
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic non-oxidative conversion of methane
Qingyun
Zhan
ab,
Yuxiang
Kong
ab,
Xinhui
Wang
ab and
Lu
Li
 *ab
*ab
aCollege of Chemistry, Jilin University, Changchun 130012, People's Republic of China. E-mail: luli@jlu.edu.cn
bState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130012, People's Republic of China
First published on 6th February 2024
Abstract
The direct conversion of methane to hydrogen and high-value hydrocarbons under mild conditions is an ideal, carbon-neutral method for utilizing natural gas resources. Compared with traditional high-temperature thermal catalytic methods, using clean light energy to activate inert C–H bonds in methane can not only significantly reduce the reaction temperature and avoid catalyst deactivation, but also surpass the limitations of thermodynamic equilibrium and provide new reaction pathways. This paper provides a comprehensive review of developments in the field of photocatalytic non-oxidative conversion of methane (PNOCM), while also highlighting our contributions, particularly focusing on catalyst design, product selectivity, and the underlying photophysical and chemical mechanisms. The challenges and potential solutions are also evaluated. The goal of this feature article is to establish a foundational understanding and stimulate further research in this emerging area.
 Qingyun Zhan | Qingyun Zhan received his BS degree in inorganic chemistry from Jilin University in 2019. He is currently a PhD candidate under the direction of Prof. Lu Li in State Key Laboratory of Inorganic Synthesis and Preparative Chemistry at Jilin University. His main research focuses on photocatalytic non-oxidation conversion of methane at low-temperature. |
 Yuxiang Kong | Yuxiang Kong received his BS in Chemistry at Qilu University of Technology (2022). He is currently a MS candidate in State Key Laboratory of Inorganic Synthesis and Preparative Chemistry at Jilin University. His research focuses on photocatalytic methane conversion. |
 Xinhui Wang | Xinhui Wang received her BS in Chemistry at Beihua University in 2022. She is currently a MS candidate in State Key Laboratory of Inorganic Synthesis and Preparative Chemistry at Jilin University. Her research focuses on photocatalytic conversion of light alkanes. |
 Lu Li | Lu Li studied chemistry at Jilin University (BS, 2006) and received a PhD in inorganic chemistry with Prof. Jiesheng Chen (2012). After postdoctoral research with Prof. Chaojun Li at McGill University in Canada (2013–2017), he returned to Jilin University as a professor in State Key Laboratory of Inorganic Synthesis and Preparative Chemistry (2017). His research interests include light-driven activation of inert small molecules (such as CH4, N2, and alkanes) and low-carbon conversion technologies. |
1. Introduction
As petroleum resources dwindle, the quest for alternative chemical raw materials intensifies. Methane (CH4) emerges as a standout candidate due to its unique characteristics.1,2 Plentifully available in natural gas, biogas, shale gas, and flammable ice, methane boasts the highest hydrogen-to-carbon (H/C) ratio among hydrocarbons. This not only positions it as a crucial fuel source but also as an essential precursor for hydrogen and various hydrocarbon products. Therefore, its efficient conversion is a key goal in the natural gas industry.Methane utilization is classified into two types: direct and indirect conversions, as depicted in Fig. 1(A). Indirect conversion, a well-established method, involves transforming methane into syngas (CO and H2) at high temperatures.3–6 This syngas then acts as a precursor for various important processes such as ammonia synthesis via the Harber–Bosch method, hydrocarbon production through the Fischer–Tropsch process, and methanol and polyol production via the Oxo-process. However, this approach, entailing the complete cleavage of methane's C–H bonds, is energy-intensive and expensive. In contrast, direct conversion of methane into high-value chemicals, circumventing syngas, presents a more efficient utilization of natural gas. This includes both oxidative and nonoxidative processes. The oxidative process often leads to overoxidation, making it challenging to balance yield and selectivity. The non-oxidative conversion of methane (NOCM) is a zero-carbon-emission pathway that efficiently uses methane's carbon and hydrogen. Unfortunately, methane's high symmetry and strong C–H bond energy (435 kJ mol−1), coupled with low polarizability (2.84 × 10−40 C2 m2 J−1),7 make its activation difficult, especially for NOCM. As shown in Fig. 1(B), there are significant positive Gibbs free energy changes across a broad temperature range in such reactions, restricting high methane conversion even at elevated temperatures.8
 | ||
| Fig. 1 (A) Direct and indirect conversion of methane. (B) Gibbs free energy changes in various NOCM reactions. (C) The kinetic progress of NOCM in thermochemical and photochemical pathways. (D) Molecular orbital diagrams of methane. (E) Schematic diagram of the principle of PNOCM. | ||
The emerging photocatalytic non-oxidative conversion of methane (PNOCM) strategy presents a contrast to traditional thermal reactions by offering novel pathways for methane conversion, potentially lowering the activation energy barrier (Fig. 1(C)). The incorporation of photon energy plays a crucial role in activating inert methane molecules, reducing the Gibbs free energy of NOCM and transcending thermodynamic limitations for efficient low-temperature conversion. However, from the molecular orbital theory standpoint (Fig. 1(D)), the photoactivation of free methane molecules requires high-energy vacuum ultraviolet light. This poses significant demands on reaction equipment and energy efficiency. Hence, the development of effective photocatalysts is imperative for methane conversion under milder conditions. As shown in Fig. 1(E), when irradiated with appropriate wavelengths, photocatalysts absorb photons, exciting electrons to the conduction band and creating holes in the valence band, facilitating further reactions with methane on the surface.9,10 These advancements in photocatalyst design can enable conversions that are unfeasible under traditional conditions or reduce the temperatures required for catalytic reactions.
Our review presents a specialized exploration of PNOCM reactions, distinguishing itself from recent broader reviews by providing a more focused examination and in-depth analysis of recent advancements, challenges, and prospects in the field of PNOCM reactions. It delves into the potential reaction pathways and C–H bond activation mechanisms within PNOCM. Concluding the review, the paper presents an outlook on the challenges and opportunities in the development of advanced and efficient photocatalysts for future applications.
2. Photocatalysts for PNOCM
Fig. 2 provide the representative achievements in PNOCM over the past few decades. Following the first report by Yoshida et al. in 1998, there has been remarkable progress in diversifying and enhancing the performance of PNOCM systems. This evolution has broadened the product scope from ethane to a range of high-value chemicals, including aromatics, olefins, propane, butane, and acetic acid. The last five years, in particular, have witnessed a rapid surge in research. | ||
| Fig. 2 Timeline (1998–2024) of representative works on photocatalytic non-oxidative conversion of methane (PNOCM). The authors’ contributions to PNOCM are highlighted with an orange background. | ||
2.1 Quantum photocatalyst
As previously mentioned, methane's high symmetry and stability make direct photoionization (CH4 + hv → CH4+ + e−) require high-energy vacuum ultraviolet light, necessitating complex equipment and substantial electricity, which limits its practical application. To reduce the photon energy needed for methane activation, Yoshida et al. in 1998 employed a composite photocatalytic material of SiO2 and Al2O3. This facilitated the first non-oxidative coupling of methane under UV light exposure. At 310 K, the conversion rate of methane reached 5.9%, with ethane selectivity around 60%.11 Subsequent research identified the AlO4 structure as the active species.12Expanding on their pioneering research, Yoshida's team developed a variety of quantum photocatalysts for the non-oxidative dehydrogenation coupling of methane.13 These catalysts are characterized by their localized photoexcited electron–hole separation processes (Fig. 3(A)), leading to enhanced selectivity in methane conversion.14–16 They primarily consist of two types: (a) catalysts with highly dispersed active centers, such as TiO2, ZnO, ZrO2, MgO, and Ce3+, embedded in a SiO2 matrix; and (b) SiO2–Al2O3–TiO2 or SiO2–CeO2–TiO2 ternary oxide systems with dual-active sites. The latter, in particular, demonstrates a synergistic effect between the two active components, optimizing the photocatalytic process. Notably, ethane often emerges as the main product, with SiO2–TiO2 showing the highest photocatalytic yield (Fig. 3(B)).
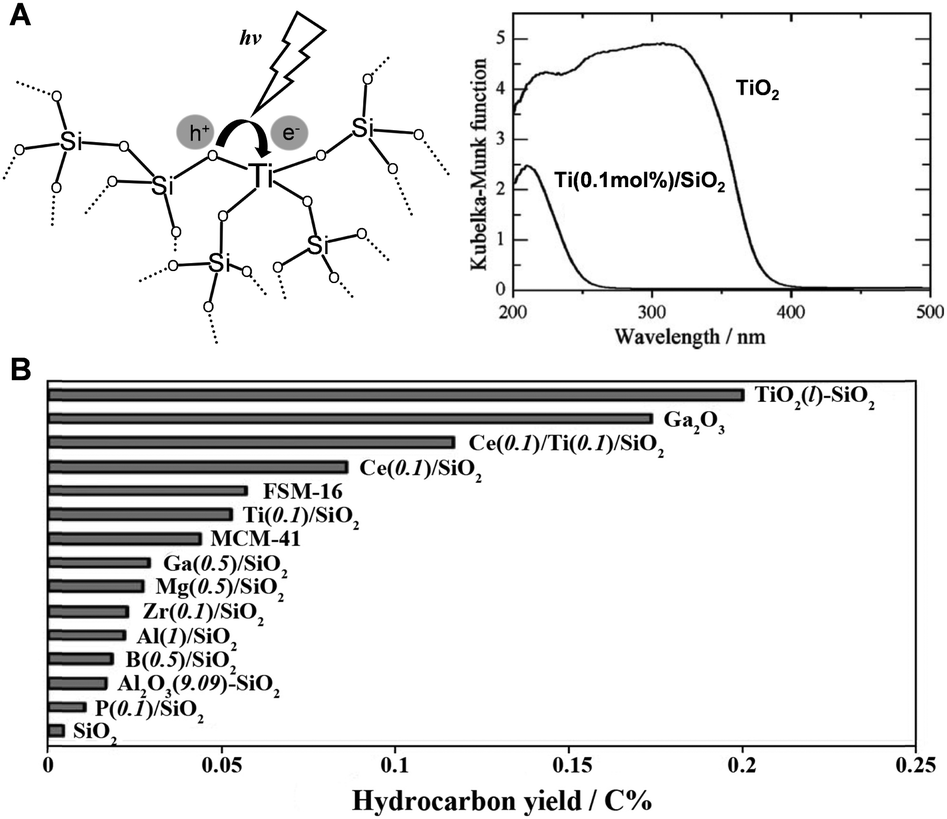 | ||
| Fig. 3 (A) Photoexcitation of highly dispersed Ti species in a silica matrix as a quantum photocatalyst and comparison of its light absorption properties with TiO2 semiconductor. (B) Activities of various quantum photocatalysts for PNOCM. Reproduced from ref. 13 with permission from The Royal Society of Chemistry. | ||
2.2 Zeolites and porous photocatalysts
Zeolites, renowned for their uniform pore structure, are widely utilized in the field of catalysis.17 Recently, zeolitic catalysts have shown promising potential in the realm of photocatalysis, where various modification strategies can enhance their photocatalytic activity.18,19 This section will discuss the use of zeolites and porous materials in photocatalytic non-oxidative coupling of methane.In the year 2002, the Yoshida research group pioneered the integration of zeolitic substrates into the investigation of PNOCM. The active sites within the H-type zeolites were verified as highly dispersed skeletal Al–O units.20 In addition to microporous zeolites, in the year 2003, the Yoshida group explored pure silica mesoporous molecular sieves such as MCM-41 and FSM-16, which displayed a certain level of reactivity towards PNOCM.21 However, these unmodified molecular sieves exhibited notably low levels of activity.
In 2011, a momentous breakthrough in the realm of zeolitic photocatalysis for PNOCM was attained by the research team led by Professor Jiesheng Chen.22 They successfully introduced a stable monovalent zinc ion (Zn+) species into ZSM-5 zeolite. The Zn+-modified ZSM-5 exhibited remarkable light-absorption capabilities and demonstrated exceptional photocatalytic efficiency in the conversion of methane. Under ambient temperature conditions and irradiation by solar light, methane underwent conversion into ethane and hydrogen, achieving a remarkable conversion exceeding 23%. This advancement marked a substantial elevation from the previous rate of 1.3 μmol h−1 g−1 to an impressive 9.8 μmol h−1 g−1. Due to the inherent selectivity conferred by zeolites, the reaction exhibited a remarkable degree of specificity towards ethane, approaching near-perfect selectivity of nearly 100%. As depicted in Fig. 4A, detailed mechanistic investigations unveiled a two-step photoexcitation process during the catalytic cycle. This process involved the transition of electrons from the molecular sieve framework to zinc ions and, subsequently, from zinc ions to the antibonding orbital of methane. In comparison to the single-step transition, the Zn+-modified ZSM-5 catalyst required remarkably reduced photon energy of only 3.2 eV to drive the methane conversion process, a substantial reduction from the previous system's demanding 4.6 eV requirement. The large reduction in the energy barrier enabled the utilization of sunlight for driving this reaction for the first time, opening up a novel avenue for harnessing solar energy more efficiently in the realm of methane conversion. It is worth noting that the electron transition from Zn+ ions to activate methane necessitates only low-energy visible light at 700 nm.
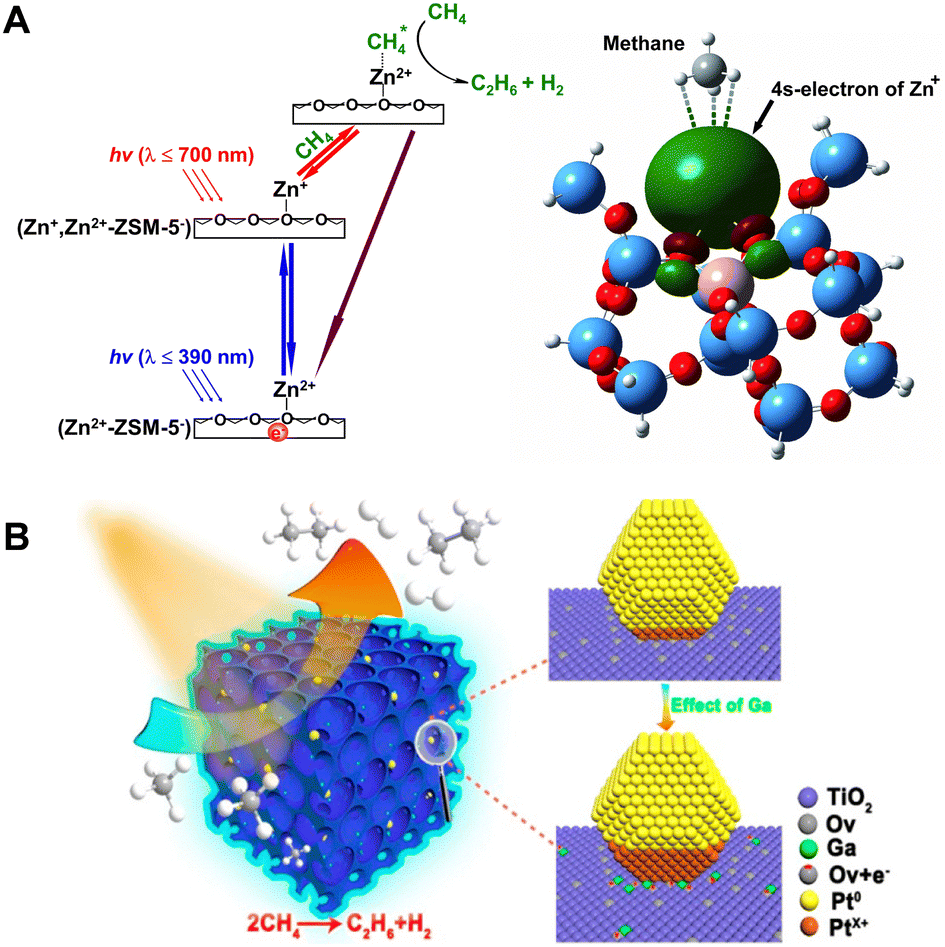 | ||
| Fig. 4 (A) Schematic energy diagram for the two-step photoexcitation processes over (Zn+,Zn2+)-ZSM-5 catalyst. Reproduced from ref. 22 with permission from John Wiley and Sons, copyright 2011. (B) Ga-doped and Pt-loaded porous TiO2–SiO2 for PNOCM. Reprinted with permission from ref. 24. Copyright 2019 American Chemical Society. | ||
The research teams led by Zhang Jinlong and Wang Lingzhi have conducted a series of works in this field.23–27 In 2022, they achieved a methane conversion rate of 4.10 μmol g−1 h−1 by using a Mo-oxide-modified SAPO-34 zeolite.23 They also developed a mesoporous TiO2–SiO2 catalyst doped with gallium and deposited with platinum (Fig. 4(B)).24 The incorporation of Ga aids in stabilizing oxygen vacancies, facilitating the formation of positively charged Pt. The presence of positively charged platinum enhances the activation of C–H bonds, while zero-valent platinum contributes to the transfer of photogenerated electrons. The synergistic interaction between the two types of Pt significantly improved the catalyst's performance in PNOCM. Moreover, they designed n-type and p-type doped hierarchical porous TiO2–SiO2 microarray catalysts. It was found that n-type doped catalysts generally exhibit higher photocatalytic activity. Taking niobium doping as an example, it not only enhances the efficiency of photogenerated charge and hole separation but also increases the density of local methyl radicals.25
2.3 Semiconductor photocatalysts
Semiconductors are the most commonly used catalysts in photocatalytic reactions. Theoretically, the photogenerated holes in the valence band of semiconductors possess strong oxidative capabilities. Selecting a semiconductor with a suitable band structure is pivotal for activating the C–H bonds. Specifically, to extract electrons from methane adsorbed on the catalyst surface, the valence band position must exceed +1.75 V relative to the standard hydrogen electrode. These can abstract electrons from methane molecules adsorbed on the catalyst surface at ambient temperatures, thereby initiating the oxidative cleavage of C–H bonds. This process results in the formation of methyl radicals and protonated hydrogen. The coupling of methyl radicals intermediates leads to the production of ethane, while the photogenerated electrons reduce the protonated hydrogen, releasing hydrogen gas. In practical applications, to enhance the efficiency of photogenerated electron–hole separation in photocatalysts, increase the adsorption polarization of methane on the catalyst surface, and inhibit excessive dehydrogenation of intermediates, semiconductors are often surface-modified, doped in the bulk phase, or integrated with noble metal cocatalysts. This section will introduce current semiconductor-based photocatalytic materials used for the NOCM reactions.The Yoshida research group pioneered the use of Ga2O3-based photocatalysts for PNOCM and demonstrated the continuous conversion of methane using Pd/Ga2O3 in a flow photocatalytic reactor.28–30 Unlike the stable Ga2O3, however, many semiconductor oxides like TiO2 and ZnO struggle to maintain their structural integrity during prolonged PNOCM reactions. The dissociated hydrogen atoms from methane tend to bind strongly with the semiconductor's lattice oxygen, releasing as water instead of hydrogen gas. This reaction leads to the deterioration of the semiconductor catalyst's structure and causes excessive oxidation of methane. To address this challenge, the Xiong group developed an atomic engineering strategy.31 They incorporated palladium single atoms (Pd1) into the surface lattice of TiO2 to from “palladium–oxygen” structural units (Pd–O4), as illustrated in Fig. 5(A). The Pd1/TiO2 material, with a high occupancy of Pd–O4 units at the valence band maximum (VBM), ensures that photogenerated holes primarily accumulate at these Pd–O4 units, not at the lattice oxygen (Fig. 5(B)). The greater the occupancy of metal Pd at the VBM, the more photogenerated holes are concentrated at the Pd sites. This is evidenced by the positive shifted binding energy in Pd 3d XPS upon light irradiation (Fig. 5(C)). This strategy facilitates methane activation and lattice oxygen stabilization, creating a synergistic effect that significantly enhances both the catalytic activity and stability. This groundbreaking approach offers a new perspective in the design of efficient semiconductor-based materials for PNOCM.
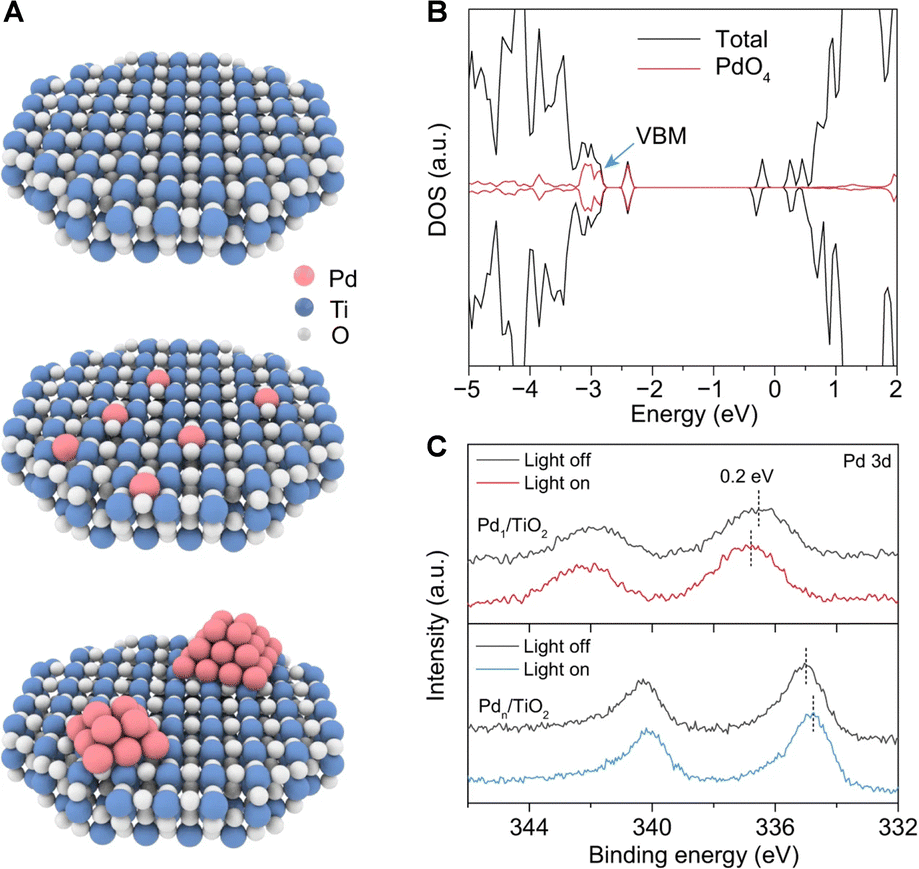 | ||
| Fig. 5 (A) Schematic illustration for TiO2(001)-based models—pristine TiO2 (top), Pd1/TiO2 (middle), and Pdn/TiO2 (bottom). (B) Total density of states (TDOS) of Pd1/TiO2. (C) Pd 3d XPS spectra of Pd1/TiO2 and Pdn/TiO2 under light irradiation. Reproduced with permission from ref. 31. Copyright 2022, Springer Nature. | ||
Noble metal nanoparticles, such as gold and silver, demonstrate significant light–matter interaction properties. The excitation of localized surface plasmon resonance (LSPR) occurs when nanostructured materials interacts with photons that match the resonance energy of the collective oscillation of the surface valence electrons.32 This process generates a potent electromagnetic field and dense energetic charge carriers (electron–hole pairs) on the nanostructures' surface.33–35 Utilizing these carriers for energy conversion and photocatalysis has recently emerged as a key research field.
The exploitation of plasmonic resonance in noble metal nanoparticles for energy conversion and photocatalysis has emerged as a hot research topic in recent years. The Long group loaded Au nanoparticles onto ZnO semiconductor nanosheets. By tuning the surface plasmon resonance (SPR) effect of the Au nanoparticles and the polarization effect of the ZnO(001) crystal face, they successfully converted methane molecules into ethane and hydrogen, achieving a photonic-to-chemical energy conversion efficiency of 0.08% (as depicted in Fig. 6(A)).36 Studies have found that under full-spectrum irradiation (320–800 nm), the conversion rate of methane was increased fivefold compared to using solely the ultraviolet (320–400 nm) or visible light (400–800 nm) spectra. This strongly indicates that the activation of methane was synergistically enhanced by the SPR of nano-sized Au particles and the polarization effect of ZnO. Furthermore, they prepared an Au–Pd/Bi2NbO5F(BNOF) bimetallic composite photocatalyst and proposed a tandem photocatalytic strategy based on Au and Pd nanoparticles.37 This strategy divides the PNOCM reaction into two tandem processes: CH4 first couples to C2H6 on Au, and then the produced C2H6 undergoes dehydrogenation to form C2H4 on Pd. The Hu group designed a continuous-flow photocatalytic system based on an Au/TiO2 catalyst, achieving a high C2H6 yield of 81.7 μmol gcatalyst−1 h−1 under weak light.38 They proposed a mechanism where methane is converted into methyl radicals through a methyl anion activation process, subsequently forming ethane. A similar mechanism is observed in the Au1–ZnO/TiO2 photocatalyst.39
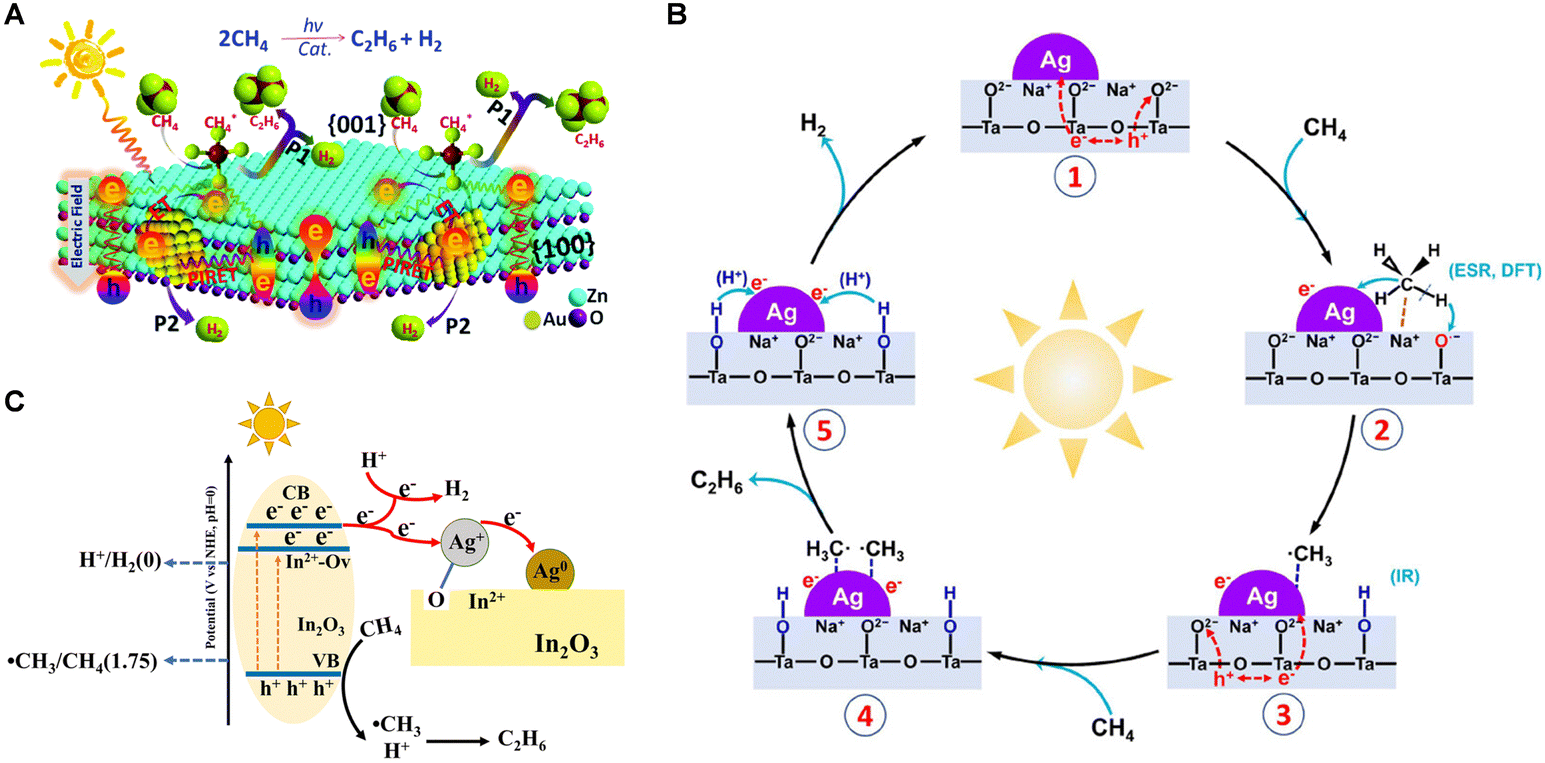 | ||
| Fig. 6 (A) Schematic description of Au modified ZnO nanosheets for PNOCM. Reproduced from ref. 36 with permission from The Royal Society of Chemistry. (B) Proposed photocatalytic mechanism of the NOCM reaction over Ag/NaTaO3. Reprinted with permission from ref. 40. Copyright 2023 American Chemical Society. (C) Schematic diagram of photoexcitation and reaction over Ag/In2O3. Reproduced from ref. 41 with permission from The Royal Society of Chemistry. | ||
In 2023, the Yang group designed an Ag-modified NaTaO3 photocatalyst, exhibiting high activity for PNOCM.40 The introduction of Ag nanoparticles significantly reduced the energy barrier for converting CH4 into ˙CH3 radicals. The Ag nanoparticles also exhibited strong affinity for adsorbing and concentrating the ˙CH3 intermediates, thereby facilitating their coupling to yield C2H6. As depicted in Fig. 6(B), the reaction mechanism initiates with CH4 adsorption at the Na sites of the Ag/NaTaO3 interface. Here, photogenerated holes on the NaTaO3 surface seize lattice oxygen to create O− species, which in turn react with a hydrogen atom from the adsorbed methane, forming OH groups. The resultant CH3 intermediates are then absorbed onto the Ag nanoparticle surface, later being released into the gas phase where they combine to produce C2H6. In a parallel development, Ag+ ions on defect-rich Ag/In2O3 catalysts function as electron acceptors, undergoing reduction to Ag0 during the reaction process.41 The presence of oxygen vacancies offers pivotal adsorption sites for O atoms from Ag2O, thus forging swift electron transfer channels and impeding the recombination of electrons with photogenerated holes, enhancing methane activation (Fig. 6(C)).
In recent years, our research group has developed a hexagonal wurtzite-structured GaN:ZnO solid solution as a photocatalyst for NOCM (illustrated in Fig. 7(A)).42 This catalyst, under room temperature illumination, is capable of converting methane molecules into stoichiometric amounts of ethane and hydrogen gas, maintaining stable catalytic activity over 70 hours and 35 cycles of testing. In situ EPR and IR spectroscopy reveal that the unique N–Zn–O structural units of the solid solution constitute its valence band, effectively capturing photogenerated holes under illumination. This process oxidizes the polarized methyl anion into a methyl free radical, which subsequently couples to form ethane. Building on this, we developed a strategy of constructing CeO2 nano-islands on the surface of a Ce–Zr solid solution (Fig. 7(B)).43 These ultra-small CeO2 nano-islands, characterized by their abundant stepped edges, exhibit unprecedented methane adsorption and activation capabilities. The highest conversion rate of methane in photocatalytic NOCM processes reaches 1517 μmol g−1 h−1. Density functional theory calculations suggest that the low-coordinated Ce–O units in the stepped CeO2(111) surfaces offer a more open planar configuration, facilitating the cleavage of C–H bonds in methane through an energetically favorable metal–CH4 σ-complex pathway rather than a radical-like pathway. Moreover, the photocatalysts display a reversible, synergistic photoactivation process, transitioning from a “resting state” to an “active state,” thus enhancing the efficiency of photogenerated charge separation and photocatalytic NOCM activity. Furthermore, the Shen Qianqian group designed single-crystal-like TiO2 nanotubes (VO-p-TNTs) to improve the separation and transport of photogenerated carriers, thereby enhancing the methane conversion efficiency.44
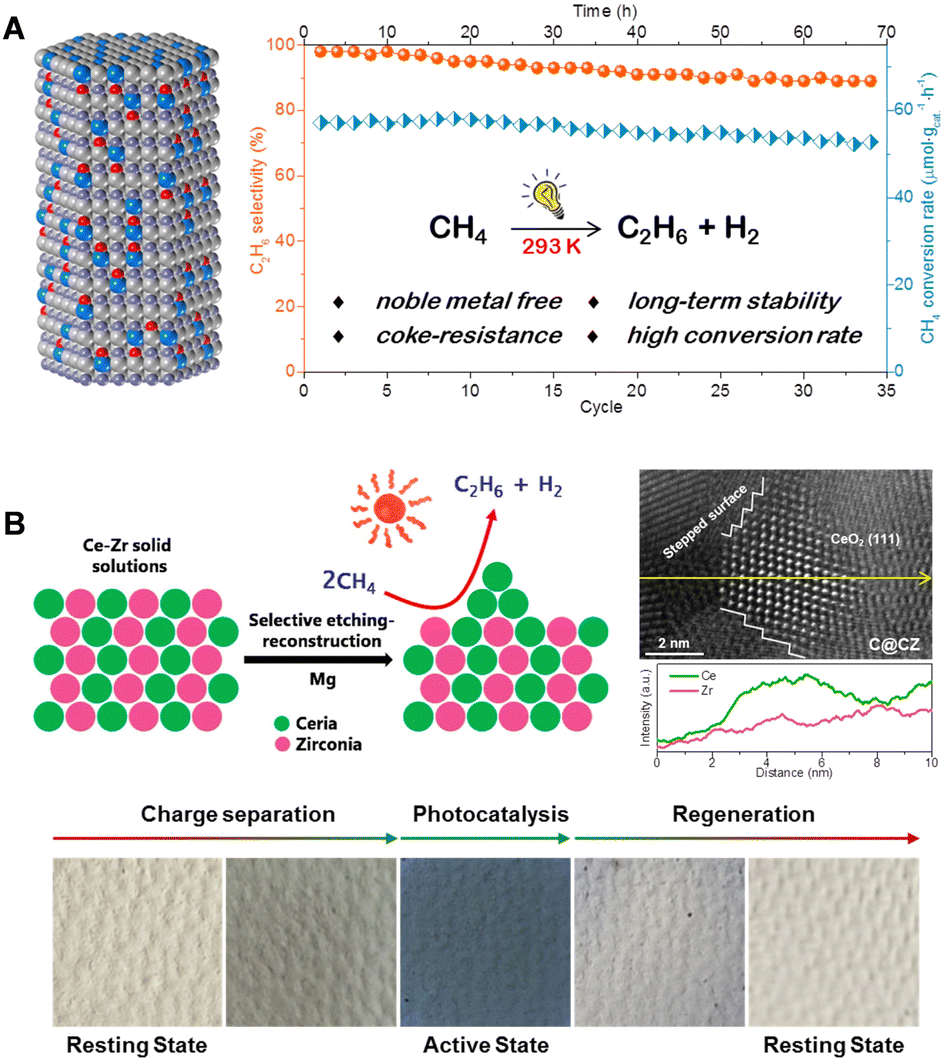 | ||
| Fig. 7 (A) Structure of GaN:ZnO solid solutions and the photocatalytic performance test of PNOCM. Reproduced from ref. 42 with permission from John Wiley and Sons, copyright 2021. (B) Preparation and characterization of CeO2 nano-islands catalysts and its photoinduced dynamic transition during PNOCM. Reproduced with permission from ref. 43. Copyright 2023 American Chemical Society. | ||
2.4 FLPs photocatalysts
American chemist Lewis, in 1923, introduced the classic concept of Lewis acids and bases, defining molecules or groups that accept electrons as Lewis acids, and those providing electrons as Lewis bases.45 In 2007, Professor Stephan proposed the concept of “Frustrated Lewis Pairs (FLPs),” denoting pairs of Lewis acids and bases that are prevented from interacting directly due to steric or other factors.46 Recently, solid FLPs, composed of spatially separated Lewis acid (LA) and Lewis base (LB) sites on solid surfaces, have been demonstrated to be effective in activating molecules like H2, CO2, and CO.47–49 In response to the challenge of polarizing the methane C–H bond, the Wang team utilized theoretical calculations to design a TiO2-based FLP with hierarchical pore structure, achieving superior C–H stretching with a high photocatalytic CH4 conversion rate (139 μmol g−1 h−1).50 Meanwhile, the Chang group explored “Single Atom”-“Frustrated Lewis Pair” (SA-FLP) dual active site catalysts through theoretical calculations, applying them in NOCM.51In a recent development, the Zhang research group employed a strategy involving thermal reduction-induced phase transition to fabricate densely packed Frustrated Lewis Pairs (FLPs) on layered Nb2O5 surfaces, rich in Nb-OH (LB sites) and low-valence Nb (LA sites).52 Initially, electrons localized near the exposed Nb sites are transferred to the Nb-OH groups under photoexcitation, thereby increasing the strength of LA and LB sites and forming a more favorable polarizing environment. Moreover, FeNi-LDO nanosheets have been developed, constructing synergistic catalytic sites of bimetallic and acid–base pairs, enhancing methane activation capability.53 The concept of Lewis acid–base pairs has shed light on the methodology of crafting a polarizing environment conducive for photocatalytic C–H activation in methane. This approach has provided an in-depth analysis of the enhancement of active sites under photoexcitation, offering fresh insights into the structural design of potent photocatalysts for the transformation of methane.
3. Product scope expansion
The PNOCM reactions described above primarily follow a radical mechanism, resulting in the production of ethane and hydrogen. In efforts to expand the applications of PNOCM and to enhance the diversity and economic value of its products, researchers have made significant strides in recent years. The team from McGill University, employing molecular beam epitaxy (MBE) technology, successfully designed and grew silicon-doped n-type GaN semiconductor nanowires, as depicted in Fig. 8(A).54 This wurtzite III–V nitride can catalyze the conversion of methane into benzene and hydrogen efficiently under ultraviolet light irradiation at room temperature. A key discovery was that a potent local electrostatic field on the m-plane surface of gallium nitride effectively polarizes methane, forming methyl radicals. These radicals then couple to produce ethane, which subsequently undergoes dehydrogenation to yield benzene. This pioneering work, for the first time, demonstrates the creation of a methane photocatalytic reaction system using semiconductor nitrides, successfully addressing the challenges of poor stability in traditional oxide semiconductors and the limited product scopes in PNOCM. Most recently, the Xiong group has engineered a PdO/Pd-WO3 heterointerface nanocomposite material, capable of directly converting CH4 into CH3COOH without the need for an additional carbon source.55 In the presence of ˙OH, methane undergoes activation at the Pd and PdO sites, leading to the formation of methyl radicals and carbonyl intermediates, respectively. These intermediates then converge at the Pd/PdO heterointerface, culminating in the synthesis of acetic acid, as illustrated by the reaction mechanism in Fig. 8(B). | ||
| Fig. 8 (A) Schematic diagram for methane C–H bond polarization on the surface of GaN m-plane. Adapted with permission from ref. 54. Copyright 2014 American Chemical Society. (B) Schematic illustration for photochemical conversion of CH4 to CH3COOH over Pd/PdO heterointerface in the presence of ˙OH radicals. Reproduced with permission from ref. 55. Copyright 2023, Springer Nature. | ||
In recent years, Pt-based catalysts have been increasingly employed in alkane dehydrogenation reactions. Research indicates that sub-nanometer Pt metal clusters display a remarkable dependency on size and structure in catalyzing alkane dehydrogenation reactions. Vajda and colleagues found that Pt8–Pt10 sub-nanometer clusters possess high catalytic activity for the oxidative dehydrogenation of propane.56 Alexandrova and her colleagues found that Pt7 clusters are more effective in catalyzing ethylene dehydrogenation than Pt4 or Pt8.57 Furthermore, Ma and co-researchers reported that clusters consisting of a few Pt atoms, with an average Pt–Pt coordination number (CNPt–Pt) of approximately 2–3, exhibit the best catalytic performance.58 The Wang research group has conducted systematic studies on the size effect of Pt particles with diameters (x) ranging from 1.5 to 2.7 nm on x-Pt/Ga2O3 for PNOCM.59
Our research group has developed a “single-atom Pt collection” modified black TiO2 (Pt@BT-O) using a “reduction–oxidation–reconstruction” strategy, as depicted in Fig. 9(A).60 The surface of this black TiO2 is densely populated with sub-nanometer Pt collections, each averaging 0.6–0.8 nm in diameter and comprising a few adjacent Pt monomers, yet without forming direct Pt–Pt bonds (Fig. 9(B)). Pt@BT-O demonstrates exceptional photocatalytic activity in methane conversion, marking a pioneering instance of utilizing pure visible light to drive PNOCM. The performance of this catalyst underscores the significant impact of the Pt species' state on its activity and selectivity (Fig. 9(C)). For the single-atom 0.04Pt@BT-O catalyst, a CH4 conversion rate of 4.9% was achieved. However, due to the isolated nature of the individual Pt active sites, there is a tendency to break all four C–H bonds in methane, resulting in hydrogen and carbon production. Conversely, 0.2Pt@BT-O, containing collections of Pt monomers, yielded a higher CH4 conversion rate of 8.2% with minimal carbon deposition. Intriguingly, in contrast to typical methane dehydrogenation coupling reactions that produce ethane, our catalyst primarily yielded propane (∼65%) and butane (12%). This unique selectivity for propane conversion from methane is unprecedented. Through comprehensive mechanistic studies, we propose a novel pathway involving light-driven intramolecular dehydrogenation of methane to form a methylcarbene intermediate (Fig. 9(D)). This novel approach transcends the constraints of traditional methyl radical mechanisms, offering a new platform for methane molecule functionalization via an array of carbene-type reactions.61
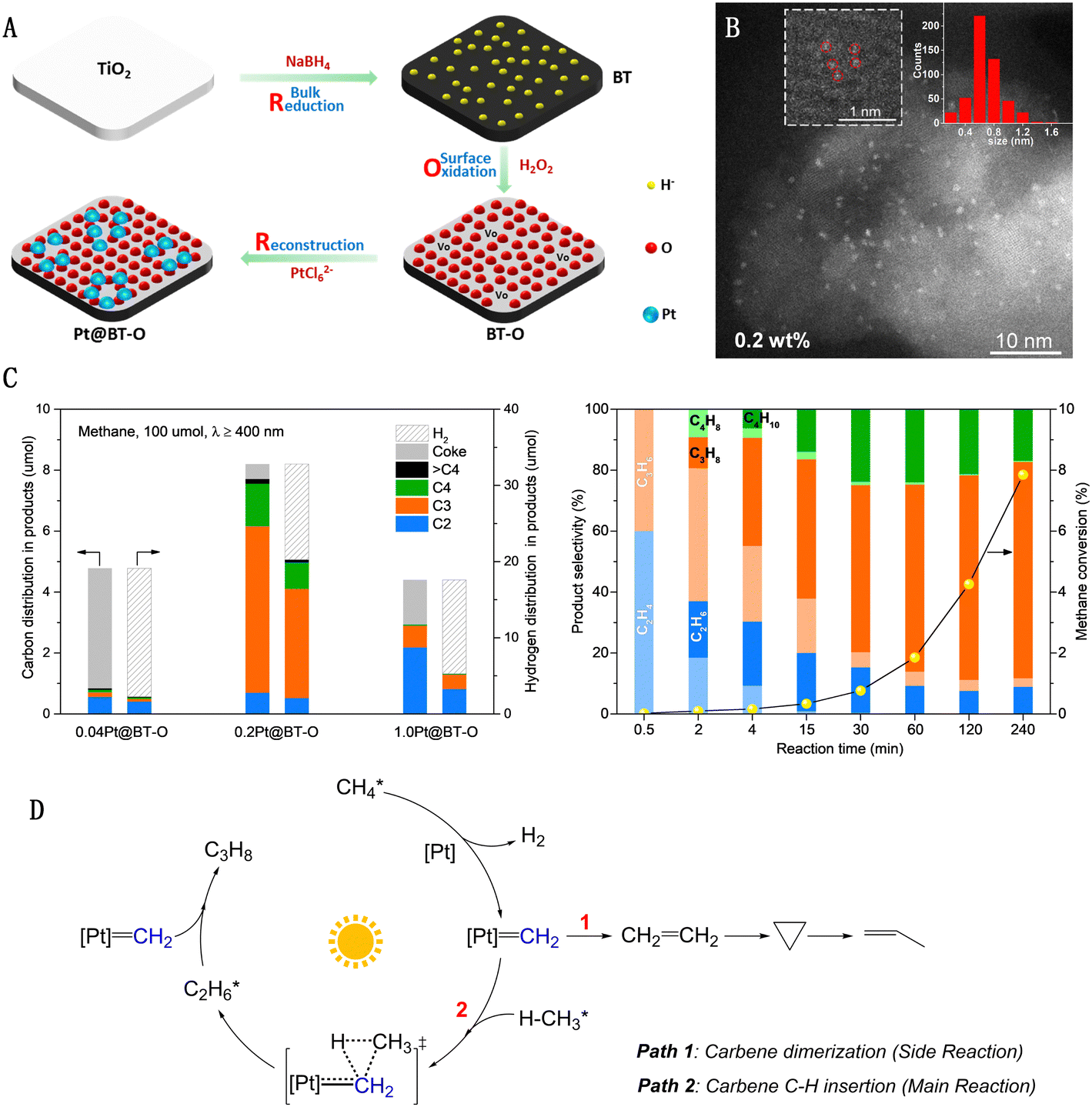 | ||
| Fig. 9 (A) Reduction–oxidation–reconstruction synthetic procedure of single atom Pt collections on black TiO2. (B) AC-HAADF-STEM images of Pt@BT-O. (C) Photocatalytic dehydrogenation of methane. (D) Schematic illustration for proposed mechanism. Reproduced with permission from ref. 60. Copyright 2022, Springer Nature. | ||
4. Chemical looping
At the end of the 19th century, Quentin and Arthur Brin pioneered an air separation technique, thereby introducing the concept of chemical looping for the first time.62–64 This innovative approach to chemical reactions involves reconfiguring reaction pathways into multiple sub-reactions that occur in distinct spatial or temporal stages. In contemporary industrial catalysis, the application of chemical looping technology has achieved remarkable results. This process utilizes the lattice oxygen present in metal oxides in a reduction bed to oxidize reactants. Following this, in an oxidation bed, the lattice oxygen within the reduced metal oxides is replenished, thereby completing the cyclic process. Recently, the Gong group published their work on tandem propane dehydrogenation (PDH). They have adeptly coupled PDH with chemical looping selective hydrogen combustion (CL-SHC) using a multifunctional FeVO4-VOx tandem reaction system.65 Leveraging the unique features of chemical looping technology, researchers have now extended this methodology to photocatalytic methane conversion. In an initial, non-oxidative environment, selective C2+ products are synthesized, aided by lattice oxygen, which effectively curbs over-oxidation. The catalyst is then regenerated in an oxygen-rich setting, ensuring the stability and continuity of the cycle.66The Chen research team reported a chemical looping strategy for PNOCM, involving the modification of ETS-10 titanium silicate zeolite with a series of metal cations through ion exchange.67 As shown in Fig. 10(A), the valence band electrons of semiconductor nano titanium chains are photoexcited to the conduction band, leading to the separation of photogenerated electrons and holes. The photogenerated electrons are captured by Ti4+ to form Ti3+, while the photogenerated holes oxidize surface hydroxyl groups into hydroxyl radicals. These highly reactive hydroxyl radicals abstract a hydrogen atom from methane to form water, and the resultant methyl radicals couple to form ethane. During the reaction, Ti-OH groups are consumed, and the photocatalyst can be rejuvenated by heat treatment in the presence of oxygen and water, restoring its photoactivity.
 | ||
| Fig. 10 (A) Reaction mechanism over Ga-ETS-10. Reproduced from ref. 67 with permission from John Wiley and Sons, copyright 2012. (B) Schematic illustration of photochemical looping process on Ag-HPW/TiO2. Reproduced with permission from ref. 68. Copyright 2020, Springer Nature. (C) Schematic illustration for photocatalytic conversion of CH4 to C2H4 over ZnO–AuPd2.7% hybrid catalyst. Reprinted with permission from ref. 71. Copyright 2019 American Chemical Society. | ||
The Khodakov team has developed a photochemical looping strategy for converting methane into ethane with high selectivity at room temperature, using a silver-heteropolyacid-titanium dioxide nanocomposite material (Ag-HPW/TiO2).68 During the photocatalytic process, a noticeable color transition in Ag-HPW/TiO2 from light gray to dark black is observed, with the rate of ethane formation starting to diminish after 3 hours. This color change signifies the reduction of silver ions to metallic silver under light, allowing the catalyst to be regenerated and maintain its methane conversion activity for up to 10 cycles, when exposed to light in air. The mechanism of this photochemical loop is depicted in Fig. 10(B). It involves two separate processes: methane conversion and catalyst regeneration. Ag+ ions are reduced to elemental silver by photogenerated electrons, while methane is activated by photogenerated holes, leading to its conversion into ethane, propane, and carbon dioxide. The metallic silver can revert back to silver ions upon exposure to ultraviolet light in air. The Ag+/Ag0 redox cycle plays a crucial role in controlling the degree of methane oxidation, thereby preventing over-oxidation. The Sun research team utilized the interaction between loaded Au nanoparticles and the ZnO carrier to induce the co-activation of C–H bonds by Auδ− and O−.69 The oxygen vacancies created during the reaction can be restored through the Mars–van Krevelen mechanism, namely by regenerating the catalyst through heating in air.
The Xiong research team integrated Pd–Zn active sites onto defect-rich WO3 nanosheets (Pd5/Zn0.35-WO3), synergistically converting CH4 into C2H4.70 Lattice oxygen plays a crucial role as an oxidant in this process, contributing to the formation of peroxide products. The lattice oxygen depleted during the reaction can be restored via photolytic oxidation in air. Furthermore, this team has developed a Pd-modified ZnO–Au hybrid catalyst (ZnO–AuPd2.7%), which demonstrates the capability of converting methane into C2+ hydrocarbons, achieving an ethylene selectivity of 39.7%.71 The mechanism of converting methane to ethylene, illustrated in Fig. 10(C), begins with methane being adsorbed and dissociated into methoxy and hydroxyl groups on the ZnO(100) surface, aided by Pd. The strong interaction between Pd and C leads to the methoxy group further dehydrogenating into a –CH2O species. This species then couples with another methyl radical, generated from methane activation, to form an ethoxy group. Subsequently, this ethoxy group undergoes further dehydrogenation and detaches from the Zn–AuPd2.7% surface, yielding C2H4. After 4 hours, the rate of C2+ hydrocarbon formation significantly decreases due to the consumption of lattice oxygen in ZnO, which can be replenished by the regeneration treatment. Similarly, the Wang research team have successfully decoupled the formation processes of ethane and hydrogen in the NOCM reaction. They developed an innovative process that merges photocatalytic methane coupling for the production of ethane with electrochemical hydrogen generation. By employing a uniquely coordinated Fe3+, they achieved high-selectivity methane conversion, while the resultant Fe2+ facilitated low-energy electrochemical hydrogen evolution, realizing solar-driven Fe3+/Fe2+ cycling and CH4 conversion to produce C2H6 and H2.72 In 2024, the Xiong group synthesized Au supported on Nb3O7(OH) with abundant surface lattice hydroxyl groups. They demonstrated that these surface lattice hydroxyl groups play a crucial role in promoting the activation of CH4 to form methoxy intermediates, significantly enhancing the catalytic performance of CH4 conversion.73
5. Summary and outlook
Contrary to the methane reforming process, characterized by high emissions and low atomic efficiency, the non-oxidative dehydrogenation of methane offers 100% utilization of the carbon and hydrogen atoms in methane. This approach enables the one-step synthesis of valuable hydrocarbons and hydrogen fuel with “zero carbon” emissions. Nonetheless, this process faces significant thermodynamic challenges, impeding its progression at ambient conditions. Advances over the past two decades have demonstrated that the introduction of light as a “green reagent” can surmount the limitations imposed by thermodynamic equilibrium, thus facilitating the catalysis of methane's non-oxidative dehydrogenation at ambient conditions. This innovation not only diminishes energy expenditures but also addresses the problem of catalyst deactivation at elevated temperatures and broadens the product scope. This review thoroughly examines the advancements in the field of photocatalytic non-oxidative conversion of methane. It encompasses the synthesis of catalytic materials, the assessment of methane conversion efficacy, and the elucidation of underlying mechanisms, all aimed at augmenting the fundamental comprehension of the photocatalytic activation and transformation of methane.The primary obstacle in advancing photocatalytic methane conversion technology is its low efficiency. To address this issue, efforts can be concentrated on four dimensions:
(1) Maximizing solar energy harnessing: the majority of PNOCM systems documented to date operate under ultraviolet light, thereby failing to harness the full potential of the solar spectrum, particularly the visible and near-infrared regions. Potential solutions include the development of narrow-band gap semiconductors or metallic photocatalysts responsive to the visible to infrared light range. This can be augmented by pairing them with plasmonic nano-metal co-catalysts and further integrating these catalysts with effective photothermal materials. In this context, semiconductors and metal co-catalysts are tailored to absorb and harness ultraviolet and visible light. Concurrently, black photothermal materials utilize the remainder of the visible and infrared spectrum for heating the catalyst, thereby maximizing the exploitation of the entire solar spectrum.
(2) Advancing C–H bond photoactivation: the non-oxidative dehydrogenation of methane necessitates direct cleavage of the inert sp3 C–H bond in methane. To enhance understanding and refine the design of photocatalysts crucial for C–H bond activation, extensive research is indispensable. An ideal photocatalyst would achieve two critical functions simultaneously: firstly, it should enhance the adsorption and polarization of methane on its surface, and secondly, it must establish a highly efficient channel for the rapid transfer of photoexcited electrons and holes from the bulk phase of the catalyst through the surface-active sites, ultimately reaching the adsorbed methane molecules. This dual functionality is essential for optimizing the catalytic process and maximizing the efficiency of methane conversion. These developments can be further supported by employing time-resolved spectroscopy and dynamic operando characterization techniques, along with density functional theory calculations, to elucidate the mechanisms and potential pathways for light-induced methane C–H bond activation.
(3) Refining photocatalytic system: the majority of PNOCM systems are currently operated in batch reactors, where the synergistic interaction between gaseous methane, solid catalysts, and photonic energy is not maximized. A significant challenge involves the development of an efficient and safe continuous flow photoreactor system that promotes optimal energy and material exchange among these components. Another concern is the limited stability of catalysts. Except for a few semiconductor catalysts such as Ga2O3, GaN, and GaN:ZnO solid solutions, most metal oxide semiconductors are susceptible to reduction under PNOCM conditions, often only enduring a limited number of cycles in durability tests. To enhance the scalability and real-world applicability of these photocatalytic processes, it is imperative to significantly extend the lifetime of catalysts, aiming for uninterrupted operation of over a month. This objective necessitates a thorough investigation into catalyst surface coking, photodegradation, and regenerative capabilities under prolonged operational conditions.
(4) Expanding product diversity: predominantly, reactions in PNOCM proceed through a coupling route, typically yielding ethane and hydrogen via methyl radical intermediates. The relatively modest commercial value of ethane necessitates its further conversion to render it industrially significant. Although there have been occasional successes in selectively producing higher-value compounds like benzene or olefins, the catalysts required for these processes often demand elaborate preparation methods, such as molecular beam epitaxy, and the use of expensive co-catalysts, including noble metals, which complicates their scalability. Emerging research suggests that exploring avenues like tandem photocatalytic strategies, chemical looping techniques, and carbene pathways could offer avenues to broaden the spectrum of products derived from methane's non-oxidative dehydrogenation.
In conclusion, with the rapid development of natural gas resources and growing interest in clean new energy sources, it is crucial to advance research in photocatalytically driven methane conversion under mild conditions. This is not only vital for exploring new methods and theories in the activation of inert C–H bonds but also in broadening the utilization of solar energy. Such research holds significant fundamental value and, if breakthroughs are achieved, undoubtedly possesses substantial economic and societal benefits.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was financially supported by the National Natural Science Foundation (NSFC) of China (Grant No. 22379050, 92061105), National Key Research and Development Program of China (2023YFA1506300), the Natural Science Foundation of Jilin Province (20210101121JC), and the Fundamental Research Funds for the Central Universities.Notes and references
- V. R. Choudhary, A. K. Kinage and T. V. Choudhary, Science, 1997, 275, 1286–1288 CrossRef CAS PubMed.
- A. Holmen, Catal. Today, 2009, 142, 2–8 CrossRef CAS.
- G. Jones, J. G. Jakobsen, S. S. Shim, J. Kleis, M. P. Andersson, J. Rossmeisl, F. Abild-Pedersen, T. Bligaard, S. Helveg, B. Hinnemann, J. R. Rostrup-Nielsen, I. Chorkendorff, J. Sehested and J. K. Nørskov, J. Catal., 2008, 259, 147–160 CrossRef CAS.
- D. Pakhare, D. Pakhare, J. Spivey and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- S. Arora, S. Arora, R. Prasad and R. Prasad, RSC Adv., 2016, 6, 108668 RSC.
- J. P. V. Hook, Catal. Rev.: Sci. Eng., 1980, 21, 1–51 CrossRef.
- R. D. Amos, Mol. Phys., 1979, 38, 33–45 CrossRef CAS.
- X. Li, C. Wang and J. Tang, Nat. Rev. Mater., 2022, 7, 617–632 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Y. Jr., Chem. Rev., 2002, 95, 735–758 CrossRef.
- A. J. Bard, Science, 1980, 207, 139–144 CrossRef CAS PubMed.
- Y. Kato, H. Yoshida and T. Hattori, Chem. Commun., 1998, 2389–2390 RSC.
- H. Yoshida, N. Matsushita, Y. Kato and T. Hattori, Phys. Chem. Chem. Phys., 2002, 4, 2459–2465 RSC.
- L. Yuliati and H. Yoshida, Chem. Soc. Rev., 2008, 37, 1592–1602 RSC.
- H. Yoshida, N. Matsushita, Y. Kato and T. Hattori, J. Phys. Chem. B, 2003, 107, 8355–8362 CrossRef CAS.
- L. Yuliati, T. Hattori and H. Yoshida, Phys. Chem. Chem. Phys., 2005, 7, 195–201 RSC.
- L. Yuliati, H. Itoh and H. Yoshida, Chem. Lett., 2006, 35, 932–933 CrossRef CAS.
- J. í Čejka, A. Corma and S. I. Zones, Zeolites and Catalysis: Synthesis, Reactions and Applications, John Wiley & Sons Inc, 2010 Search PubMed.
- S. Murcia-López, M. C. Bacariza, K. Villa, J. M. Lopes, C. Henriques, J. R. Morante and T. Andreu, ACS Catal., 2017, 7, 2878–2885 CrossRef.
- F. Sastre, V. Fornés, A. Corma and H. García, J. Am. Chem. Soc., 2011, 133, 17257–17261 CrossRef CAS PubMed.
- Y. Kato, H. Yoshida, A. Satsuma and T. Hattori, Microporous Mesoporous Mater., 2002, 51, 223–231 CrossRef CAS.
- L. Yuliati, M. Tsubota, A. Satsuma, H. Itoh and H. Yoshida, J. Catal., 2006, 238, 214–220 CrossRef CAS.
- L. Li, G.-D. Li, C. Yan, X.-Y. Mu, X.-L. Pan, X.-X. Zou, K.-X. Wang and J.-S. Chen, Angew. Chem., Int. Ed., 2011, 50, 8299–8303 CrossRef CAS PubMed.
- L. Pan, S. Wu, Z. Huang, S. Zhang, L. Wang and J. Zhang, Catal. Sci. Technol., 2022, 12, 3322–3327 RSC.
- S. Wu, X. Tan, J. Lei, H. Chen, L. Wang and J. Zhang, J. Am. Chem. Soc., 2019, 141, 6592–6600 CrossRef CAS PubMed.
- Z. Chen, S. Wu, J. Ma, S. Mine, T. Toyao, M. Matsuoka, L. Wang and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 11901–11909 CrossRef CAS PubMed.
- X. Feng, K. Kang, Y. Wu, J. Zhang and L. Wang, Chem. Commun., 2021, 57, 13000–13003 RSC.
- J. Ma, X. Zhang, Q. Zhang, K. Kang, J. Zhang and L. Wang, Res. Chem. Intermed., 2022, 48, 3247–3258 CrossRef CAS.
- L. Yuliati, T. Hattori, H. Itoh and H. Yoshida, J. Catal., 2008, 257, 396–402 CrossRef CAS.
- S. P. Singh, A. Anzai, S. Kawaharasaki, A. Yamamoto and H. Yoshida, Catal. Today, 2021, 375, 264–272 CrossRef CAS.
- S. P. Singh, A. Yamamoto, E. Fudo, A. Tanaka, H. Kominami and H. Yoshida, ACS Catal., 2021, 11, 13768–13781 CrossRef CAS.
- W. Zhang, C. Fu, J. Low, D. Duan, J. Ma, W. Jiang, Y. Chen, H. Liu, Z. Qi, R. Long, Y. Yao, X. Li, H. Zhang, Z. Liu, J. Yang, Z. Zou and Y. Xiong, Nat. Commun., 2022, 13, 2806 CrossRef CAS PubMed.
- L.-W. Chou, N. Shin, S. V. Sivaram and M. A. Filler, J. Am. Chem. Soc., 2012, 134, 16155–16158 CrossRef CAS PubMed.
- Q. Xiao, E. Jaatinen and H. Zhu, Chem. – Asian J., 2014, 9, 3046–3064 CrossRef CAS PubMed.
- H. Zhang and A. O. Govorov, J. Phys. Chem. C, 2014, 118, 7606–7614 CrossRef CAS.
- M. J. Kale, T. Avanesian and P. Christopher, ACS Catal., 2013, 4, 116–128 CrossRef.
- L. Meng, Z. Chen, Z. Ma, S. He, Y. Hou, H.-H. Li, R. Yuan, X.-H. Huang, X. Wang, X. Wang and J. Long, Energy Environ. Sci., 2018, 11, 294–298 RSC.
- C. Tang, S. Du, H. Huang, S. Tan, J. Zhao, H. Zhang, W. Ni, X. Yue, Z. Ding, Z. Zhang, R. Yuan, W. Dai, X. Fu, M. B. J. Roeffaers and J. Long, ACS Catal., 2023, 13, 6683–6689 CrossRef CAS.
- J. Lang, Y. Ma, X. Wu, Y. Jiang and Y. H. Hu, Green Chem., 2020, 22, 4669–4675 RSC.
- N. Zhang, Y. Fu, W. Kong, B. Pan, C. Yuan, S. Li, H. Zhu and J. Zhang, J. Environ. Chem. Eng., 2023, 11, 110665 CrossRef CAS.
- J. Zhang, J. Shen, D. Li, J. Long, X. Gao, W. Feng, S. Zhang, Z. Zhang, X. Wang and W. Yang, ACS Catal., 2023, 13, 2094–2105 CrossRef CAS.
- M. Xiao, L. Wang, H. Wang, J. Yuan, X. Chen, Z. Zhang, X. Fu and W. Dai, Catal. Sci. Technol., 2023, 13, 4148–4155 RSC.
- G. Wang, X. Mu, J. Li, Q. Zhan, Y. Qian, X. Mu and L. Li, Angew. Chem., Int. Ed., 2021, 60, 20760–20764 CrossRef CAS PubMed.
- G. Wang, X. Mu, R. Tan, Z. Pan, J. Li, Q. Zhan, R. Fu, S. Song and L. Li, ACS Catal., 2023, 13, 11666–11674 CrossRef CAS.
- J. Xue, J. Li, Z. Sun, H. Li, H. Chang, X. Liu, H. Jia, Q. Li and Q. Shen, J. Adv. Ceram., 2023, 12, 1577–1592 CrossRef CAS.
- G. N. Lewis, Valence And The Structure Of Atoms And Molecules, American Chemical Society Monograph Series, 1923 Search PubMed.
- J. S. J. McCahill, G. C. Welch and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 4968–4971 CrossRef CAS PubMed.
- D. W. Stephan, Science, 2016, 354 DOI:10.1126/science.aaf7229.
- X. Wang, L. Lu, B. Wang, Z. Xu, Z. Xin, S. Yan, Z. Geng and Z. Zou, Adv. Funct. Mater., 2018, 28, 1804191 CrossRef.
- X. Zheng, I. Zulkifly, A. Heilmann, C. McManus and S. Aldridge, Angew. Chem., Int. Ed., 2021, 133, 16552–16555 CrossRef.
- J. Ma, Q. Zhang, Z. Chen, K. Kang, L. Pan, S. Wu, C. Chen, Z. Wu, J. Zhang and L. Wang, Chem. Catal., 2022, 2, 1775–1792 CrossRef CAS.
- T. Ban, X.-Y. Yu, H.-Z. Kang, Z.-Q. Huang, J. Li and C.-R. Chang, ACS Catal., 2023, 13, 1299–1309 CrossRef CAS.
- Z. Chen, Y. Ye, X. Feng, Y. Wang, X. Han, Y. Zhu, S. Wu, S. Wang, W. Yang, L. Wang and J. Zhang, Nat. Commun., 2023, 14, 2000 CrossRef CAS PubMed.
- L. Pan, J. Zhang and L. Wang, Res. Chem. Intermed., 2022, 48, 2903–2913 CrossRef CAS.
- L. Li, S. Fan, X. Mu, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2014, 136, 7793–7796 CrossRef CAS PubMed.
- W. Zhang, D. Xi, Y. Chen, A. Chen, Y. Jiang, H. Liu, Z. Zhou, H. Zhang, Z. Liu, R. Long and Y. Xiong, Nat. Commun., 2023, 14, 3047 CrossRef CAS PubMed.
- S. Vajda, M. J. Pellin, J. P. Greeley, C. L. Marshall, L. A. Curtiss, G. A. Ballentine, J. W. Elam, S. Catillon-Mucherie, P. C. Redfern, F. Mehmood and P. Zapol, Nat. Mater., 2009, 8, 213–216 CrossRef CAS PubMed.
- M.-A. Ha, E. T. Baxter, A. C. Cass, S. L. Anderson and A. N. Alexandrova, J. Am. Chem. Soc., 2017, 139, 11568–11575 CrossRef CAS PubMed.
- Y. Deng, Y. Guo, Z. Jia, J.-C. Liu, J. Guo, X. Cai, C. Dong, M. Wang, C. Li, J. Diao, Z. Jiang, J. Xie, N. Wang, H. Xiao, B. Xu, H. Zhang, H. Liu, J. Li and D. Ma, J. Am. Chem. Soc., 2022, 144, 3535–3542 CrossRef CAS PubMed.
- J. Ma, X. Tan, Q. Zhang, Y. Wang, J. Zhang and L. Wang, ACS Catal., 2021, 11, 3352–3360 CrossRef CAS.
- L. Zhang, L. Liu, Z. Pan, R. Zhang, Z. Gao, G. Wang, K. Huang, X. Mu, F. Bai, Y. Wang, W. Zhang, Z. Cui and L. Li, Nat. Energy, 2022, 7, 1042–1051 CrossRef CAS.
- X. Mu and L. Li, Nat. Energy, 2022, 7, 1011–1012 CrossRef.
- L.-S. Fan, Chemical Looping Systems for Fossil Energy Conversions, John Wiley & Sons Inc., 2010 Search PubMed.
- L. Zeng, Z. Cheng, J. A. Fan, L.-S. Fan and J. Gong, Nat. Rev. Chem., 2018, 2, 349–364 CrossRef CAS.
- T. C. Hepworth, Nature, 1892, 47, 176–177 CrossRef.
- W. Wang, S. Chen, C. Pei, R. Luo, J. Sun, H. Song, G. Sun, X. Wang, Z.-J. Zhao and J. Gong, Science, 2023, 381, 886–890 CrossRef CAS PubMed.
- Y. Jiang, S. Li, X. Fan and Z. Tang, Nano Res., 2023, 16, 12558–12571 CrossRef CAS.
- L. Li, Y.-Y. Cai, G.-D. Li, X.-Y. Mu, K.-X. Wang and J.-S. Chen, Angew. Chem., Int. Ed., 2012, 51, 4702–4706 CrossRef CAS PubMed.
- X. Yu, V. L. Zholobenko, S. Moldovan, D. Hu, D. Wu, V. V. Ordomsky and A. Y. Khodakov, Nat. Energy, 2020, 5, 511–519 CrossRef CAS.
- K. Zheng, X. Zhang, J. Hu, C. Xu, J. Zhu, J. Li, M. Wu, S. Zhu, L. Li, S. Wang, Y. Lv, X. He, M. Zuo, C. Liu, Y. Pan, J. Zhu, Y. Sun and Y. Xie, Sci. China: Chem., 2023 DOI:10.1007/s11426-023-1792-8.
- Y. Liu, Y. Chen, W. Jiang, T. Kong, P. H. C. Camargo, C. Gao and Y. Xiong, Research, 2022, 2022 DOI:10.34133/2022/9831340.
- W. Jiang, J. Low, K. Mao, D. Duan, S. Chen, W. Liu, C.-W. Pao, J. Ma, S. Sang, C. Shu, X. Zhan, Z. Qi, H. Zhang, Z. Liu, X. Wu, R. Long, L. Song and Y. Xiong, J. Am. Chem. Soc., 2021, 143, 269–278 CrossRef CAS PubMed.
- H. Zhang, W. Zhong, Q. Gong, P. Sun, X. Fei, X. Wu, S. Xu, Q. Zhang, G. Fu, S. Xie and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202303405 CrossRef CAS PubMed.
- Z. Ma, Y. Chen, C. Gao and Y. Xiong, Chem. Commun., 2024, 60, 1132–1135 RSC.
| This journal is © The Royal Society of Chemistry 2024 |