, 1006–1014 CrossRef CAS.
- H. G. Alt and C. E. Denner, J. Organomet. Chem., 1990, 390, 53–60 CrossRef CAS.
- B. Hessen, A. Meetsma, F. Van Bolhuis, J. H. Teuben, G. Helgesson and S. Jagner, Organometallics, 1990, 9, 1925–1936 CrossRef CAS.
- (a) R. Alvarez, E. Carmona, A. Galindo, E. Gutierrez, J. M. Marin, A. Monge, M. L. Poveda, C. Ruiz and J. M. Savariault, Organometallics, 1989, 8, 2430–2439 CrossRef CAS; (b) R. Alvarez, E. Carmona, D. J. Cole-Hamilton, A. Galindo, E. Gutierrez-Puebla, A. Monge, M. L. Poveda and C. Ruiz, J. Am. Chem. Soc., 1985, 107, 5529–5531 CrossRef CAS.
- (a) T. T. Adamson, S. P. Kelley and W. H. Bernskoetter, Organometallics, 2020, 39, 3562–3571 CrossRef CAS; (b) S. M. Rummelt, H. Zhong, I. Korobkov and P. J. Chirik, J. Am. Chem. Soc., 2018, 140, 11589–11593 CrossRef CAS PubMed; (c) H. Hoberg, K. Jenni, K. Angermund and C. Krüger, Angew. Chem., Int. Ed. Engl., 1987, 26, 153–155 CrossRef.
- T. Ito, K. Takahashi and N. Iwasawa, Organometallics, 2019, 38, 205–209 CrossRef CAS.
- (a) S. Takegasa, M. M. Lee, K. Tokuhiro, R. Nakano and M. Yamashita, Chem. – Eur. J., 2022, 28, e202201870 CrossRef CAS PubMed; (b) M. Aresta and E. Quaranta, J. Organomet. Chem., 1993, 463, 215–221 CrossRef CAS.
- (a) K. Takahashi, K. Cho, A. Iwai, T. Ito and N. Iwasawa, Chem. – Eur. J., 2019, 25, 13504–13508 CrossRef CAS PubMed; (b) I. Knopf, D. Tofan, D. Beetstra, A. Al-Nezari, K. Al-Bahily and C. C. Cummins, Chem. Sci., 2017, 8, 1463–1468 RSC; (c) N. Huguet, I. Jevtovikj, A. Gordillo, M. L. Lejkowski, R. Lindner, M. Bru, A. Y. Khalimon, F. Rominger, S. A. Schunk, P. Hofmann and M. Limbach, Chem. – Eur. J., 2014, 20, 16858–16862 CrossRef CAS PubMed; (d) C. Hendriksen, E. A. Pidko, G. Yang, B. Schäffner and D. Vogt, Chem. – Eur. J., 2014, 20, 12037–12040 CrossRef CAS PubMed; (e) M. L. Lejkowski, R. Lindner, T. Kageyama, G. É. Bódizs, P. N. Plessow, I. B. Müller, A. Schäfer, F. Rominger, P. Hofmann, C. Futter, S. A. Schunk and M. Limbach, Chem. – Eur. J., 2012, 18, 14017–14025 CrossRef CAS PubMed; (f) H. Hoberg, Y. Peres, C. Krüger and Y.-H. Tsay, Angew. Chem., Int. Ed. Engl., 1987, 26, 771–773 CrossRef.
- S. C. E. Stieber, N. Huguet, T. Kageyama, I. Jevtovikj, P. Ariyananda, A. Gordillo, S. A. Schunk, F. Rominger, P. Hofmann and M. Limbach, Chem. Commun., 2015, 51, 10907–10909 RSC.
- V. N. Solov’ev, E. V. Polikarpov, A. V. Nemukhin and G. B. Sergeev, J. Phys. Chem. A, 1999, 103, 6721–6725 CrossRef.
- (a) C. Bakewell, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2018, 57, 6638–6642 CrossRef CAS PubMed; (b) C. Bakewell, A. J. P. White and M. R. Crimmin, Chem. Sci., 2019, 10, 2452–2458 RSC.
- R. Y. Kong and M. R. Crimmin, Chem. Commun., 2019, 55, 6181–6184 RSC.
- (a) J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 11000–11003 CrossRef CAS PubMed; (b) K. Sugita, R. Nakano and M. Yamashita, Chem. – Eur. J., 2020, 26, 2174–2177 CrossRef CAS PubMed.
- R. J. Schwamm, M. D. Anker, M. Lein and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 1489–1493 CrossRef CAS PubMed.
- M. J. Evans, S. E. Neale, M. D. Anker, C. L. McMullin and M. P. Coles, Angew. Chem., Int. Ed., 2022, 61, e202117396 CrossRef CAS PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- (a) F. Haftbaradaran, G. Mund, R. J. Batchelor, J. F. Britten and D. B. Leznoff, Dalton Trans., 2005, 2343–2345 RSC; (b) F. Haftbaradaran, A. M. Kuchison, M. J. Katz, G. Schatte and D. B. Leznoff, Inorg. Chem., 2008, 47, 812–822 CrossRef CAS PubMed.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- M. D. Anker, M. Lein and M. P. Coles, Chem. Sci., 2019, 10, 1212–1218 RSC.
- R. J. Schwamm and M. P. Coles, Chem. – Eur. J., 2019, 25, 14183–14191 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra; experimental, crystallographic and computational details; displacement ellipsoid plots. CCDC 2310396–2310398. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc05785b |
| This journal is © The Royal Society of Chemistry 2024 |