
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModular synthesis of congested β2,2-amino acids via the merger of photocatalysis and oxidative functionalisations†
Khadijah
Anwar
a,
Luca
Capaldo
 b,
Ting
Wan
b,
Timothy
Noël
b and
Adrián
Gómez-Suárez
*a
b,
Ting
Wan
b,
Timothy
Noël
b and
Adrián
Gómez-Suárez
*a
aOrganic Chemistry, Bergische Universität Wuppertal, Gaußstr. 20, 42119, Wuppertal, Germany. E-mail: gomezsuarez@uni-wuppertal.de
bFlow Chemistry Group, Van’t Hoff Institute for Molecular Sciences (HIMS), University of Amsterdam, Science Park 904, Amsterdam 1098 XH, The Netherlands
First published on 5th January 2024
Abstract
A two-step protocol for the modular synthesis of β2- and α-quaternary β2,2-amino acid derivatives is reported. The key steps are a photocatalytic hydroalkylation reaction, followed by an oxidative functionalisation to access N-protected β-amino acids, esters, and amides. This strategy can be effectively scaled up via continuous-flow technology.
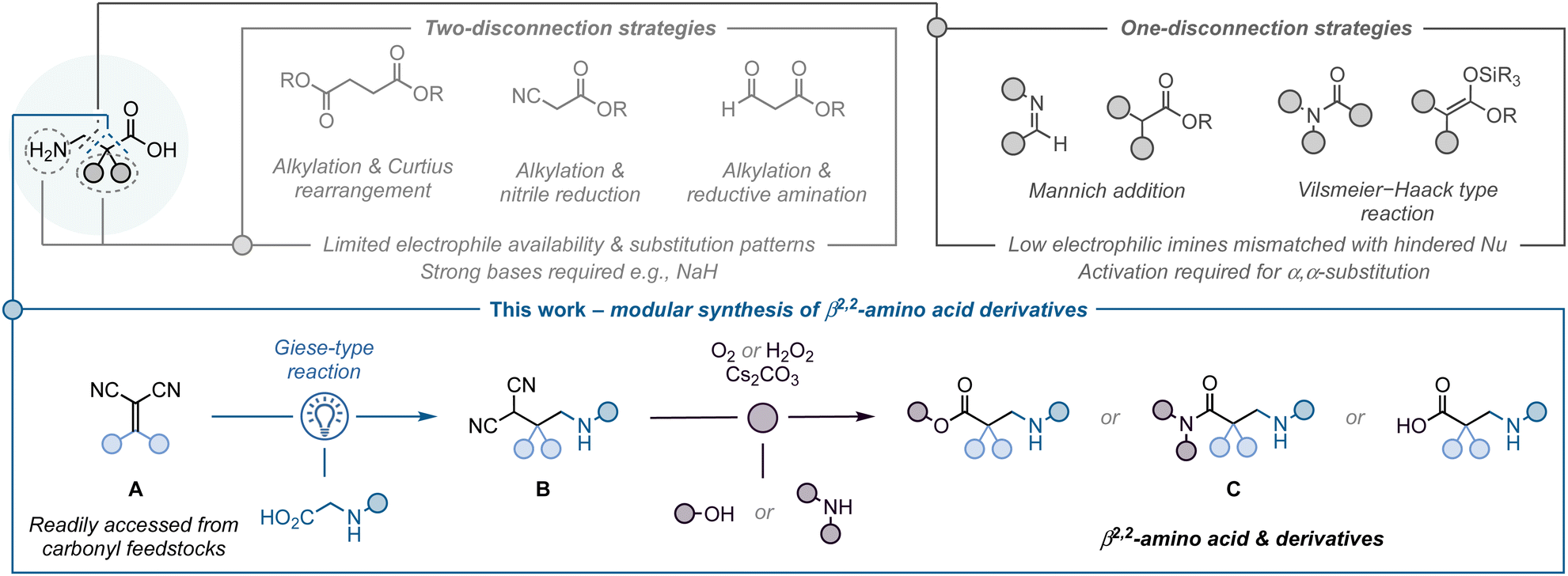
β-Amino acids are key components in a wide range of biologically active molecules.1 Their incorporation into peptides results in strong modifications to the peptide secondary structure, leading to an increased resistance to proteases and peptidases.1c,d Compared to the more abundant α-amino acids, the addition of another C–C bond in β-amino acids provides new opportunities to modify the substituent pattern on the C2 and C3 positions. This leads to an extra level of complexity and depending on this, it is possible to have β2-, β2,2-, β3-, or β2,3-amino acids. Among them, the β2- and β3-variants are the most prominent in the life sciences, which is reflected by the numerous methodologies available to access them.2 The less explored α-quaternary β2,2-amino acids have been shown to be particularly effective at affecting peptide conformation by inducing the formation of more rigid secondary structures, enhancing stability towards proteolytic degradation.1a In addition, they can be used as intermediates to access pharmaceutically relevant structures such as β-lactams, γ-amino alcohols, or azetidines. The synthetic challenges associated with the construction of quaternary centres means there are few methods available for the preparation of β2,2-amino acids, hampering the exploration of their biological properties. Therefore, the development of new approaches to access α-quaternary β2-amino acids is important to unlock their full potential.
Current approaches primarily proceed via two-electron disconnections (Fig. 1). These include multi-step sequences, such as alkylation followed by Curtius rearrangement,3 nitrile reductions,4 reductive aminations;5,6 or the Arndt–Eistert homologation.7 The most common way to prepare β-amino acids is through Mannich addition reactions, which are limited to certain reactive electrophiles (e.g., iminium species such as Vilsmeier–Hack type reagents), or the use of unsubstituted, or very specifically substituted, enolate equivalents as nucleophiles.8,9 As a consequence, there are major restrictions in the type of substituents which can be incorporated into the final product, limiting these approaches to the synthesis of β2-, β3- and β2,3-amino acid derivatives. In contrast to these processes governed by ionic intermediates, radical reactions can enable a more facile construction of quaternary centres. The comparatively high energy of radicals can be used to overcome kinetic barriers associated with steric hindrance, which may be more difficult to accomplish with a two-electron approach. This has been extensively demonstrated, for example, in the application of radical-based strategies for the synthesis of complex natural products bearing quaternary centres.10
 | ||
| Fig. 1 Access to quaternary β2-amino esters – state-of-the-art vs. our strategy. | ||
With this in mind, we have designed a modular strategy to access α-quaternary β2,2-amino acid analogues from readily available feedstocks using radical chemistry.11 The proposed approach proceeds in a two-step sequence (Fig. 1B): (1) light-mediated Giese-type reaction12 between alkylidenemalononitriles (A)13,14 – readily synthesised via Knoevenagel condensation –15 and α-amino acids,16 and (2) oxidation of the resulting β-quaternary malononitriles17 (B) to access the targeted β2,2-amino acid analogues (C).
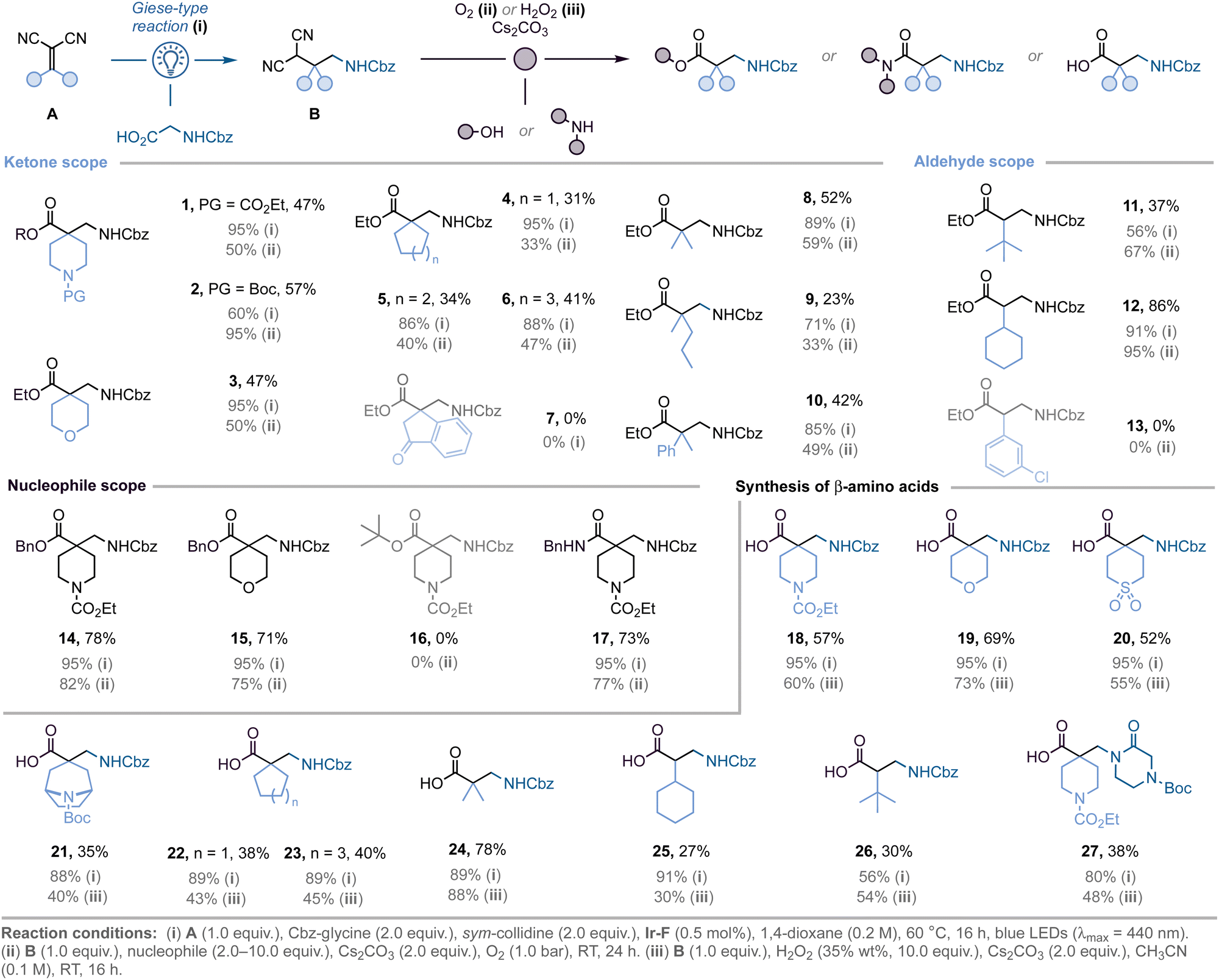
Optimisation of the light-mediated Giese-type reaction to access malononitrile derivatives B revealed that the best conditions were: irradiation (blue LEDs, 2 × 32 W, λmax = 440 nm) of a mixture of alkylidenemalononitrile A (1.0 equiv.) and Cbz-glycine (2.0 equiv.) in the presence of Ir[(dF(CF3)ppy)2(dtbbpy)]PF6 (Ir-F, 0.5 mol%) and sym-collidine (2.0 equiv.), in 1,4-dioxane (0.2 M) at 60 °C.18 This was followed by aerobic oxidation (O2 balloon) of malononitrile B with Cs2CO3 (2.0 equiv.), and EtOH (10.0 equiv.) as nucleophile for 24 h, affording the targeted β2,2-amino esters in optimal yields.17,18
With the optimised conditions in hand, we explored the scope and limitations of this strategy (Scheme 1). Aliphatic heterocycles are well-tolerated, as shown by the successful preparation of quaternary β2-amino esters bearing piperidine (1–2), and tetrahydropyran (3) motifs (two-step yields: 47%, 57%, and 47%, respectively). Modification of the size of the ring system at the α-position is achievable, as exemplified by the incorporation of cyclopentane (4), cyclohexane (5), and cycloheptane (6) motifs (31%, 34%, and 41% yield, respectively). The Giese-type reaction was unsuccessful when employing dihydroindenone-derived alkylidene malononitrile (7). This might be due to the increased steric hindrance afforded by the dihydroindenone-core, making the key radical addition step more difficult. Additionally, the methodology is not restricted to cyclic ketones, as shown by the synthesis of β2,2-amino esters bearing acyclic substituents (8–10). Monosubstituted β2-amino esters can also be obtained when using aldehydes in the Knoevenagel condensation step. The use of sterically demanding tert-butyl (11) and cyclohexyl (12) substituents led to the desired products in high yields, however benzylidene malononitriles gave complex mixtures of products (13, not isolated). Moreover, while the use of benzyl alcohol instead of EtOH allowed for the preparation of the corresponding benzyl esters 14 and 15 in good yields (78% and 71%, respectively), the corresponding tert-butyl ester (16) could not be isolated; probably due to the increased steric hindrance afforded by the tert-butyl group. Finally, the use of benzyl amine afforded the corresponding amide (17) in good yields after two steps (73%).
 | ||
| Scheme 1 Scope & limitations of the methodology. Giese reactions carried out in 0.5–1.0 mmol scale, oxidative functionalisations in 0.2 mmol scale. | ||
To expand the practicality of the method, N-protected β-amino acids were prepared directly using H2O2 as the oxidant (18–23).19,20 Both acyclic ketones (24), as well as aldehydes (25–26), can be used to access the corresponding β-amino acids. In addition, the modularity of our strategy was further highlighted by replacing Cbz-glycine by an unnatural amino acid bearing a piperazinone core in the first step, thus enabling the synthesis of highly polar β2-amino acid 27 in 38% yield.
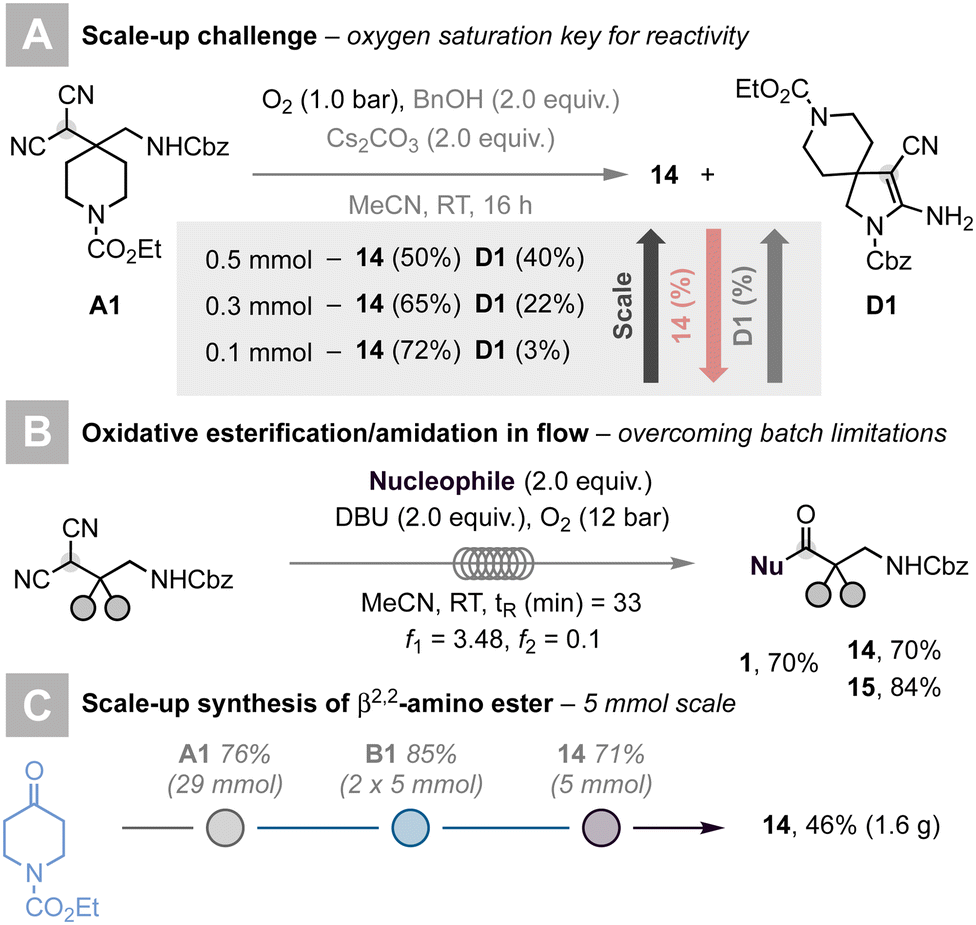
Attempts to scale up the reaction were frustrated by the competitive formation of spirocyclic compound D1 (Scheme 2A).21 We hypothesised that due to poor solubility of oxygen in organic solvents, the formation of ester 14 was hampered, thus favouring the pathway leading to D1. We reasoned that the selective formation of the targeted β2-amino ester should be achieved by increasing the O2 content in solution. In batch, this calls for high O2 pressures, increasing safety hazards, and limiting the applicability of our methodology; flow technology stands as an ideal solution to this challenge.
 | ||
| Scheme 2 Scale-up synthesis of quaternary β2-amino esters. | ||
In recent years, flow chemistry22 has emerged as a powerful technology to handle gas/liquid reaction mixtures. The possibility to control pressure using cheap back-pressure regulators (BPRs) and the advantages offered by Taylor recirculation for improved mass-transfer have changed paradigms in this field.23 Flow chemistry has proven incredibly beneficial to perform oxidation reactions using molecular oxygen as a simple, green, and mild oxidant.24 Other advantages of flow chemistry include shorter reaction times, enhanced heat-transfer, straightforward scalability, and improved safety standards and reproducibility.
Thus, we decided to tackle the challenges outlined above and scale our protocol up by adopting continuous-flow conditions. After a short re-optimisation of reaction conditions, which led to the replacement of insoluble carbonates with DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) in the role of the base to avoid possible clogging issues, we succeeded in translating our reaction from batch to flow.18 A feed containing the malononitrile, DBU, and the nucleophile was merged with an O2 feed via a PEEK T-mixer. The resulting slug-flow was pressurised at 12 bar by means of an adjustable BPR and pumped through a PFA coil (perfluoroalkoxy, V = 25 mL, ID: 2 mm) kept at 50 °C. A residence time (tR) of 33 min was required to ensure reproducible access to β2-amino esters. This new process proved effective to access ester 1, 14, and 15 in high yields (Scheme 2B). The use of this continuous-flow process, in combination with the readily scalable batch reactions to access alkylidenemalononitrile A1 and α-quaternary malononitrile B1, enabled the synthesis of β2-amino ester 14 in 46% yield (1.6 g) after three steps (Scheme 2C).
In summary, we have developed a modular two-step strategy for the synthesis of α-quaternary β-amino acid derivatives from readily available ketones, malononitrile, and α-amino acids. The method tolerates the introduction of (hetero)cyclic and acyclic substituents, both aliphatic and aromatic, at the β-position and allows for the synthesis of monosubstituted β2-amino acids when aldehydes are employed instead of ketones. Moreover, by tuning the last step of the strategy, it is possible to selectively access β-amino esters, β-amino amides, or N-protected β-amino acids. Finally, to overcome limitations associated with mass transfer in the scale-up of the oxidative esterification step, we have developed a continuous-flow process that enables reproducible and scalable access to α-quaternary β2,2-amino esters in gram scale.
This work was supported by the Fonds der Chemischen Industrie (Liebig fellowship to A. G. S.), by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 443074366, and the Bergische Universität Wuppertal. Umicore A. G. is acknowledged for its generous donation of materials. We thank Dr Sebastian Govaerts for fruitful discussions and proof reading of the manuscript. Prof. Stefan Kirsch (BUW) is greatly acknowledged for his continuous support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) G. Lelais and D. Seebach, Biopolymers, 2004, 76, 206–243 CrossRef CAS; (b) P. Spiteller and F. von Nussbaum, Enantioselective Synthesis of β-Amino Acids, 2005, pp. 19–91 Search PubMed; (c) C. Cabrele, T. A. Martinek, O. Reiser and L. Berlicki, J. Med. Chem., 2014, 57, 9718–9739 CrossRef CAS PubMed; (d) M. P. Del Borgo, K. Kulkarni and M. I. Aguilar, Curr. Pharm. Des., 2017, 23, 3772–3785 CrossRef CAS PubMed; (e) U. Bąchor and M. Mączyński, Molecules, 2021, 26, 438 CrossRef.
- (a) D. Seebach, A. K. Beck, S. Capone, G. Deniau, U. Grošelj and E. Zass, Synthesis, 2009, 1–32 CrossRef CAS; (b) V. Chhiba, M. Bode, K. Mathiba and D. Brady, Cascade Biocatalysis, 2014, pp. 297–314 Search PubMed.
- J. J. W. Duan, L. Chen, Z. Lu, B. Jiang, N. Asakawa, J. E. Sheppeck, R.-Q. Liu, M. B. Covington, W. Pitts, S.-H. Kim and C. P. Decicco, Bioorg. Med. Chem. Lett., 2007, 17, 266–271 CrossRef CAS.
- (a) C. Cativiela, M. D. Diaz-de-Villegas and J. A. Galvez, J. Org. Chem., 1994, 59, 2497–2505 CrossRef CAS; (b) T. Hansen, T. Alst, M. Havelkova and M. B. Strøm, J. Med. Chem., 2010, 53, 595–606 CrossRef CAS PubMed; (c) M. H. Paulsen, M. Engqvist, D. Ausbacher, M. B. Strøm and A. Bayer, Org. Biomol. Chem., 2016, 14, 7570–7578 RSC.
- D. Lin, G. Deng, J. Wang, X. Ding, H. Jiang and H. Liu, J. Org. Chem., 2010, 75, 1717–1722 CrossRef CAS.
- For alternative two-step approaches see: (a) J.-S. Yu, H. Noda and M. Shibasaki, Angew. Chem., Int. Ed., 2018, 57, 818–822 CrossRef CAS; (b) P. Tovillas, C. D. Navo, P. Oroz, A. Avenoza, F. Corzana, M. M. Zurbano, G. Jimenez-Oses, J. H. Busto and J. M. Peregrina, J. Org. Chem., 2022, 87, 8730–8743 CrossRef CAS PubMed.
- D. Seebach, M. Overhand, F. N. M. Kühnle, B. Martinoni, L. Oberer, U. Hommel and H. Widmer, Helv. Chim. Acta, 1996, 79, 913–941 CrossRef CAS.
- (a) N. S. Chowdari, J. T. Suri and C. F. Barbas, Org. Lett., 2004, 6, 2507–2510 CrossRef CAS; (b) T. Akiyama, Y. Saitoh, H. Morita and K. Fuchibe, Adv. Syn. Catal., 2005, 347, 1523–1526 CrossRef CAS; (c) A. Hasegawa, Y. Naganawa, M. Fushimi, K. Ishihara and H. Yamamoto, Org. Lett., 2006, 8, 3175–3178 CrossRef CAS; (d) A. Ting, S. Lou and S. E. Schaus, Org. Lett., 2006, 8, 2003–2006 CrossRef CAS PubMed; (e) A. L. Tillman, J. Ye and D. J. Dixon, Chem. Commun., 2006, 1191–1193, 10.1039/B515725K; (f) R. Moumné, B. Denise, A. Parlier, S. Lavielle, H. Rudler and P. Karoyan, Tetrahedron Lett., 2007, 48, 8277–8280 CrossRef.
- A. Romanens and G. Bélanger, Org. Lett., 2015, 17, 322–325 CrossRef CAS.
- (a) M. J. Schnermann and L. E. Overman, Angew. Chem., Int. Ed., 2012, 51, 9576–9580 CrossRef CAS; (b) C. R. Jamison and L. E. Overman, Acc. Chem. Res., 2016, 49, 1578–1586 CrossRef CAS PubMed; (c) Y. Slutskyy, C. R. Jamison, G. L. Lackner, D. S. Muller, A. P. Dieskau, N. L. Untiedt and L. E. Overman, J. Org. Chem., 2016, 81, 7029–7035 CrossRef CAS; (d) J. Li, F. Li, E. King-Smith and H. Renata, Nat. Chem., 2020, 12, 173–179 CrossRef CAS; (e) Z. Xin, H. Wang, H. He and S. Gao, Tetrahedron Lett., 2021, 71, 153029 CrossRef CAS.
- (a) H. Jia and T. Ritter, Angew. Chem., Int. Ed., 2022, 61, e202208978 CrossRef CAS; (b) N. Tanaka, J. L. Zhu, O. L. Valencia, C. R. Schull and K. A. Scheidt, J. Am. Chem. Soc., 2023, 145, 24486–24492 CAS.
- A. L. Gant Kanegusuku and J. L. Roizen, Angew. Chem., Int. Ed., 2021, 60, 21116–21149 CrossRef CAS PubMed.
- (a) H.-L. Cui and Y.-C. Chen, Chem. Commun., 2009, 4479–4486 RSC; (b) B. Farajpour and A. Alizadeh, Org. Biomol. Chem., 2022, 20, 8366–8394 RSC.
- Examples of radical additions to alkylidenemalononitriles: (a) R. A. Holton, A. D. Williams and R. M. Kennedy, J. Org. Chem., 2002, 51, 5480–5482 CrossRef; (b) Z. Tu, C. Lin, Y. Jang, Y.-J. Jang, S. Ko, H. Fang, J.-T. Liu and C.-F. Yao, Tetrahedron Lett., 2006, 47, 6133–6137 CrossRef CAS; (c) D. Dondi, D. Ravelli, M. Fagnoni, M. Mella, A. Molinari, A. Maldotti and A. Albini, Chem. – Eur. J., 2009, 15, 7949–7957 CrossRef CAS PubMed; (d) X. Z. Fan, J. W. Rong, H. L. Wu, Q. Zhou, H. P. Deng, J. D. Tan, C. W. Xue, L. Z. Wu, H. R. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef CAS; (e) L. Li, L. Fang, W. Wu and J. Zhu, Org. Lett., 2020, 22, 5401–5406 CrossRef CAS PubMed; (f) D. Kuwana, Y. Komori, M. Nagatomo and M. Inoue, J. Org. Chem., 2022, 87, 730–736 CrossRef CAS.
- K. van Beurden, S. de Koning, D. Molendijk and J. van Schijndel, Green Chem. Lett. Rev., 2020, 13, 349–364 CrossRef CAS.
- (a) L. Chu, C. Ohta, Z. Zuo and D. W. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS; (b) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed; (c) Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257–5260 CrossRef CAS PubMed; (d) S. B. Lang, K. M. O'Nele and J. A. Tunge, J. Am. Chem. Soc., 2014, 136, 13606–13609 CrossRef CAS PubMed; (e) Y. Yin, Y. Dai, H. Jia, J. Li, L. Bu, B. Qiao, X. Zhao and Z. Jiang, J. Am. Chem. Soc., 2018, 140, 6083–6087 CrossRef CAS; (f) T. Shao, X. Ban and Z. Jiang, Chem. Rec., 2023, 23, e202300122 CrossRef CAS.
- (a) J. Li, M. J. Lear and Y. Hayashi, Angew. Chem., Int. Ed., 2016, 55, 9060–9064 CrossRef CAS PubMed; (b) Y. Hayashi, J. Li, H. Asano and D. Sakamoto, Eur. J. Org. Chem., 2018, 675–677 Search PubMed.
- See ESI† for further details.
- Y. Sun, Z. Qiao, D. Li, J. Ni, J. Wang, P. Wang, N. Song and M. Li, Org. Lett., 2022, 24, 7944–7949 CrossRef CAS PubMed.
- For compound 20, 4-thianone was used as the starting ketone.
- K. Anwar, F. J. A. Troyano, A. H. Abazid, O. El Yarroudi, I. Funes-Ardoiz and A. Gomez-Suarez, Org. Lett., 2023, 25, 3216–3221 CrossRef CAS PubMed.
- (a) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed; (b) B. Gutmann and C. O. Kappe, J. Flow Chem., 2017, 7, 65–71 CrossRef CAS; (c) M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2020, 24, 1802–1813 CrossRef CAS; (d) L. Capaldo, Z. Wen and T. Noël, Chem. Sci., 2023, 14, 4230–4247 RSC.
- (a) G. Laudadio, S. Govaerts, Y. Wang, D. Ravelli, H. F. Koolman, M. Fagnoni, S. W. Djuric and T. Noël, Angew. Chem., Int. Ed., 2018, 57, 4078–4082 CrossRef CAS; (b) F. Raymenants, T. M. Masson, J. Sanjosé-Orduna and T. Noël, Angew. Chem., Int. Ed., 2023, 62, e202308563 CrossRef CAS PubMed.
- (a) A. Gavriilidis, A. Constantinou, K. Hellgardt, K. K. Hii, G. J. Hutchings, G. L. Brett, S. Kuhn and S. P. Marsden, Reaction Chem. Eng., 2016, 1, 595–612 RSC; (b) H. P. L. Gemoets, Y. Su, M. Shang, V. Hessel, R. Luque and T. Noël, Chem. Soc. Rev., 2016, 45, 83–117 RSC; (c) C. A. Hone and C. O. Kappe, Top. Curr. Chem., 2018, 377, 2 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc06172h |
| This journal is © The Royal Society of Chemistry 2024 |