
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stapling of leu-enkephalin analogs with bifunctional reagents for prolonged analgesic activity†
Monika
Kijewska‡
 *a,
Grzegorz
Wołczański‡
*a,
Piotr
Kosson
b,
Robert
Wieczorek
a,
Marek
Lisowski
a and
Piotr
Stefanowicz
a
*a,
Grzegorz
Wołczański‡
*a,
Piotr
Kosson
b,
Robert
Wieczorek
a,
Marek
Lisowski
a and
Piotr
Stefanowicz
a
aFaculty of Chemistry, University of Wrocław, Joliot-Curie 14, 50-383 Wrocław, Poland. E-mail: monika.kijewska@uwr.edu.pl; grzegorz.wolczanski87@gmail.com
bMossakowski Medical Research Institute, Polish Academy of Sciences, 5 Pawinskiego Street, 02-106 Warszawa, Poland
First published on 6th February 2024
Abstract
The design and synthesis of leu-enkephalin analogs by replacing the glycine residues with N-(2-thioethyl)glycines and opening the cyclisation potential is presented. The cyclization (stapling) was achieved using bifunctional reagents (hexafluorobenzene and trithiocyanuric acid derivatives). The CD conformational studies of the stapled analogs suggest that the peptides adopt the type I β-turn conformation, which is in agreement with the theoretical analysis. The analog containing a trithiocyanuric acid derivative with a benzyl substituent shows potent analgesic activity.
In 1975, Hughes et al. discovered the endogenous opioid pentapeptides leu-enkephalin and met-enkephalin, which, in addition to their antinociceptive effects, have many other biological activities.1 Structure–function studies have shown that two aromatic residues, Tyr and Phe, play a key role in the interaction with opioid receptors.2,3 This recognition is closely related to conformational preferences, which have been studied in detail, showing that the μ receptor requires the two rings to be on opposite sides of the peptide backbone, whereas δ receptors prefer aromatic residues on the same side.4 The structures of the enkephalins have been studied by X-ray crystallography, spectroscopic methods, and molecular modelling.5–9 These reports indicate that enkephalins are flexible and occur mainly in extended7 and folded conformations.6,8,9 A single turn stabilized by a β-turn located in the peptide region Gly2-Leu5 and a double turn in which the γ-turn is located on Gly2 and the β-turn is centred on Gly3-Phe4, depending on the biomimetic environment used, are prominent folded conformations. Enkephalins' flexibility makes finding their active conformation tough. Studies suggest that in opioid peptides, the residues in turn position not only restrict conformation but also directly interact with receptors.
The use of enkephalins in the treatment of pain is limited by their low metabolic stability and bioavailability, and their inability to cross the blood–brain barrier (BBB). Moreover, flexibility of enkephalins might cause side effects by binding to various receptors.10 The design of new analogs was aided by molecular modelling and receptor structure determination.11–13 Therefore, the synthesis of conformationally constrained leu-enkephalin analogs has been developed over many decades in the search for new peptidomimetics with improved stability and biological activity. Numerous analogs were obtained by cyclization,14 substitution of single amino acid residues with natural or unnatural amino acids,15 incorporation of cyclopropane-based scaffolds,16 introduction of linear and oligoheterocyclic motifs,17 introduction of amide bond isosters (ester, N-methylamide, triazole, alkene, trifluoroethylamine, azapeptide and fluoroalkene),18 introduction of sugar moieties,19 β-turn mimetic synthesis20 and retropeptide synthesis.21
Stapling has been used to cross-link the peptide chains between i, i + 4 and i, i + 7 residues, stabilizing the secondary, mostly helical structure.22 In our approach, two glycine residues, placed in positions i and i + 1, were replaced by N-(2-thioethyl)glycine derivatives resulting in the leu-enkephalin peptoid (1) which was further cyclized through reactive –SH groups. The analog 1, which was synthesized based on the modified protocol developed by our group,23 was subjected to a cyclization reaction using two different reagents: hexafluorobenzene (Scheme 1a) and the trithiocyanuric acid derivative (Scheme 1b). As reported previously, cyclization through SH groups resulted in promising cyclic opioid peptides – DPDPE ((D-Pen2,D-Pen5)-enkephalin) in which the Gly2 and Leu5 residues were replaced by the unnatural amino acid – Pen (penicillamine).14c Details of our synthesis are provided in the ESI† (S2.1 and S2.3). The cyclization reagents were chosen to investigate different geometries of the target product, since hexafluorobenzene prefers 1,4-substitution,24 while trithiocyanuric acid derivative prefers 1,3-substitution.25 Hexafluorobenzene stapling has so far been applied to –SH groups located in the side chain of amino acid residues at positions i and i + 4 of the peptide chain, resulting in a stable helical structure.24 The stapling strategy based on sulfhydryl groups catching with 2-alkylsulfanyl-1,3,5-triazin-4,6-diyl linkers has been applied for the model peptide by the method inspired by recently published metathesis reactions of tris(alkyl) thiocyanurates.25 These chemoselective reactions, occurring under mild basic conditions, are promising in designing agents for site-selective peptide or protein modification. Product 1 was characterized by LC-MS/MS and LC-UV. The LC-MS spectrum showed an expected signal at m/z 676.2865, corresponding to the molecular formula C32H45N5O7S2 (Fig. S3A–C and S4, ESI†), which was confirmed by MS/MS via peptide sequence fragment ions (Fig. S3D, ESI†). The hexafluorobenzene reaction adapted from Spokoyny et al.24 was modified by the introduction of a reducing reagent (TCEP) due to rapid intermolecular oxidation in the presence of TRIS, resulting in 65% yield of the product (Fig. S5, ESI†). Attempts at intramolecular disulphide bridge formation led to the formation of the dimeric product 1a from the linear precursor with –SH groups. The detailed analysis of the dimer structure (parallel and antiparallel) was not continued. Stapled analogue 2 was purified by HPLC. The homogeneity and identity of the product were confirmed by LC-MS/MS (Fig. S6, ESI†), LC-UV (Fig. S15, ESI†) and NMR (Fig. S34–S39, ESI†) and the LC-MS spectrum showed a strong signal at m/z 822.2703 corresponding to the desired product with molecular formula C38H43N5O7S2F4 (Fig. S7, ESI†). Tandem mass spectrometry confirmed cyclic product formation, with identified ions derived from the linear portion of the peptide chain (Fig. S5E, ESI†). The expected 1,4 substitution of hexafluorobenzen26 was confirmed by a single resonance registered at −73.3 ppm in the 19F NMR and consistent with a high symmetry (Fig. S39, ESI†). The cyclization towards 3a was achieved with 2 eq. of TMT(Acm)3 in 0.5 M TEAB H2O buffer and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of DMF/20 mM TCEP (see the ESI,† S2.1.5). The reaction is immediate and completes within 3 hours to give 3a quantitatively as documented in the LC-MS analysis. The reference 3b derivative showed a limited solubility causing a need for extended reaction time and a full conversion after 24 hours of incubation (40 °C). The identities of both stapled analogues (3a and 3b) were confirmed by ESI-MS and MS/MS analysis (Fig. S10–S13, ESI†). Products were purified chromatographically and the purity was confirmed by LC-UV analysis (Fig. S17 and S18, ESI†). CID fragmentation spectra of triazinyl analogs (3a and 3b, Fig. S13, ESI†) are more complicated than those of the 4FB counterpart (2, Fig. S5E, ESI†). The most intense signals correspond to fragments derived by cleavage of bonds in the linear part. However, we also identified ions created by direct fragmentation of the linker or β elimination in one of stapled N-(2-SEt)Gly residues followed by further fragmentation of the thiocyanurate moiety. The 1H NMR spectrum of 2 and 3a is complex due to presence of all four possible isomers (cis–trans isomerization of two tertiary amide bonds). In addition, DFT analysis of four possible isomers of analogs 2 and 3a showed slight energy differences, confirming that all forms are equally probable. 1H NMR comparative analysis of analogues 2 and 3a (Fig. S42 and S43, ESI†) in most informative regions (amide, aromatic, aliphatic – Leu) shows clearly the existence of 2–4 forms. Well documented isomerization of peptidyl-proline27 or peptoid28 amide bonds allows conclusion of isomerization in the case of acyl-N-(2-mercaptoethyl)glycine, which fit to the observed spectra. We observed an increase in the number of forms in the triazine-stapled analogue. Analysis of the Tyr residue's –OH group (9.4–9.3 ppm) shows signal splitting into two for analogue 2 and into four for analogue 3a (Fig. S43A, ESI†). The fluorobenzene-stapled peptide exhibits doubled para signals of tyrosine in the aromatic region (6.5–7 ppm), likely due to isomerization of the tyrosyl-N-[2-SEt]Gly bond (Fig. S43C, ESI†). In the triazine-stapled analogue, a more conformationally labile molecule is indicated by a complex multiplet at 6.7 ppm and partially separated doublets from 6.95–7.15 ppm (two in each range: 6.9–7 ppm and 7.05–7.15 ppm). Similar complexity is observed for the –CH3 groups of the leucine residue, identified as a doublet of doublets in Leu-enkephalin and a complex multiplet in stapled analogs. VT NMR analysis shows a temperature-dependent equilibrium in cis–trans isomerization of cyclic systems (Fig. S44–S46, ESI†).
:1 mixture of DMF/20 mM TCEP (see the ESI,† S2.1.5). The reaction is immediate and completes within 3 hours to give 3a quantitatively as documented in the LC-MS analysis. The reference 3b derivative showed a limited solubility causing a need for extended reaction time and a full conversion after 24 hours of incubation (40 °C). The identities of both stapled analogues (3a and 3b) were confirmed by ESI-MS and MS/MS analysis (Fig. S10–S13, ESI†). Products were purified chromatographically and the purity was confirmed by LC-UV analysis (Fig. S17 and S18, ESI†). CID fragmentation spectra of triazinyl analogs (3a and 3b, Fig. S13, ESI†) are more complicated than those of the 4FB counterpart (2, Fig. S5E, ESI†). The most intense signals correspond to fragments derived by cleavage of bonds in the linear part. However, we also identified ions created by direct fragmentation of the linker or β elimination in one of stapled N-(2-SEt)Gly residues followed by further fragmentation of the thiocyanurate moiety. The 1H NMR spectrum of 2 and 3a is complex due to presence of all four possible isomers (cis–trans isomerization of two tertiary amide bonds). In addition, DFT analysis of four possible isomers of analogs 2 and 3a showed slight energy differences, confirming that all forms are equally probable. 1H NMR comparative analysis of analogues 2 and 3a (Fig. S42 and S43, ESI†) in most informative regions (amide, aromatic, aliphatic – Leu) shows clearly the existence of 2–4 forms. Well documented isomerization of peptidyl-proline27 or peptoid28 amide bonds allows conclusion of isomerization in the case of acyl-N-(2-mercaptoethyl)glycine, which fit to the observed spectra. We observed an increase in the number of forms in the triazine-stapled analogue. Analysis of the Tyr residue's –OH group (9.4–9.3 ppm) shows signal splitting into two for analogue 2 and into four for analogue 3a (Fig. S43A, ESI†). The fluorobenzene-stapled peptide exhibits doubled para signals of tyrosine in the aromatic region (6.5–7 ppm), likely due to isomerization of the tyrosyl-N-[2-SEt]Gly bond (Fig. S43C, ESI†). In the triazine-stapled analogue, a more conformationally labile molecule is indicated by a complex multiplet at 6.7 ppm and partially separated doublets from 6.95–7.15 ppm (two in each range: 6.9–7 ppm and 7.05–7.15 ppm). Similar complexity is observed for the –CH3 groups of the leucine residue, identified as a doublet of doublets in Leu-enkephalin and a complex multiplet in stapled analogs. VT NMR analysis shows a temperature-dependent equilibrium in cis–trans isomerization of cyclic systems (Fig. S44–S46, ESI†).
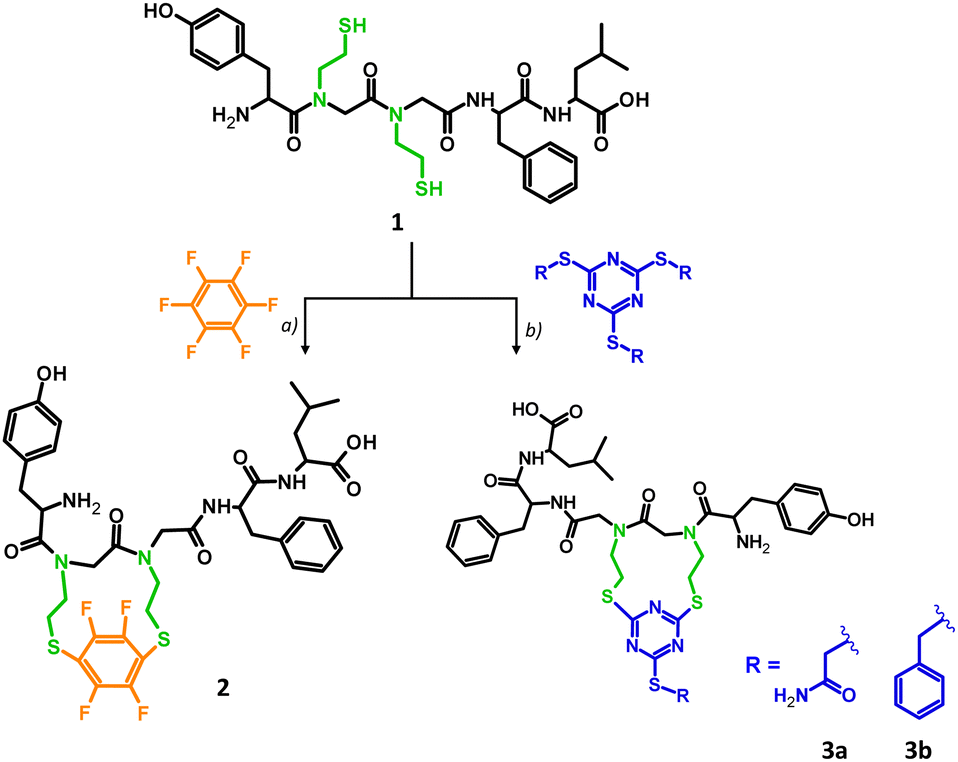 | ||
| Scheme 1 Synthesis scheme of the stapled analogs of leu-enkephalin (reaction conditions: (a) C6F6 (25 eq.), TCEP (5 eq.), 50 mM TRIS in DMF; (b) TMT(R)3 (2 eq.), TCEP (5 eq.), 0.5 M TEAB (pH = 8.5) in DMF). | ||
The CD conformational studies were performed for leu-enkephalin, stapled analogues (2, 3a, and 3b), and the side product 1a (dimer) in the far-UV region in three different solvents: MeCN, TFE, and H2O (Fig. S19–S23, ESI†). The aromatic side chains of Tyr1, Phe4 and bifunctional reagents used for stapling in the CD contributions obscure the peptide chromophore bands, making it hard to draw conformational conclusions. The CD spectra of 3a and 3b in TFE are quite similar to each other. They show negative bands at 250–260 nm, negative shoulders at 220–225 nm, and negative bands at 207 nm. The spectrum of 2 is somewhat similar to the two above ones below 220 nm, whereas at the longer wavelengths, there are only positive ellipticities, contrary to 3a and 3b. The CD spectra of all the analogues suggest that the peptides adopt the type I β-turn conformation in TFE. It was found that the CD spectrum typical of a type I β-turn is helical29 and follows two CD patterns – one with a negative band at 224 nm, a negative shoulder at 210 nm, and a positive band at 193 nm, and another with a negative shoulder at 224 nm, a negative band at 205 nm, and a positive band at 193 nm.30 The conclusion concerning a type I β-turn in the analogues studied is consistent with the results of the theoretical calculations (Table S10, ESI†). The Φ and Ψ angle values found for Gly2 and Gly3 residues of 2, 3a, and 3b in the low-energy conformers correspond to the standard values of the type I β-turn.31 This specific type of β-turn has been found for leu-enkephalin by the NMR studies in DMSO-d6.32 Moreover, also found in all the analogues were the intramolecular 4 → 1 hydrogen bonds, between N–H of Phe4 and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of Tyr1, stabilizing the β-turn conformation. The calculations showed also another conformational feature of 2, 3a, and 3b, namely the presence of a γ-turn on the Phe4 residue, stabilized by the 3 → 1 hydrogen bond, between Leu5 N–H and Gly3 CO. A γ-turn gives a positive CD band at 230 nm.33 Such a band can be seen in the spectrum of 2 but not in the spectra of 3a and 3b. In the latter two peptides, this band is probably overlapped by neighbouring negative ones. The CD spectra of 3b in MeCN and water are similar to those in TFE, except for the longwave negative band. It suggests that in these two solvents, the conformation of 3b is the same as in TFE (Fig. S23, ESI†).
O of Tyr1, stabilizing the β-turn conformation. The calculations showed also another conformational feature of 2, 3a, and 3b, namely the presence of a γ-turn on the Phe4 residue, stabilized by the 3 → 1 hydrogen bond, between Leu5 N–H and Gly3 CO. A γ-turn gives a positive CD band at 230 nm.33 Such a band can be seen in the spectrum of 2 but not in the spectra of 3a and 3b. In the latter two peptides, this band is probably overlapped by neighbouring negative ones. The CD spectra of 3b in MeCN and water are similar to those in TFE, except for the longwave negative band. It suggests that in these two solvents, the conformation of 3b is the same as in TFE (Fig. S23, ESI†).
In contrast, the spectra of 3a are more difficult to interpret in MeCN and water, suggesting that the peptide experiences greater conformational flexibility in these solvents. Yet another situation is observed in the case of 2. While one can suppose, on the basis of the CD spectra in the region of 200–220 nm, that the conformation of this analogue in somehow similar to that in TFE, in MeCN it seems to be distinctly different. The spectrum of 2 in MeCN in the far-UV region is more similar to the spectrum of leu-enkephalin in the same solvent rather than to spectra in TFE and water. As confirmed from the CD spectra, the presented solvent dependence of the tested analogues shows that the introduced bridge on the adjacent substituted glycine residues still leaves the conformational freedom of the entire system, but clearly affects the secondary structure. Computational methods have been used as a useful tool to predict the structure of the molecules, ligands and complexes.34 Molecular orbital studies on 2, 3a, 3b and Leu-Enk have been done at the DFT level of theory with the IEFPCM (integral equation formalism for polarizable continuum model) solvent (water) approach. The starting structure of the peptides for DFT calculations was generated on the basis of the amino acid sequence after 45 ps simulation at 300 K, without cutoffs using BIO+ implementation of the CHARMM force field. The results of the conformational studies correlate very well with the DFT analysis of the stapled analogues. At the DFT level of theory, we have found four thermodynamically stable conformations 2, 3a, 3b and Leu-Enk displaying terminus–terminus interaction with PA⋯PD distances 2.649 Å, 2.532 Å, 2.534 Å and 2.668 Å respectively (Fig. S47–S50, ESI†). Despite their limited size, these molecules form consistent intramolecular hydrogen bonds, as shown in Table S11 (ESI†). The structures of stapled 2, 3a and 3b are stabilized by hydrogen bonds between Tyr1 and Phe4 residues resulting in a 10-member ring with values 2.879 Å, 2.974 Å and 2.794 Å, respectively. An additional gamma-bend stabilized by hydrogen bonds (2.885 Å and 2.888 Å) between N-(2-SEt)Gly3 and Leu5 is observed in the triazine analogues (3a and 3b). The reference compound (Leu-Enk) displays a 7-member HB bonded ring formed between Tyr1 and Gly2 with 2.878 Å PA⋯PD distance as presented in Fig. S49 (ESI†). The distance between the most remote carbon atoms in the aromatic rings of 3b is ∼20 Å, while that in 3a is only ∼10 Å. The cartesian coordinates of presented molecules can be found in the ESI† (Table S12).
Biological tests involved two stapled analogues (2 and 3a) and unaltered leu-enkephalin as the reference compound. An analogue featuring a trithiocyanuric acid derivative was tested alongside another with an added benzyl system. Leu-enkephalin was synthesized following the standard Fmoc protocol; analytical data are provided in the ESI.† Antinociceptive activity of analogues 2 and 3b was assessed in Wistar rats via intrathecal administration (i.t.) using a tail flick test, and the results are shown in Fig. 1. All analogues displayed time-dependent analgesic effects at 16 nmol kg−1 i.t. dose, with distinct differences between 2 and 3b. Analog 2 peaked at 30 min, exhibiting a comparable analgesic effect to 3b (∼50% MPE) and leu-enkephalin. Its lowest efficacy (∼20% MPE) was observed at 120 min.
 | ||
| Fig. 1 Analgesic responses of analogs 2 and 3b as well as Leu-Enk (standard). Results were analyzed using two-way repeated measures of ANOVA followed by Tukey's multiple comparison test and plotted as the mean ± SEM (n = 7–8). Asterisks (*) indicate a significant difference *p < 0.03, **p < 0.001. | ||
A different profile of analgesic activity was shown by 3b. The lowest activity was at 5 min (20.72% MPE); at 30 min the effect was similar to that of 2, and the highest effect was at 120 min, which was higher than that of Leu-Enkephalin (20.72% MPE to 65.2% MPE). At 120 min post-administration, 2 exhibited weak activity where 3b reached the highest peak suggesting this compound may have longer duration of action than 120 min. Overall, the antinociceptive action of the tested analogues was relatively high, comparable to the standard leu-enkephalin. It should be noted that 3b has other analgesic profile response than 2 and leu-enkephalin. Its activity increased in the following minutes to reach its maximum at 120 min. The effect of the obtained derivatives (2 and 3b) is analogous to that of the action of leu-enkephalin, as demonstrated by analgesic tests. The prolonged action of the stapled analog can be explained by higher enzymatic stability for cyclic peptides. This observation requires further investigation because it may be important for the development of tolerance with chronic administration for this compound.
Biological studies confirmed the activity of the stapled analogs, suggesting that their rigid structure interacts effectively with the receptor. Identification of the bioactive conformer remains challenging due to isomerization induced by tertiary amide bonds observed by NMR analysis. Theoretical and CD analyses show structural similarities among the analogs, suggesting the importance of aromatic ring orientation. However, the determination of the privileged structural motif for effective receptor interaction remains elusive. The presence and orientation of the additional benzyl substituent likely enhance the prolonged analgesic effect of analog 3b.
MK thanks the “Excellence initiative – research university” for the years 2020–2026 for the University of Wrocław (BPI-DUB.4610.658.2021) for the financial support. The authors would like to thank Andrzej Reszka (Shim-Pol, Poland) for providing the Shimadzu LCMS-IT-TOF. The Wrocław Supercomputer Centre (KDM WCSS) is kindly acknowledged for sharing computation resources necessary for DFT calculations.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) P. W. Schiller, Prog. Med. Chem., 1991, 28, 301–340 CrossRef CAS PubMed; (b) J. E. Holden, Y. Jeong and J. M. Forrest, AACN Adv. Crit. Care, 2005, 16, 291–301 Search PubMed; (c) M. S. Henry, L. Gendron, M.-E. Tremblay and G. Drolet, Neural Plast., 2017, 1546125 Search PubMed; (d) I. I. Shcheniavsky, Probl. Cryobiol. Cryomed., 2021, 31, 003–013 CrossRef; (e) C. V. Borlongan, Y. Wang and T. P. Su, Front Biosci., 2004, 9, 3392–3398 CrossRef CAS PubMed.
- V. J. Hurby and C. A. Gerhring, Med. Res. Rev., 1989, 9, 343–401 CrossRef PubMed.
- (a) H. Kimura, C. H. Stammer, Y. Shimohigashi, C. Ren-Lin and J. Stewart, Biochem. Biophys. Res. Commun., 1983, 115, 112–115 CrossRef CAS PubMed; (b) Y. Shimohigashi, T. Costa, A. Pfeiffer, A. Herz, H. Kimura and C. H. Stammer, FEBS Lett., 1987, 222, 71–74 CrossRef CAS PubMed.
- D. Tourwé, K. Verschueren, A. Frycia, P. Davis, F. Porreca, V. J. Hruby, G. Toth, H. Jaspers, P. Verheyden and G. Van Binst, Biopolymers, 1996, 38, 1–12 CrossRef.
- D. Mastropalo, A. Camerman, L. Y. Y. Ma and N. Camerman, Life Sci., 1987, 40, 1995–1999 CrossRef PubMed.
- C. Garbay-Jaureguiberry, B. P. Roques, R. Oberlin, M. Anteunis, S. Combrisson and J. Y. Lallemand, FEBS Lett., 1977, 76, 93–98 CrossRef CAS PubMed.
- S. Rudolph-Bohner, D. Quarzago, M. Czisch, U. Ragnarsson and L. Moroder, Biopolymers, 1997, 41, 591–606 CrossRef CAS.
- D. Picone, A. D'Ursi, A. Motta, T. Tancredi and P. A. Temussi, Eur. J. Biochem., 1990, 192, 433–439 CrossRef CAS PubMed.
- A. Milon, T. Miyazawa and T. Higashijima, Biochemistry, 1990, 29, 65–75 CrossRef CAS PubMed.
- A. Janecka, J. Fichna and T. Janecki, Curr. Top. Med. Chem., 2004, 4, 1–17 CrossRef CAS PubMed.
- I. Efremenko and R. H. Fish, Organometallics, 2015, 34, 4117–4126 CrossRef CAS.
- H. S. Park, B. J. Byun and Y. K. Kang, ACS Omega, 2022, 7(31), 27755–27768 CrossRef CAS PubMed.
- F. Wieberneit, A. Korste, H. B. Albada, N. Metzler-Nolte and R. Stoll, Dalton Trans., 2013, 42, 9799 RSC.
- (a) G. Toth, T. H. Kramer, R. Knapp, G. Lui, P. Davis, T. F. Burks, H. I. Yamamura and V. J. Hruby, J. Med. Chem., 1990, 33, 249–253 CrossRef CAS PubMed; (b) M. H. Hannah, O. E. Shainnel, M. L. Ganno, J. C. Davis, C. T. Dooley, J. P. McLaughlina and A. Nefzi, Org. Biomol. Chem., 2019, 17, 5305–5315 RSC; (c) G. Toth, T. H. Kramer, R. Knapp, G. Lui, P. Davis, T. F. Burks, H. I. Yamamura and V. J. Hruby, J. Med. Chem., 1990, 33, 249–253 CrossRef CAS PubMed.
- (a) H. Xue, M. Guo, Ch Wang, Y. Shen, R. Qi, Y. Wu, Z. Xu and M. Chang, Org. Chem. Front., 2020, 7, 2426–2431 RSC; (b) S. Maricic, U. Berg and T. Frejd, Tetrahedron, 2002, 58, 3085–3093 CrossRef CAS; (c) Y. Sasaki, M. Hirabuki, A. Ambo, H. Ouchi and Y. Yamamoto, Bioorg. Med. Chem. Lett., 2001, 11, 327–329 CrossRef CAS PubMed; (d) H. Xue, M. Guo, Ch Wang, Y. Shen, R. Qi, Y. Wu, Z. Xu and M. Chang, Org. Chem. Front., 2020, 7, 2426–2431 RSC.
- A. Mizuno, K. Matsui and S. Shuto, Chem. – Eur. J., 2017, 23, 14394–14409 CrossRef CAS PubMed.
- S. Hammami, Z. Mighri, C. T. Dooley and A. Nefzi, Bioorg. Med. Chem. Lett., 2014, 24, 4482–4485 CrossRef CAS PubMed.
- (a) K. Rochon, A. Proteau-Gagne, P. Bourassa, J.-F. Nadon, J. Cote, V. Bournival, F. Gobeil, Jr., B. Guerin, Y. L. Dory and L. Gendron, ACS Chem. Neurosci., 2013, 4, 1204–1216 CrossRef CAS PubMed; (b) S. Sun, Q. Jia and Z. Zhang, Bioorg. Med. Chem. Lett., 2019, 29(18), 2535–2550 CrossRef CAS PubMed; (c) J.-L. Beaudeau, V. Blais, B. J. Holleran, A. Bergeron, G. Piñeyro, B. Guerin, L. Gendron and Y. L. Dory, ACS Chem. Neurosci., 2019, 10, 1615–1626 CrossRef CAS PubMed; (d) S. N. Karad, M. Pal, R. S. Crowley, T. E. Prisinzano and R. A. Altman, Chem. Med. Chem., 2017, 12, 571–576 CrossRef CAS PubMed; (e) R. Sinisi, A. Ghilardi, S. Ruiu, P. Lazzari, L. Malpezzi, M. Sani, L. Pani and M. Zanda, Chem. Med. Chem., 2009, 4, 1416–1420 CrossRef CAS PubMed; (f) V. Eeda, M. Selvaraju and R. A. Altman, J. Fluorine Chem., 2019, 218, 90–98 CrossRef CAS PubMed; (g) M. O. Bowles and C. Proulx, Org. Lett., 2022, 24, 1768–1773 CrossRef CAS PubMed.
- M. J. Slusarz, Comput. Biol. Chem., 2022, 101, 107783 CrossRef CAS PubMed.
- C. Xiong, J. Zhang, P. Davis, W. Wang, J. Ying, F. Porreca and V. J. Hruby, Chem. Commun., 2003, 1598–1599 RSC.
- N. Soumanou, D. Lybye, T. Hjelmgaard and S. Faure, Green Chem., 2023, 25, 3615–3623 RSC.
- (a) X. Li, S. Chen, W.-D. Zhang and H.-G. Hu, Chem. Rev., 2020, 120, 10079–10144 CrossRef CAS PubMed; (b) M. Kijewska, G. Wołczański, M. Światowska, K. Kędziora, M. Pawlicki and P. Stefanowicz, Chem. – Eur. J., 2023, 29, e202301370 CrossRef CAS PubMed; (c) M. Kijewska, A. Czerwińska, S. Al-Harthi, G. Wołczański, M. Waliczek, A.-H. Emwas, M. Jaremko, Ł. Jaremko, P. Stefanowicz and Z. Szewczuk, Chem. Commun., 2020, 56, 8814–8817 RSC.
- M. Wierzbicka, M. Waliczek, A. Dziadecka and P. Stefanowicz, J. Org. Chem., 2021, 86, 12292–12299 CrossRef CAS PubMed.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y.-S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135(16), 5946–5949 CrossRef CAS PubMed.
- G. Wołczański, W. Gil, J. Cichos, M. Lisowski and P. Stefanowicz, J. Org. Chem., 2023, 88, 8192–8202 CrossRef PubMed.
- P. Dognin, P. M. Killoran, G. S. Hanson, L. Halsall, T. Chaudhry, Z. Islam, F. Giuntini and Ch. R. Coxon, Peptide Sci., 2021, 113, e24182 CrossRef.
- U. Reimer, G. Scherer, M. Drewello, S. Kruber, M. Schutkowsk and G. Fischer, J. Mol. Biol., 1998, 279, 449–460 CrossRef CAS PubMed.
- D. Kalita, B. Sahariah, S. P. Mookerjee and B. K. Sarma, Chem. – Asian J., 2022, 17, e2022001 CrossRef PubMed.
- J. Bandekar, D. J. Evans, S. Krimm, S. J. Leach, S. Lee, J. R. McQuie, E. Minasian, G. Nemethy, M. S. Pottle, H. A. Scheraga, E. R. Stimson and R. W. Woody, Int. J. Peptide Protein Res., 1982, 19, 187–205 CrossRef CAS PubMed.
- A. Perczel and G. D. Fasman, Protein Sci., 1992, 1, 378–395 CrossRef CAS PubMed.
- M. Migliore, A. Bonvicini, V. Tognetti, L. Guilhaudis, M. Baaden, H. Oulyadi, L. Jouberta and I. Segalas-Milazzo, Phys. Chem. Chem. Phys., 2020, 22, 1611–1623 RSC.
- (a) C. Garbay-Jaureguiberry, B. P. Roques, R. Oberlin, M. Anteunis, S. Combrisson and J. Y. Lallemand, FEBS Lett., 1977, 76, 93–98 CrossRef CAS PubMed; (b) E. R. Stimson, Y. C. Meinwald and H. A. Scheraga, Biochemistry, 1979, 18, 1661–1671 CrossRef CAS PubMed.
- E. Vass, Z. Majer, K. Kőhalmy and M. Hollósi, Chirality, 2010, 22, 762–771 CrossRef CAS PubMed.
- (a) Z. Mielke, Z. Latajka, A. Olbert-Majkut and R. Wieczorek, J. Phys. Chem. A, 2000, 104, 3764–3769 CrossRef CAS; (b) R. Wieczorek, Z. Latajka and L. Lundell, J. Phys. Chem. A, 1999, 103, 6234–6239 CrossRef CAS; (c) T. K. Olszewski, E. Wojaczyńska, R. Wieczorek and J. Bąkowicz, Tetrahedron: Asymmetry, 2015, 26, 601–607 CrossRef CAS; (d) P. Salvador, R. Wieczorek and J. J. Dannenberg, J. Phys. Chem. B, 2007, 111, 2398–2403 CrossRef CAS PubMed; (e) M. Rudowska, R. Wieczorek, A. Kluczyk and P. Stefanowicz, JASMS, 2013, 24, 846–856 CAS; (f) E. Gumienna-Kontecka, G. Berthon, I. O. Fritsky, R. Wieczorek, Z. Latajka and H. Kozłowski, J. Chem. Soc., Dalton Trans., 2000, 4201–4208 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc06345c |
| ‡ M. K. and G. W. contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |