
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiels–Alder cycloaddition polymerization for porous poly-phenylenes with exceptional gas uptake properties†
Timur
Ashirov
 a,
Patrick W.
Fritz
a,
Taner
Yildirim
*b and
Ali
Coskun
*a
a,
Patrick W.
Fritz
a,
Taner
Yildirim
*b and
Ali
Coskun
*a
aDepartment of Chemistry, University of Fribourg, Chemin du Musée 9, Fribourg 1700, Switzerland. E-mail: ali.coskun@unifr.ch
bNIST Center for Neutron Research, National Institute of Standards and Technology, Gaithersburg, MD 20899, USA. E-mail: taner.yildirim@nist.gov
First published on 8th February 2024
Abstract
We report the synthesis of two-dimensional and three-dimensional porous polyphenylenes (2D/3D-pPPs) via the Diels–Alder cycloaddition polymerization reaction. The resulting 2D and 3D-pPPs showed surface areas up to 1553 m2 g−1, pore volumes of 1.45 cm3 g−1 and very high H2 uptake capacities of 7.4 and 7.1 wt% at 77 K, respectively, along with a competitive high-pressure CO2 and CH4 uptake performance.
Diamond, renowned as the hardest known material, boasts remarkable physiochemical stability due to the fact that its carbon atoms are interconnected by robust covalent bonds.1 In the pursuit of replicating its chemical structure, researchers have uncovered a class of materials known as porous aromatic frameworks (PAFs) as a subclass of porous organic polymers (POPs), which not only emulate the molecular arrangement of diamond but also exhibit exceptional porosity. PAFs represent a distinct category of porous materials, featuring covalently linked phenyl groups that endow them with ultrahigh thermal stabilities often exceeding 400 °C,2 chemical robustness, and high surface areas.3 For instance, PAF-1 exhibits a surface area of 5600 m2 g−1 along with a high thermal stability up to 450 °C and chemical robustness.2 Despite their amorphous structure, PAFs exhibit significantly superior physiochemical stability. These features have led to their utilization in various applications, including gas storage and capture,2–5 pollutant removal,4,6,7 catalysis,4,8–10 and sensing.4,8 Furthermore, two-dimensional PAFs hold great promise in separation applications owing to their graphene-like structures.11,12 Despite their promising features, PAFs have been predominantly synthesized through cross-coupling reactions, such as the Yamamoto-type Ullman,2 Sonogashira–Hagihara,13 and Suzuki–Miyaura14 reactions, which require the use of either costly precious-metals or other metals in stoichiometric amounts as catalysts.15–17
For example, the PAF-1 synthesis requires the use of 5.2 equivalents of the bis(1,5-cyclooctadiene)nickel(0) catalyst ([Ni(cod)2]).2 Moreover, the earlier iterations of PAFs synthesized by employing 3–6 equivalents of the same Ni catalyst exhibited varying surface areas.5,18,19 In the cases of Sonogashira–Hagihara and Suzuki–Miyaura cross-coupling reactions, the most prevalent choice of catalysts are Pd(PPh3)4, and analogous Pd-based catalysts.7,13,20–22 Efforts to synthesize PAFs using cost-effective catalysts, such as AlCl3, and FeCl3, via Friedel–Crafts23 and Scholl reactions,24 have been relatively limited. These catalysts, are also used in a significant excess, thus increasing the environmental impact of the process. Moreover, these alternative catalysts have yielded PAFs characterized by notably reduced thermal stability and complex chemical structures, arising from uncontrolled side reactions.23–25 More recently, methanesulfonic acid has been employed both as a catalyst and a solvent in the synthesis of PAFs via an aldol triple condensation reaction26 starting from acetylbenzenes. However, porous polymer networks (PPNs) prepared using this approach exhibited substantial oxygen contents. These results highlight the fact that PAF synthesis often relies on the use of either expensive precious-metal catalysts or leads to the formation of polymer networks with lower stability owing to incomplete polymerization and/or side reactions. In this direction, while the catalyst-free synthesis of linear polyphenylenes through the Diels–Alder cycloaddition polymerization reaction presents an interesting strategy,27 the synthesis of porous polyphenylenes using this approach has yet to be realized. Here, we introduced a new strategy for the synthesis PAFs via Diels–Alder cycloaddition polymerization reaction by employing 5,5′-(1,4-phenylene)bis(2H-pyran-2-one) (Bispyrone) as a diene, and 1,3,5-triethynylbenzene (TEB) and tetrakis(4-ethynylphenyl)methane (TETPM) as dienophiles (Scheme 1). The resulting 2D and 3D porous polyphenylenes (pPPs) exhibited high surface areas of up to 1553 m2 g−1 with a pore volume of 1.45 cm3 g−1, and a remarkable high-pressure H2 uptake capacities of 7.4 and 7.1 wt% at 77 K, respectively.
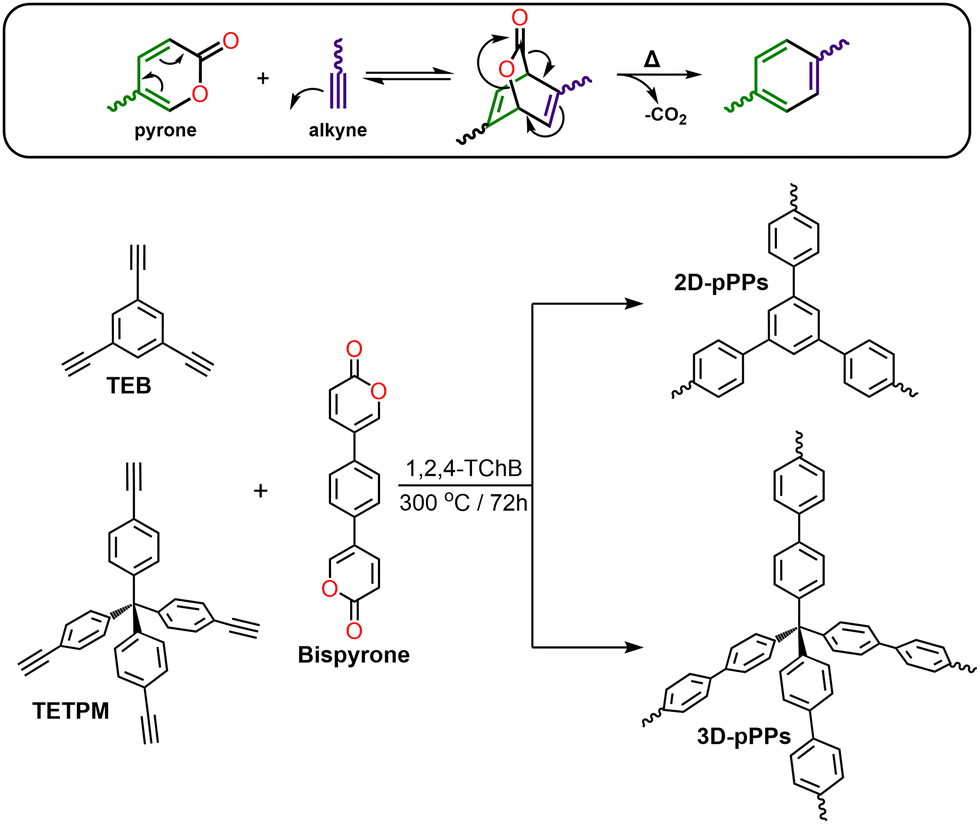 | ||
| Scheme 1 Diels–Alder cycloaddition reaction of pyrone with an alkyne (top). Synthetic scheme for the preparation of 2D-pPPs and 3D-pPPs via Diels–Alder cycloaddition polymerization reaction (bottom). | ||
Pyrones are recognized for their ability to undergo in situ diene formation via thermal CO2 elimination, subsequently engaging in Diels–Alder cycloaddition reaction with suitable dienophiles (see Scheme 1, top). For the synthesis of two-dimensional (2D) and three-dimensional (3D) poly(phenylenes) (pPPs), we synthesized the corresponding bispyrone starting from 1,4-dibromobenzene over five steps (see ESI†). The pPPs were synthesized using 1,2,4-trichlorobenzene (1,2,4-TChB) as a solvent at 300 °C for 72 h (for the experimental details, see the ESI†). The use of N-methyl-2-pyrrolidone as a solvent at 200 °C resulted in incomplete polymerization (Fig. S1, ESI†).
The successful synthesis of porous polyphenylenes was verified by the significant decrease in the intensity of characteristic –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –C–O-stretching vibrations of bispyrone at 1711, 1225, and 1130 cm−1 in the FT-IR spectra of 2D and 3D-pPPs (Fig. 1a and b). Moreover, the complete disappearance of the alkyne stretching bands originating from TEB and TETPM at 3275 and 3282 cm−1, respectively, served as an additional evidence for the successful polymerization reaction (Fig. 1a and b). Additionally, the most prominent vibration observed in the FTIR spectra of 2D and 3D-pPPs, occurring at 816 and 808 cm−1, respectively, pertains to the p-substituted benzene, suggesting the absence of side reactions. The chemical structure of the polymers was further verified by the solid-state cross-polarization magic angle spinning 13C nuclear magnetic resonance (CP-MAS NMR) spectroscopy. In the CP-MAS 13C NMR spectrum of 2D-pPPs, only two peaks at 139.8 and 120.7 ppm were observed, corresponding to the aromatic and ipso carbons (Fig. 1c). The CP-MAS 13C NMR spectrum of 3D-pPPs exhibited four peaks at 145.5, 139.5, 126.3, and 64.0 ppm, which were attributed to bridged and aromatic carbons, and the one at 64.0 ppm assigned to the tertiary carbon at the core of tetraphenylmethane moiety (Fig. 1c). The solid-state NMR spectra also corroborated with the successful formation of the polyphenylenes and the complete polymerization owing to the absence of peaks associated with –CO.
O and –C–O-stretching vibrations of bispyrone at 1711, 1225, and 1130 cm−1 in the FT-IR spectra of 2D and 3D-pPPs (Fig. 1a and b). Moreover, the complete disappearance of the alkyne stretching bands originating from TEB and TETPM at 3275 and 3282 cm−1, respectively, served as an additional evidence for the successful polymerization reaction (Fig. 1a and b). Additionally, the most prominent vibration observed in the FTIR spectra of 2D and 3D-pPPs, occurring at 816 and 808 cm−1, respectively, pertains to the p-substituted benzene, suggesting the absence of side reactions. The chemical structure of the polymers was further verified by the solid-state cross-polarization magic angle spinning 13C nuclear magnetic resonance (CP-MAS NMR) spectroscopy. In the CP-MAS 13C NMR spectrum of 2D-pPPs, only two peaks at 139.8 and 120.7 ppm were observed, corresponding to the aromatic and ipso carbons (Fig. 1c). The CP-MAS 13C NMR spectrum of 3D-pPPs exhibited four peaks at 145.5, 139.5, 126.3, and 64.0 ppm, which were attributed to bridged and aromatic carbons, and the one at 64.0 ppm assigned to the tertiary carbon at the core of tetraphenylmethane moiety (Fig. 1c). The solid-state NMR spectra also corroborated with the successful formation of the polyphenylenes and the complete polymerization owing to the absence of peaks associated with –CO.
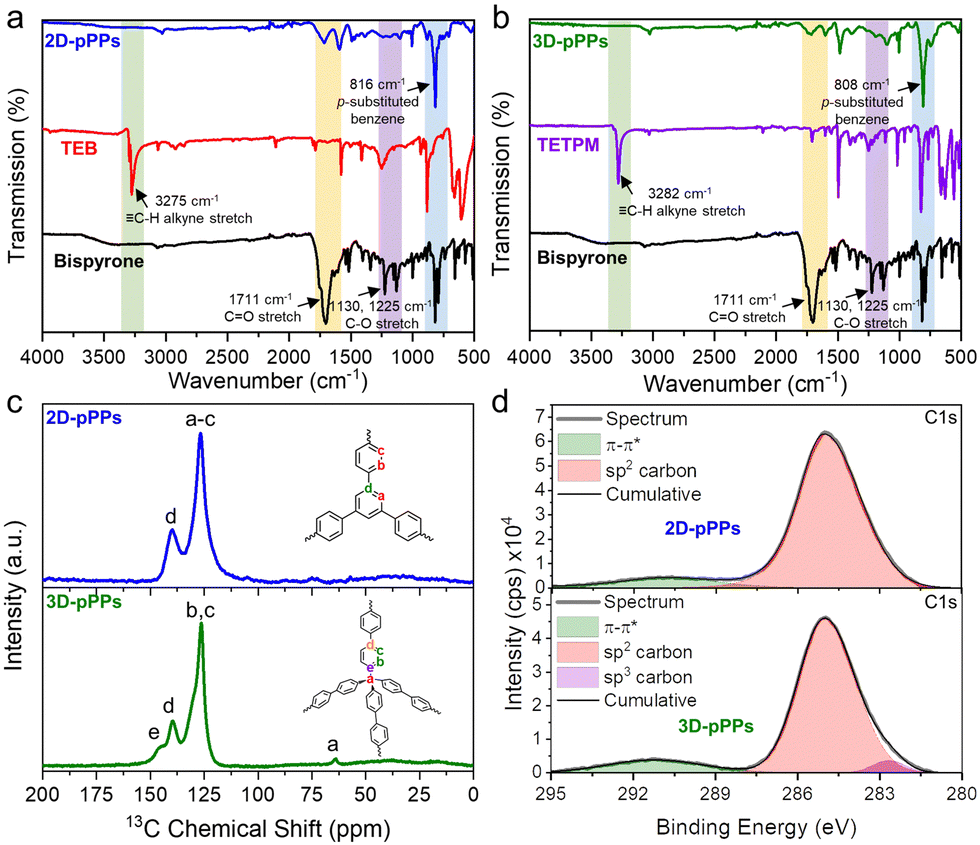 | ||
| Fig. 1 Spectroscopic characterization data of porous polyphenylenes. (a) FTIR spectra of 2D-pPPs and (b) 3D-pPPs along with the starting materials. (c) CP MAS 13C NMR spectra of 2D and 3D-pPPs. (d) The deconvoluted C1s XPS spectra of 2D and 3D-pPPs. | ||
The chemical bonding characteristics of 2D and 3D-pPPs were elucidated through X-ray photoelectron spectroscopy (XPS) analysis. As expected, the survey spectra of both 2D and 3D-pPPs exhibited significant carbon contents of 92.55% and 90.45%, respectively, (Fig. S2 and Table S1, ESI†), which agree well with the theoretical values. In the high-resolution C1s spectrum of 2D-pPPs, two principal peaks at 285.0 and 290.9 eV were identified and attributed to sp2 carbons and π–π* interactions,28 respectively (Fig. 1d). The high-resolution C1s spectrum of 3D-pPPs on the other hand displayed three peaks at 282.7, 285.0, and 291.2 eV, corresponding to sp3, sp2 carbons, and π–π* interactions, respectively (Fig. 2d). Detailed peak ratios are provided in Table S2 (ESI†). The survey spectra also unveiled the presence of 4.70 and 6.95 wt% of oxygen content in the 2D- and 3D-pPPs, respectively (Fig. S2 and Table S1, ESI†), which were attributed primarily to –CO end groups in the polymers along with the metal oxides and surface oxygens (Fig. S3 and Table S3, ESI†). These structural characterization data collectively provide strong evidence for the successful formation of both 2D- and 3D-pPPs. Moreover, we also probed the composition of polyphenylenes using elemental analysis (EA). The EA results revealed 90.68% carbon for the 2D and 88.41% for the 3D-pPPs, along with 4.07% and 5.00% of hydrogen, respectively (Table S1, ESI†). Both 2D- and 3D-pPPs exhibited high thermal stabilities, exhibiting no apparent mass loss up to 450 °C, followed by a mere 20% mass reduction at 800 °C, and retaining 60% of their initial mass even at 1000 °C under a N2 atmosphere (Fig. S4, ESI†). When subjected to TGA under air, a residual mass of 5–10% was observed, likely due to incomplete combustion.27 The X-ray diffraction analysis of the pPPs revealed their amorphous nature similar to previously reported PAFs (Fig. S5, ESI†). The morphological features of the 2D- and 3D-pPPs were investigated by scanning-electron microscopy (SEM) analysis. The resulting SEM micrographs unveiled that 2D-pPPs composed of agglomerated small flakes, while 3D-pPPs exhibited distinct microparticles (Fig. S6, ESI†). The porosity of pPPs was examined by measuring N2 adsorption–desorption isotherms at 77 K (Fig. 2a). Both 2D- and 3D-pPPs exhibited a type II isotherm, a characteristic typically observed in carbon materials featuring a combination of micro and mesoporosity. The emergence of mesopores was expected due to the length of the employed organic linkers. However, as the linker size increases, the interpenetration of the polymer networks has been commonly observed, which results in the formation of smaller pores and the co-existence of both micro- and mesopores in the pPPs. While 2D-pPPs displayed a small hysteresis in the desorption branch, the 3D-pPPs exhibited a distinctive H2 type hysteresis loop, indicative of the presence of inkbottle-shaped pores (see Fig. 2a). In order to assess the surface area, the Brunauer–Emmett–Teller (BET) theory was employed within the specified pressure range, determined from the Rouquerol plots (Fig. S7 and Table S4, ESI†). The 2D- and 3D-pPPs exhibited similar BET surface areas of 1553 and 1536 m2 g−1, respectively. Notably, 2D-pPP displayed a higher pore volume of 1.45 cm3 g−1 in contrast to the 1.27 cm3 g−1 observed in 3D-pPPs (Table S4, ESI†). This difference can be attributed to the reduced accessibility of pores within 3D-pPPs arising from the interpenetration of the polymer network, as is commonly observed for the 3D-POPs and COFs.29 The pore size distribution (PSD) of the polymers was calculated by using non-local density functional theory (NLDFT). PSD plots of both porous polyphenylenes exhibited micropores with an average pore size of 1.2 nm, which agrees well with the significant N2 uptake in the low-pressure range (Fig. 2b). The evaluation of mesopores was performed by employing the Barrett–Joyner–Halenda (BJH) model, which indicated the existence of mesopores in both polymers (Fig. S8, ESI†).
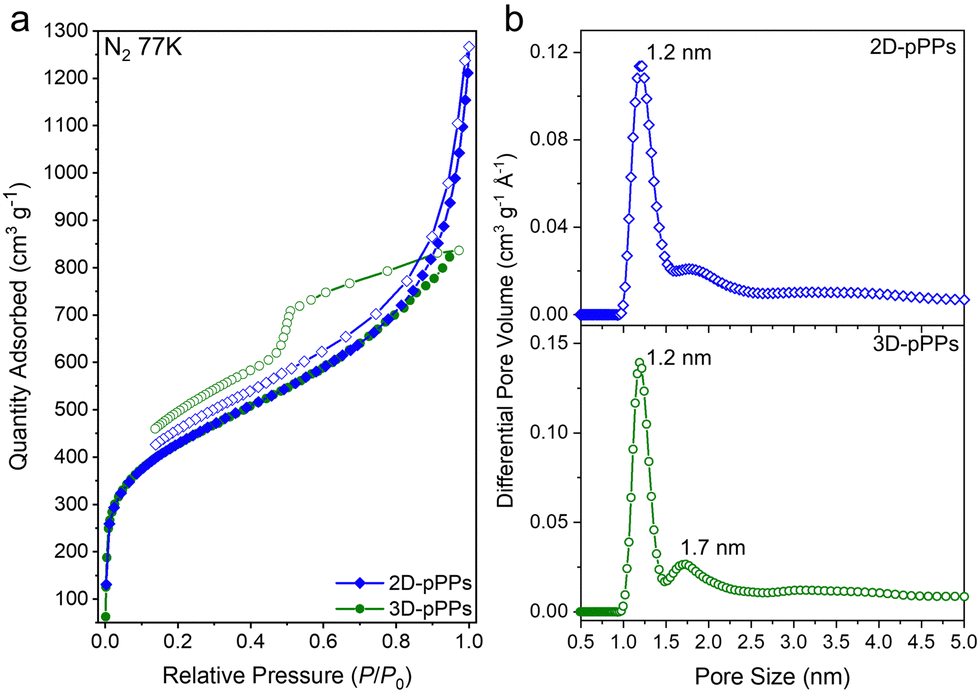 | ||
| Fig. 2 Porosity analysis of polyphenylenes, pPPs. (a) N2 uptake isotherms of 2D and 3D-pPPs measured at 77 K. Filled and empty symbols represent adsorption and desorption branches, respectively. (b) NLDFT pore size distribution of 2D-pPPs (top) and 3D-pPPs (bottom). | ||
The high surface areas and pore volumes of 2D/3D-pPPs prompted us to investigate their high-pressure small gas uptake performance. Both 2D- and 3D-pPPs showed very high H2 uptake capacities of 7.4% and 7.1%, respectively, at 77 K, 110 bar (Fig. 3a and b). These results position 2D and 3D-pPPs as highly promising materials when compared to other porous polymers such as PAFs and PPNs, especially considering their higher surface areas (Fig. S9 and Table S5, ESI†). This high H2 uptake capacity is attributed to the combined effect of the large abundance of micropores30 and high BET surface area. Whereas all the previously reported PAFs showed a linear correlation between their surface areas and H2 uptake capacities, both 2D/3D-pPPs showed a significant deviation from this and outperformed their counterparts with similar surface areas (Fig. S9, ESI†). 3D-pPPs exhibited slightly higher H2 excess uptake of 2.9 wt% compared to 2D-pPPs’ 2.7 wt% at the same temperature (Fig. S10, ESI†). This difference in total uptake can be attributed to the varying pore volumes in the two structures, while the higher excess uptakes in 3D-pPPs could be due to the swelling of the polymer network.31 The difference in gas uptake between the two structures becomes less significant at high temperatures (Fig. 3a and b and Table S7, ESI†). Additionally, both 2D and 3D-pPPs exhibited very similar H2 heats of adsorption (Qst) values. The 2D-pPPs showed a H2Qst value of 6.6 kJ mol−1 at zero coverage, while 3D-pPPs had a slightly lower Qst of 6.3 kJ mol−1 (Fig. 3c). We also investigated the high-pressure CH4 uptake performance of both polymers. 2D and 3D-pPPs exhibited good high-pressure CH4 uptake capacities of 25.6 and 24.6 wt%, respectively, at 296 K (Fig. 3d and e). The presence of an aromatic backbone along with the high pore volumes of both polymers are likely to contribute to the high affinity of towards CH4.32 The heats of adsorption for CH4 were 18.7 and 17.9 kJ mol−1 for 2D- and 3D-pPPs, respectively, indicative of a physisorption mechanism (Fig. 3f). 2D-pPPs consistently demonstrated higher total uptake, while 3D-pPPs displayed greater excess uptake (Fig. S10 and Table S7, ESI†).
Finally, considering the substantial micropore content, we also explored the CO2 uptake performance of 2D- and 3D-pPPs. 2D-pPPs exhibited an impressive CO2 uptake capacity of 248.6 wt% at 220 K, 25 bar, while the 3D-pPPs showed 214.5 wt% (Fig. 3g and h). Notably, 3D-pPPs displayed a higher uptake of 119.3 wt% at 270 K and 65.2 wt% at 296 K, in contrast to 2D-pPPs, which showed uptake capacities of 100.4 wt% and 60.7 wt%, respectively (Fig. 3g and h). These values align favorably with those reported for other PAFs and POPs, following the trend of CO2 and CH4 uptake in relation to surface area (Fig. S9, S10 and Table S6, ESI†). Furthermore, the heats of adsorption for CO2 for both 2D and 3D-pPPs were found to be 25.1 and 24.1 kJ mol−1, respectively. These values position polyphenylenes as potential candidates for CO2 storage (Fig. 3i).33
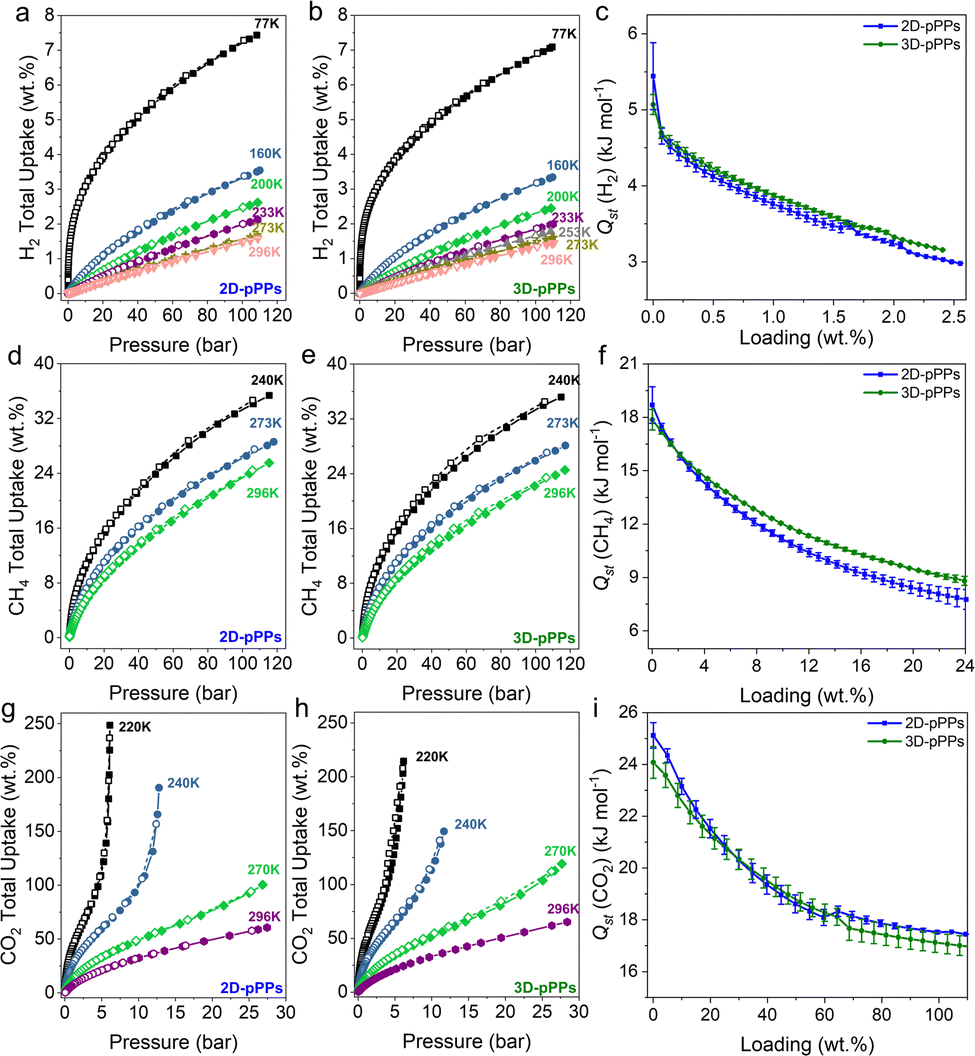 | ||
| Fig. 3 High pressure gas uptake performance of polyphenylenes. H2 uptake isotherms obtained at different temperatures for (a) 2D-pPPs and (b) 3D-pPPs. CH4 uptake isotherms obtained at different temperatures for (d) 2D-pPPs and (e) 3D-pPPs. CO2 uptake isotherms obtained at different temperatures for (g) 2D-pPPs and (h) 3D-pPPs. The heat of adsorption (Qst) for 2D and 3D-pPPs for (c) H2, (f) CH4 and (i) CO2. | ||
In conclusion, we introduced a new approach for the synthesis of porous polyphenylenes through Diels–Alder cycloaddition polymerization reaction, effectively eliminating the need for metal catalysts in the preparation of carbon-based frameworks. This approach yielded porous polyphenylenes that exhibit exceptional H2 uptake capacities along with good CH4 and CO2 uptake performance. In a broader context, these results will stimulate the development of environmentally friendly approaches for the synthesis of porous organic polymers.
T. A. synthesized the precursors and polymers and conducted all characterizations. P. W. F. assisted in synthesizing the precursors and characterizing the polymers. T. Y. performed high-pressure gas uptake measurements. A. C. secured funding, supervised the project, and co-wrote the paper with all the authors.
Prof. Ali Coskun acknowledges the Swiss National Science Foundation (SNF) for grant 200021-175947. Dr Timur Ashirov thanks Anne Schuwey, Emmanuel Morand and their apprentices in the Synthesis Laboratory at the University of Fribourg, Department of Chemistry for their assistance in the precursor synthesis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. H. Wentorf, R. C. DeVries and F. P. Bundy, Science, 1980, 208, 873–880 CrossRef CAS PubMed.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- C. Pei, T. Ben and S. Qiu, Mater. Horiz., 2015, 2, 11–21 RSC.
- Y. Tian and G. Zhu, Chem. Rev., 2020, 120, 8934–8986 CrossRef CAS PubMed.
- M. Li, H. Ren, F. Sun, Y. Tian, Y. Zhu, J. Li, X. Mu, J. Xu, F. Deng and G. Zhu, Adv. Mater., 2018, 30, 1804169 CrossRef PubMed.
- Z. Wang, Q. Meng, R. Ma, Z. Wang, Y. Yang, H. Sha, X. Ma, X. Ruan, X. Zou, Y. Yuan and G. Zhu, Chemistry, 2020, 6, 1683–1691 CrossRef CAS.
- L. Zhang, J.-S. Sun, F. Sun, P. Chen, J. Liu and G. Zhu, Chem. – Eur. J., 2019, 25, 3903–3908 CrossRef CAS PubMed.
- U. Díaz and A. Corma, Coord. Chem. Rev., 2016, 311, 85–124 CrossRef.
- E. Verde-Sesto, M. Pintado-Sierra, A. Corma, E. M. Maya, J. G. de la Campa, M. Iglesias and F. Sánchez, Chem. – Eur. J., 2014, 20, 5111–5120 CrossRef CAS PubMed.
- L. Căta, N. Terenti, C. Cociug, N. D. Hădade, I. Grosu, C. Bucur, B. Cojocaru, V. I. Parvulescu, M. Mazur and J. Čejka, ACS Appl. Mater. Interfaces, 2022, 14, 10428–10437 CrossRef PubMed.
- Y. Li, Z. Zhou, P. Shen and Z. Chen, Chem. Commun., 2010, 46, 3672–3674 RSC.
- Y. Ma, F. Cui, H. Rong, J. Song, X. Jing, Y. Tian and G. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202113682 CrossRef CAS PubMed.
- Z. Yan, H. Ren, H. Ma, R. Yuan, Y. Yuan, X. Zou, F. Sun and G. Zhu, Microporous Mesoporous Mater., 2013, 173, 92–98 CrossRef CAS.
- S. Meng, X. Zou, C. Liu, H. Ma, N. Zhao, H. Ren, M. Jia, J. Liu and G. Zhu, ChemCatChem, 2016, 8, 2393–2400 CrossRef CAS.
- M. Ammon, T. Sander and S. Maier, J. Am. Chem. Soc., 2017, 139, 12976–12984 CrossRef CAS PubMed.
- K. Xu, J. I. Urgel, K. Eimre, M. Di Giovannantonio, A. Keerthi, H. Komber, S. Wang, A. Narita, R. Berger, P. Ruffieux, C. A. Pignedoli, J. Liu, K. Müllen, R. Fasel and X. Feng, J. Am. Chem. Soc., 2019, 141, 7726–7730 CrossRef CAS PubMed.
- M. Bieri, M. Treier, J. Cai, K. Aït-Mansour, P. Ruffieux, O. Gröning, P. Gröning, M. Kastler, R. Rieger, X. Feng, K. Müllen and R. Fasel, Chem. Commun., 2009, 6919–6921, 10.1039/B915190G.
- T. Ben, C. Pei, D. Zhang, J. Xu, F. Deng, X. Jing and S. Qiu, Energy Environ. Sci., 2011, 4, 3991–3999 RSC.
- H. Ren, T. Ben, F. Sun, M. Guo, X. Jing, H. Ma, K. Cai, S. Qiu and G. Zhu, J. Mater. Chem., 2011, 21, 10348–10353 RSC.
- Y. Yuan, F. Sun, H. Ren, X. Jing, W. Wang, H. Ma, H. Zhao and G. Zhu, J. Mater. Chem., 2011, 21, 13498–13502 RSC.
- J.-S. Sun, L.-P. Jing, Y. Tian, F. Sun, P. Chen and G. Zhu, Chem. Commun., 2018, 54, 1603–1606 RSC.
- M. Remmers, B. Müller, K. Martin, H.-J. Räder and W. Köhler, Macromolecules, 1999, 32, 1073–1079 CrossRef CAS.
- L. Meng, X. Zou, S. Guo, H. Ma, Y. Zhao and G. Zhu, ACS Appl. Mater. Interfaces, 2015, 7, 15561–15569 CrossRef CAS PubMed.
- S. Meng, H. Ma, L. Jiang, H. Ren and G. Zhu, J. Mater. Chem. A, 2014, 2, 14536–14541 RSC.
- K. J. Msayib and N. B. McKeown, J. Mater. Chem. A, 2016, 4, 10110–10113 RSC.
- C. Wang, C. Li, E. R. C. Rutledge, S. Che, J. Lee, A. J. Kalin, C. Zhang, H.-C. Zhou, Z.-H. Guo and L. Fang, J. Mater. Chem. A, 2020, 8, 15891–15899 RSC.
- J. K. Stille and Y. Gilliams, Macromolecules, 1971, 4, 515–517 CrossRef CAS.
- P. W. Fritz, T. Chen, T. Ashirov, A.-D. Nguyen, M. Dincă and A. Coskun, Angew. Chem., Int. Ed., 2022, 61, e202116527 CrossRef CAS PubMed.
- X. Liu, J. Li, B. Gui, G. Lin, Q. Fu, S. Yin, X. Liu, J. Sun and C. Wang, J. Am. Chem. Soc., 2021, 143, 2123–2129 CrossRef CAS PubMed.
- N. C. Gallego, L. He, D. Saha, C. I. Contescu and Y. B. Melnichenko, J. Am. Chem. Soc., 2011, 133, 13794–13797 CrossRef CAS PubMed.
- D. Fairen-Jimenez, Y. J. Colón, O. K. Farha, Y.-S. Bae, J. T. Hupp and R. Q. Snurr, Chem. Commun., 2012, 48, 10496–10498 RSC.
- V. Rozyyev, D. Thirion, R. Ullah, J. Lee, M. Jung, H. Oh, M. Atilhan and C. T. Yavuz, Nat. Energy, 2019, 4, 604–611 CrossRef CAS.
- Z. Chen, P. Li, R. Anderson, X. Wang, X. Zhang, L. Robison, L. R. Redfern, S. Moribe, T. Islamoglu, D. A. Gómez-Gualdrón, T. Yildirim, J. F. Stoddart and O. K. Farha, Science, 2020, 368, 297–303 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc06162k |
| This journal is © The Royal Society of Chemistry 2024 |