
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pd-catalysed C–H alkynylation of benzophospholes†
Yu
Tokura
a,
Shibo
Xu
b,
Kosuke
Yasui
a,
Yuji
Nishii
a and
Koji
Hirano
 *ab
*ab
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University, Suita, Osaka 565-0871, Japan. E-mail: k_hirano@chem.eng.osaka-u.ac.jp
First published on 9th February 2024
Abstract
A palladium-catalysed C2–H alkynylation of benzophospholes with alkynyl bromides has been developed to afford the corresponding phosphole-alkyne conjugations in good to high yields. The C–C triple bond as well as terminal alkyne C–H bond in the obtained products is a good synthetic handle for further manipulations, thus giving the versatile π-conjugated benzophosphole derivatives. The optoelectronic properties of the newly synthesized conjugated benzophospholes are also described.
Because of its unique optoelectronic and physical properties, the phosphole nucleus has received significant attention as an important heteroaromatic core in the design of phosphorus-containing organic functional materials.1 In particular, highly π-conjugated benzophospholes are frequently occurring in organic light-emitting diodes, solar cells, imaging dyes, and memory devices. Among them, the C2-alkynylated (benzo)phospholes are attractive owing to their effective phosphole-alkyne π-conjugation as well as the versatile reactivity of the C–C triple bond and terminal alkyne C–H bond, which are additional synthetic handles for further functionalisations.2 In 2013, Matano and Imahori reported the synthesis of C2-alkynylated benzophospholes by the palladium-catalysed Sonogashira coupling of C2-brominated benzophosphole, which was prepared from trimethylsilyl-protected phenylacetylene in a three-step manner (Scheme 1a).3 However, there are few examples in the literature probably because of the somewhat tedious synthetic procedure for the preparation of the starting substrate. Thus, the development of a more concise and versatile synthetic methodology for C2-alkynylated benzophospholes is greatly appealing.
 | ||
| Scheme 1 Approaches to C2-alkynylated benzophospholes. (a) Sonogashira coupling approach and (b) C–H functionalisation approach. | ||
On the other hand, our research group recently focused on the synthetic potential of benzophosphole C–H activation4 and developed the palladium-catalysed C2–H arylation and alkenylation reactions.5 During continuing interest in this chemistry, we here report a palladium-catalysed C2–H alkynylation with alkynyl bromides to directly deliver the corresponding C2-alkynylated benzophospholes in good yields (Scheme 1b). Different from the related benzoheteroles including indoles and benzothiophenes,6 to the best of our knowledge, the C–H alkynylation of the phosphole nucleus has not been reported yet. The starting C2–H benzophospholes are readily prepared from 1,1-diarylethenes in one synthetic operation.7 Thus, the present C–H activation protocol can offer a convergent approach to the C2-alkynylated benzophospholes with high structural diversity. In addition, based on the established alkyne chemistry, the obtained products can be rapidly manipulated into more versatile π-conjugated benzophosphole derivatives. Their optoelectronic properties are also described.
We selected the C2–H benzophosphole oxide 1a and triisopropylsilyl (TIPS)-substituted alkynyl bromide 2a as model substrates and started optimisation studies (Table 1). In an early experiment, treatment of 1a (0.10 mmol) with 2a (2.0 equiv.) in the presence of 10 mol% Pd(OAc)2 catalyst and CsOPiv base (2.0 equiv.) in heated 1,4-dioxane (1.0 mL at 110 °C) for 20 h afforded the desired 3aa in 7% NMR yield (entry 1). Notably, the reaction occurred selectively at the C2–H position over the ortho C–Hs on the phenyl ring despite the potential directing ability of the phosphole P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety.8 This preliminary but promising result prompted us to investigate basic additives in detail. While KOPiv showed a similar performance (entry 2), NaOPiv increased the yield to 38% (entry 3). Other sodium bases such as NaOAc, Na2CO3, and NaHCO3 were less effective (entries 4–6). Thus, with NaOPiv as the optimal base, we then screened other reaction parameters. The lower reaction temperature (60 °C) further improved the reaction efficiency (entry 7). The Pd source also affected the yield of 3aa to some extent (entries 8–11): as far as we tested, Pd(OPiv)2 was found to be the best precursor (entry 8). After additional fine-tuning, the C2-alkynylated 3aa was finally obtained in 84% NMR yield (72% isolated yield) under slightly diluted conditions for prolonged reaction periods (1.5 mL 1,4-dioxane, 48 h; entry 12). The reaction could also be conducted on a 1.0 mmol scale with a comparable product yield. We also tested other bases, solvents, and alkynyl coupling partners, but better yields of 3aa were not observed. The use of the corresponding phosphole sulfide 1a-S and P(III) phosphole 1a-III instead of phosphole oxide 1a furnished no C–H alkynylated products (see the ESI† for more details).
O moiety.8 This preliminary but promising result prompted us to investigate basic additives in detail. While KOPiv showed a similar performance (entry 2), NaOPiv increased the yield to 38% (entry 3). Other sodium bases such as NaOAc, Na2CO3, and NaHCO3 were less effective (entries 4–6). Thus, with NaOPiv as the optimal base, we then screened other reaction parameters. The lower reaction temperature (60 °C) further improved the reaction efficiency (entry 7). The Pd source also affected the yield of 3aa to some extent (entries 8–11): as far as we tested, Pd(OPiv)2 was found to be the best precursor (entry 8). After additional fine-tuning, the C2-alkynylated 3aa was finally obtained in 84% NMR yield (72% isolated yield) under slightly diluted conditions for prolonged reaction periods (1.5 mL 1,4-dioxane, 48 h; entry 12). The reaction could also be conducted on a 1.0 mmol scale with a comparable product yield. We also tested other bases, solvents, and alkynyl coupling partners, but better yields of 3aa were not observed. The use of the corresponding phosphole sulfide 1a-S and P(III) phosphole 1a-III instead of phosphole oxide 1a furnished no C–H alkynylated products (see the ESI† for more details).
|
|
|||
|---|---|---|---|
| Entry | Pd | Base | Yield (%)b |
a Conditions: 1a (0.10 mmol), 2a (0.20 mmol), Pd (0.010 mmol on Pd), base (0.20 mmol), 1,4-dioxane (1.0 mL), 110 °C, 20 h, N2.
b Estimated by 31P{1H} NMR with P(O)(OEt)3 as the internal standard. Isolated yield of 3aa is in parentheses.
c At 60 °C.
d In 1,4-dioxane (1.5 mL) for 48 h.
e On a 1.0 mmol scale.
|
|||
| 1 | Pd(OAc)2 | CsOPiv | 7 |
| 2 | Pd(OAc)2 | KOPiv | 8 |
| 3 | Pd(OAc)2 | NaOPiv | 38 |
| 4 | Pd(OAc)2 | NaOAc | 20 |
| 5 | Pd(OAc)2 | Na2CO3 | 11 |
| 6 | Pd(OAc)2 | NaHCO3 | 0 |
| 7c | Pd(OAc)2 | NaOPiv | 60 |
| 8c | Pd(OPiv)2 | NaOPiv | 73 |
| 9c | Pd(TFA)2 | NaOPiv | 65 |
| 10c | PdCl2 | NaOPiv | 64 |
| 11c | Pd2(dba)3 | NaOPiv | 64 |
| 12cd | Pd(OPiv)2 | NaOPiv | 84 (72, 68e) |

With the optimal conditions (Table 1, entry 12), we examined the scope of the C–H alkynylation reaction (Scheme 2). Several substituted benzophosphole oxides 1 participated in the reaction: both electron-donating (methyl, tert-butyl, and methoxy) and -withdrawing substituents (chloro, fluoro, and trifluoromethyl) were equally compatible under the standard reaction conditions to produce the corresponding C2-alkynylated benzophospholes 3ca–ga in 63–97% yields. The reaction of the more π-conjugated naphthalene-fused phosphole derivative also occurred with a comparable efficiency (3ha). Moreover, the carbon-bridged tricyclic system 3ia was formed in 97% yield. On the other hand, the C2,3-free benzophosphole provided an inseparable mixture of C2-, C3-, and C2,C3-alkynylated products in ca. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio, which was assigned by 1H and 31P{1H} NMR analysis as well as HRMS (see the ESI† for more details). The structure of 3ca was unambiguously confirmed by X-ray analysis (CCDC 2279968†). More detailed functional group compatibility was investigated by the robustness screen9 (see the ESI†). In addition to 2a, some sterically congested silyl-substituted alkynyl bromides underwent the C–H coupling reaction: triphenylsilyl- and tert-butyldimethylsilyl (TBS)-protected C2-alkynylphospholes 3ab and 3ac were obtained in acceptable yields. The bulky tert-butyl- and tert-propargylic alcohol derivatives could also be employed albeit with moderate efficiency (3ad and 3ae). On the other hand, the less sterically demanding phenyl acetylene derivative did not form the coupling product 3af probably because of self-dimerisation reactions under the optimal conditions.
:1:1 ratio, which was assigned by 1H and 31P{1H} NMR analysis as well as HRMS (see the ESI† for more details). The structure of 3ca was unambiguously confirmed by X-ray analysis (CCDC 2279968†). More detailed functional group compatibility was investigated by the robustness screen9 (see the ESI†). In addition to 2a, some sterically congested silyl-substituted alkynyl bromides underwent the C–H coupling reaction: triphenylsilyl- and tert-butyldimethylsilyl (TBS)-protected C2-alkynylphospholes 3ab and 3ac were obtained in acceptable yields. The bulky tert-butyl- and tert-propargylic alcohol derivatives could also be employed albeit with moderate efficiency (3ad and 3ae). On the other hand, the less sterically demanding phenyl acetylene derivative did not form the coupling product 3af probably because of self-dimerisation reactions under the optimal conditions.
 | ||
| Scheme 2 Pd-catalysed C–H alkynylation of benzophospholes 1 with alkynyl bromides 2. Reaction conditions: 1 (0.10 mmol), 2 (0.20 mmol), Pd(OPiv)2 (0.010 mmol), NaOPiv (0.20 mmol), 1,4-dioxane (1.5 mL), 60 °C, 48 h, N2. Isolated yields are shown. aOn a 1.0 mmol scale. | ||
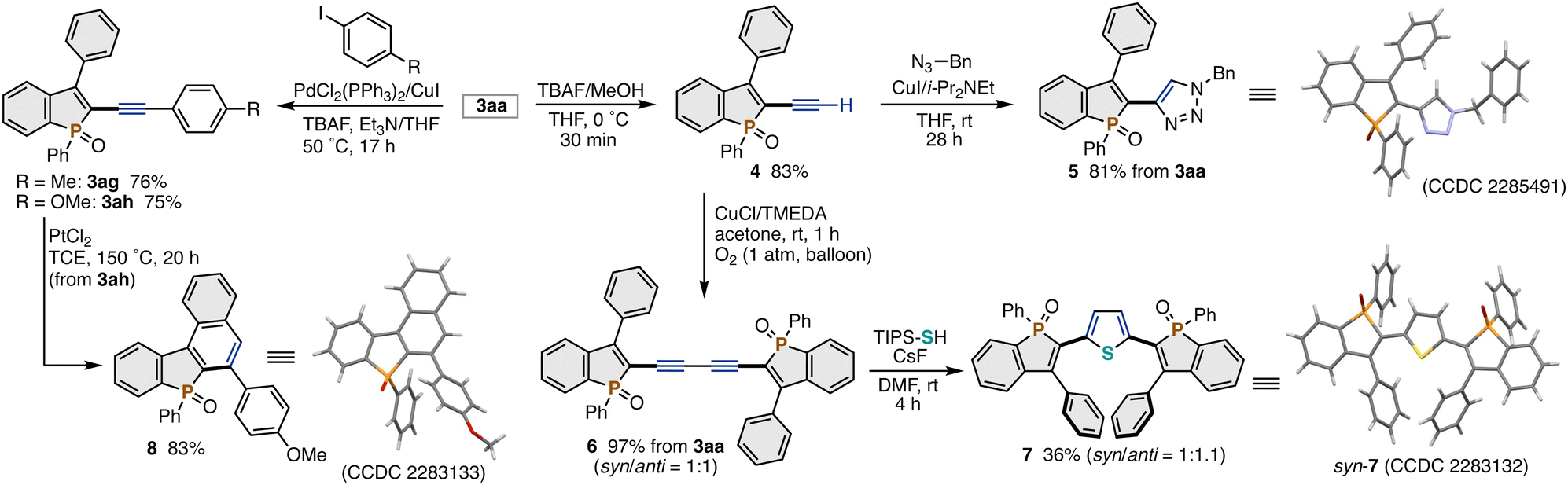
The TIPS-protected C2-alkynylated benzophosphole 3aa can be readily and variously derivatised (Scheme 3). The simple protodesilylation proceeded upon treatment with TBAF and MeOH in THF to afford the terminal alkyne 4 in 83% yield. In addition, the crude 4 directly underwent the copper-catalysed azide–alkyne cycloaddition and Glaser coupling without any additional purification, delivering the benzophosphole-triazole hybrid 52d and two phosphole-containing 1,3-diynes 6,2c respectively, in acceptable 2-step yields. The 1,3-diyne moiety in 6 was further transformed by the cyclisation with TIPS-SH/CsF to the phosphole-thiophene-phosphole ter(hetero)aryl 7. On the other hand, the desilylative Sonogashira coupling reaction of 3aa with aryl iodides was also possible to give the corresponding arylalkyne-substituted phospholes 3ag and 3ah in good yields, which can complement the unsuccessful reaction using the aryl-substituted alkynyl bromide (3af in Scheme 2). Subsequent PtCl2-catalysed cycloisomerisation of 3ah with concomitant aryl migration furnished the acene-type molecule 8 in a synthetically useful yield.10 The structures of 5, 7, and 8 were determined by X-ray analysis (CCDC 2285491, 2283132, and 2283133†).
 | ||
| Scheme 3 Derivatisation of C2-alkynylated benzophosphole 3aa. | ||
To get some mechanistic insight into the C–H cleavage step of 1a, we performed the H/D exchange reaction of 1a with D2O (Scheme 4). In the presence of Pd(OPiv)2 or NaOPiv alone, no deuterium incorporation was observed even after 48 h. In sharp contrast, the combination of Pd(OPiv)2 and NaOPiv gave the deuterated benzophosphole 1a-D albeit with a moderate deuterium content. These results suggest the cooperative effect of Pd and Na in the C–H metalation process, where the PO moiety coordinates to the Lewis acidic Na cation to increase the acidity of the proximal C2–H bond11 and it then can be cleaved by the Pd(OPiv)2-involved concerted metalation-deprotonation (CMD).12 In addition, the H/D exchange was observed selectively at the C2 position but not at the ortho position of the phenyl ring on P, which is consistent with the observed high regioselectivity in Table 1 and suggests that the regioselectivity in the alkynylation can be determined in the C–H cleavage step. On the basis of the aforementioned findings, the reaction of benzophosphole 1a with alkynyl bromide 2a is believed to proceed through (1) the Pd(OPiv)2-promoted C2–H metalation of NaOPiv-coordinated 1a to form a benzophosphole-Pd intermediate, (2) addition–elimination with 2a, giving 3aa along with Pd–Br species, and (3) ligand exchange with NaOPiv to regenerate the catalytically active starting Pd(OPiv)2. We cannot completely exclude the possibility of the Pd(0)/(II) redox cycle including the oxidative addition of 2a to Pd(0),5a but the control experiment with the independently prepared Pd(II)-alkynyl complex did not form the alkynylated product at all (Schemes S4 and S5, ESI†). The proposed C–H activation mechanism is also consistent with the reactivity trend dependent on the phosphorus moiety (Table 1 and Scheme S1, ESI†), in which only the most electron-withdrawing PO successfully gave the product: the less electron-withdrawing PS and P(III) groups cannot ensure the acidity enough for the successful C–H cleavage.13 However, at present, the base-assisted internal electrophilic substitution (BIES) mechanism14 is also plausible. Further studies remain to be elucidated.
 | ||
| Scheme 4 Deuterium incorporation experiments. | ||
We finally investigated the optoelectronic properties of some newly synthesized π-conjugated benzophosphole derivatives (Fig. S1–S13 and Tables S10 and 11 in the ESI†). Compared to the C3–H C2-alkynylated benzophosphole previously reported by Matano and Imahori,3 both the absorption and emission maxima of the C3-phenylated 3ah were red-shifted by ca. 20 nm. On the other hand, the differential pulse voltammetry (DPV) analysis of 3ah showed the uniquely increased reduction potential (−1.86 V vs. −2.02 V3) even with the maintenance of the oxidation potential, thus suggesting that the Ph group at the C3 position mainly affects the LUMO level. The properties of benzophosphole-triazole hybrid 5 were generally similar to the corresponding non-benzofused phosphole-triazole conjugation analogue.2d The two phosphole-linked 1,3-diynes 6 showed a remarkably low LUMO level (−3.41 eV). The phosphole-thiophene-phosphole conjugations syn-7 and anti-7 exhibited large bathochromic shifts of the emission maxima (532–537 nm) and distinctly smaller Stokes shifts, which are reflected by their relatively rigid structures associated with the less steric repulsion between the five-membered phosphole and thiophene heteroaromatic skeletons. Almost all values of syn-7 and anti-7 are identical, thus indicating that the optoelectronic properties are less dependent on the stereochemistry on phosphorus. Among them, the fluorescence quantum yields of MeO-substituted 3ah and its annulated derivative 8 were relatively good (0.74 and 0.52, respectively).
In conclusion, we have developed a palladium-catalysed C–H alkynylation of benzophosphole with alkynyl bromides. The resulting alkyne moiety could be a good synthetic handle for further manipulations. Combined with the ready availability of the starting C2–H benzophospholes and the straightforwardness of the C–H activation protocols, the present strategy can provide a modular approach to various phosphole-based π-conjugated molecules of potent interest in materials chemistry.15
This work was supported by JSPS KAKENHI Grant No. JP 22H02077 (Grant-in-Aid for Scientific Research(B), to K. H.) as well as by JST FOREST Program, Grant Number JPMJFR211X to K. H.
Conflicts of interest
The authors declare no competing financial interest.Notes and references
- Reviews and accounts: (a) T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681 CrossRef CAS PubMed; (b) Y. Matano and H. Imahori, Org. Biomol. Chem., 2009, 7, 1258 RSC; (c) M. P. Duffy, W. Delaunay, P.-A. Bouit and M. Hissler, Chem. Soc. Rev., 2016, 45, 5296 RSC; (d) Y. Matano, Chem. Rec., 2015, 15, 636 CrossRef CAS PubMed; (e) H. Tsuji, K. Sato, L. Ilies, Y. Itoh, Y. Sato and E. Nakamura, Org. Lett., 2008, 10, 2263 CrossRef CAS PubMed; (f) C. Wang, M. Taki, Y. Sato, A. Fukazawa, T. Higashiyama and S. Yamaguchi, J. Am. Chem. Soc., 2017, 139, 10374 CrossRef CAS PubMed; (g) P. Demay-Drouhard, J. R. Gaffen, C. B. Caputo and T. Baumgartner, Can. J. Chem., 2023, 101, 146 CrossRef CAS.
- (a) E. Y.-H. Hong, C.-T. Poon and V. W.-W. Yam, J. Am. Chem. Soc., 2016, 138, 6368 CrossRef CAS PubMed; (b) A. F.-F. Cheung, E. Y.-H. Hong and V. W.-W. Yam, Chem. – Eur. J., 2018, 24, 1383 CrossRef CAS PubMed; (c) Y. Matano, M. Nakashima and H. Imahori, Chem. Lett., 2021, 50, 1581 CrossRef; (d) Y. Matano, M. Nakashima, A. Saito and H. Imahori, Org. Lett., 2009, 11, 3338 CrossRef CAS PubMed.
- Y. Matano, Y. Hayashi, K. Suda, Y. Kimura and H. Imahori, Angew. Chem., Int. Ed., 2009, 48, 4002 CrossRef CAS PubMed.
- Selected recent reviews on C–H activation: (a) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC; (b) P. Gandeepan, T. Muller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192 CrossRef CAS PubMed; (c) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788 CrossRef CAS PubMed; (d) N. Y. S. Lam, K. Wu and J.-Q. Yu, Angew. Chem., Int. Ed., 2021, 60, 15767 CrossRef CAS PubMed.
- (a) S. Xu, K. Nishimura, K. Saito, K. Hirano and M. Miura, Chem. Sci., 2022, 13, 10950 RSC; (b) Y. Tokura, S. Xu, Y. Kojima, M. Miura and K. Hirano, Chem. Commun., 2022, 58, 12208 RSC.
- For examples of C–H alkynylation of indoles and benzothiophenes, see: (a) I. V. Seregin, V. Ryabova and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 7742 CrossRef CAS PubMed; (b) J. P. Brand, J. Charpentier and J. Waser, Angew. Chem., Int. Ed., 2009, 48, 9346 CrossRef CAS PubMed; (c) J. P. Brand and J. Waser, Angew. Chem., Int. Ed., 2010, 49, 7304 CrossRef CAS PubMed; (d) T. de Haro and C. Nevado, J. Am. Chem. Soc., 2010, 132, 1512 CrossRef CAS PubMed; (e) L. Yang, L. Zhao and C.-J. Li, Chem. Commun., 2010, 46, 4184 RSC; (f) X. Jie, Y. Shang, P. Hu and W. Su, Angew. Chem., Int. Ed., 2013, 52, 3630 CrossRef CAS PubMed . For a recent review on the C-H alkynylation of arenes, see: ; (g) J. A. S. Sarala, S. Bhowmick, R. L. de Carvalho, S. A. Al-Thabati, M. Mokhtar, E. N. da Silva Júnior and D. Maiti, Adv. Synth. Catal., 2021, 363, 4994 CrossRef.
- K. Nishimura, K. Hirano and M. Miura, Org. Lett., 2020, 22, 3185 CrossRef CAS PubMed.
- Y. Unoh, T. Satoh, K. Hirano and M. Miura, ACS Catal., 2015, 5, 6634 CrossRef CAS.
- K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597 CrossRef CAS PubMed.
- Attempts to apply the double cyclisation reaction of 6 under the same PtCl2-mediated conditions remained unsuccessful.
- The C2–H of phosphole is innately relatively acidic, but the coordination to the Na cation can further increase its acidity, thus accelerating the C–H metalation via CMD-type process. For pKa values of phosphole derivatives and related heteroaromatics, see: (a) D. Delaere, N. Pham-Tran and M. T. Nguyen, J. Phys. Chem. A, 2003, 107, 7514 CrossRef CAS; (b) L. D. Quin, J. G. Bryson and C. G. Moreland, J. Am. Chem. Soc., 1969, 91, 3308 CrossRef CAS; (c) K. Shen, Y. Fu, J.-N. Li, L. Liu and Q.-X. Guo, Tetrahedron, 2007, 63, 1568 CrossRef CAS.
- For selected reviews on CMD, see: (a) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118 CrossRef; (b) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (c) D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649 CrossRef CAS PubMed . The use of NaBr instead of NaOPiv did not promote any D incorporation reaction (vs.Scheme 4), thus suggesting an additional role of NaOPiv as the OPiv donor.
- Ph2PO shows a larger Hammett substituent constant than Ph2PS and Ph2P (σm = 0.38, 0.29, and 0.11, respectively), see: C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- T. Rogge, J. C. A. Oliveira, R. Kuniyil, L. Hu and L. Ackermann, ACS Catal., 2020, 10, 10551 CrossRef CAS.
- We preliminarily tried the reaction with (D)-N-Ac-alanine as the chiral ligand. Successful conversion (76%) was observed, and the product 3aa was obtained with 65:35 er, thus suggesting the possibility of asymmetric catalysis. See the ESI† for more details.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2279968, 2283132, 2283133, and 2285491. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc05994d |
| This journal is © The Royal Society of Chemistry 2024 |