
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDifunctionalization of gem-difluoroalkenes for amination and heteroarylation via metal-free photocatalysis†
Yuanchen
Zhong‡
ab,
Zhen
Zhuang‡
bc,
Xiaofei
Zhang
 b,
Bin
Xu
*a and
Chunhao
Yang
*bc
b,
Bin
Xu
*a and
Chunhao
Yang
*bc
aDepartment of Chemistry, Shanghai University, Shanghai 200444, China. E-mail: xubin@shu.edu.cn
bState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai, 201203, China. E-mail: chyang@simm.ac.cn
cSchool of Pharmacy, University of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing, 100049, China
First published on 15th April 2024
Abstract
gem-Difluoroalkenes are widely used building blocks in fluorine chemistry. Herein, a metal-free photocatalytic amination and heteroarylation method of gem-difluoroalkenes with heteroaryl carboxylic acid oxime esters as substrates is reported. This environmentally benign reaction proceeds via radical–radical cross-coupling in energy-transfer-mediated photocatalysis and can be used in the rapid construction of heteroaryl difluoroethylamine scaffolds and late-stage modification of complex pharmaceutical structures.
Fluorine-containing compounds have attracted wide attention in the fields of materials science and pharmaceutical chemistry due to their unique physical and chemical properties.1–3 Among them, difluoromethyl units are favored in drug development due to the significant improvement in their metabolic properties such as oral bioavailability and long plasma half-lives.4 In particular, the (hetero)aryl difluoroethylamine structures show great potential value for multiple drug targets. For example, researchers from Merck & Co. developed thrombin inhibitor (DP4088) I with a difluoroethylamine scaffold, which has a good oral bioavailability based on metabolism-directed optimization.4 Moreover, antifungal agent II,5 nitric oxide synthase inhibitor III6 and Nav 1.7 blocker IV7 all contain this bioactive moiety (Fig. 1).
 | ||
| Fig. 1 Bioactive molecules containing (hetero)aryl difluoroethylamine structures. | ||
gem-Difluoroalkenes are very easily obtainable building blocks for synthesizing several fluorine-containing compounds and have sparked interest in recent years.8–10 Recent research on functionalization of gem-difluoroalkenes has mainly focused on the construction of C–X (X = OR, SR, etc.) or C(sp3)–C(sp3) bonds, involving the introduction of some functional groups such as alkyl,11–13 alkoxyl,14,15 alkylthiol,16,17 sulfonyl18 and other groups (Scheme 1a).9,10,19,20 However, there are only a few reports on the introduction of aromatic groups to gem-difluoroalkenes, and previous reports focused on transitional-metal catalyzed coupling reactions (Scheme 1a).21,22 A gentle and environmentally friendly method for the difunctionalization of gem-difluoroalkenes would thus be a great complement to the construction of heteroaryl difluoroethylamine structures.
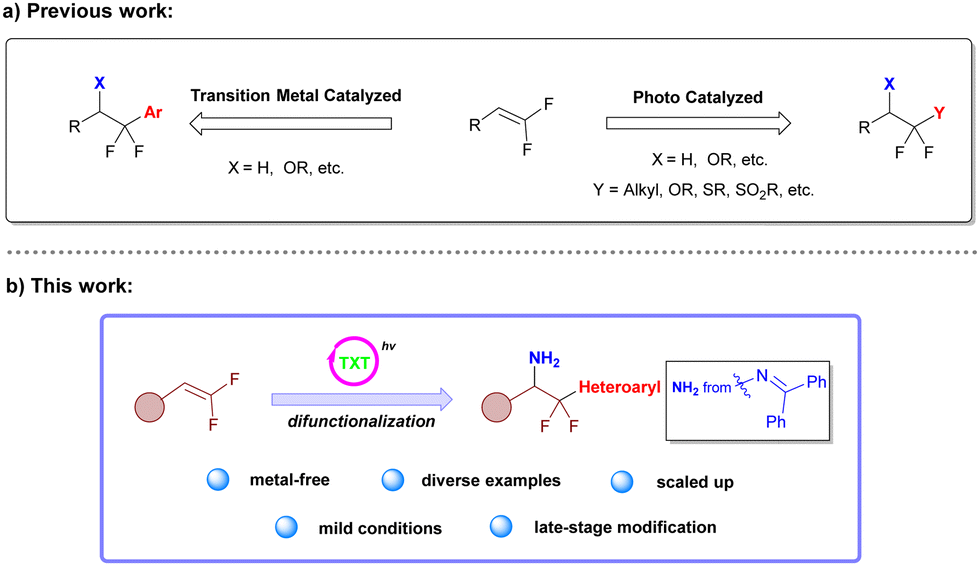 | ||
| Scheme 1 Functionalization of gem-difluoroalkenes and our reaction design. | ||
Since 2019, oxime derivatives have been widely used in various reactions, such as decarboxylative functionalization of aryl and alkyl carboxylic acids,23 decarboxylative imination,24,25 decarboxylative fluorosulfonylation of aryl and alkyl carboxylic acids26,27 and dual functionalization of various structures.28–34 These reactions involve the generation of N- and O-centered radicals through energy-transfer (EnT) in the presence of suitable photocatalysts with high triplet excited state energy and appropriate light sources. Based on the previous work from our group on functionalization of gem-difluoroalkenes and heteroaryl carboxylic oxime esters,12,27 we attempted to utilize readily available oxime derivatives to provide imino and heteroaryl radicals for amination and heteroarylation of gem-difluoroalkenes simultaneously (Scheme 1b).
To confirm our hypothesis, we explored the optimal conditions for the reaction with the substrate models of gem-difluoroalkene 1a and oxime ester 2a (for more details, see the ESI†). Initially, we screened common photocatalysts under a 405 nm wavelength LED and determined thioxanthone (TXT) as the optimal catalyst for the reaction with a yield of 46% (Table 1, entries 1–4). Subsequent solvent screening revealed that DMSO as a solvent was more favorable for the reaction, increasing the yield from 46% to 62% (Table 1, entries 4–7). We further explored the influence of different wavelength LEDs on the reaction and found a slight improvement in yield under 365 nm irradiation (Table 1, entries 7–9). Considering the occurrence of imino radical self-coupling during the reaction process, increasing the ratio of oxime ester ultimately resulted in a yield of 71% (Table 1, entry 10). The control experiment performed at the end demonstrated the necessity of a light source, a photocatalyst, and an inert atmosphere for the reaction (Table 1, entries 11–13).
|
|
||||
|---|---|---|---|---|
| Entrya | Photocatalystb | Solvent | Wavelength | Yieldc (%) |
| a Reaction conditions: 1a (0.15 mmol, 1.0 equiv.), 2a (0.15 mmol, 1.0 equiv.), photocatalyst (5 mol%), 30 W LED, argon atmosphere, at room temperature in solvent (0.75 mL, 0.2 M) for 12 h. b PC-1: [Ir(dF(CF3)ppy)2(dtbbpy)](PF6); PC-2: fac-Ir(dFppy)3; PC-3: 4CzlPN; TXT: thioxanthone. c Isolated yield. d 2a (0.45 mmol, 3 equiv.). e Under the air atmosphere. f In the dark. | ||||
| 1 | PC-1 | EtOAc | 405 | 26 |
| 2 | PC-2 | EtOAc | 405 | 20 |
| 3 | PC-3 | EtOAc | 405 | 30 |
| 4 | TXT | EtOAc | 405 | 46 |
| 5 | TXT | MeCN | 405 | 40 |
| 6 | TXT | DCE | 405 | 23 |
| 7 | TXT | DMSO | 405 | 62 |
| 8 | TXT | DMSO | 440 | Trace |
| 9 | TXT | DMSO | 365 | 65 |
| 10d | TXT | DMSO | 365 | 71 |
| 11 | — | DMSO | 365 | Trace |
| 12e | TXT | DMSO | 365 | Trace |
| 13f | TXT | DMSO | — | n.d. |
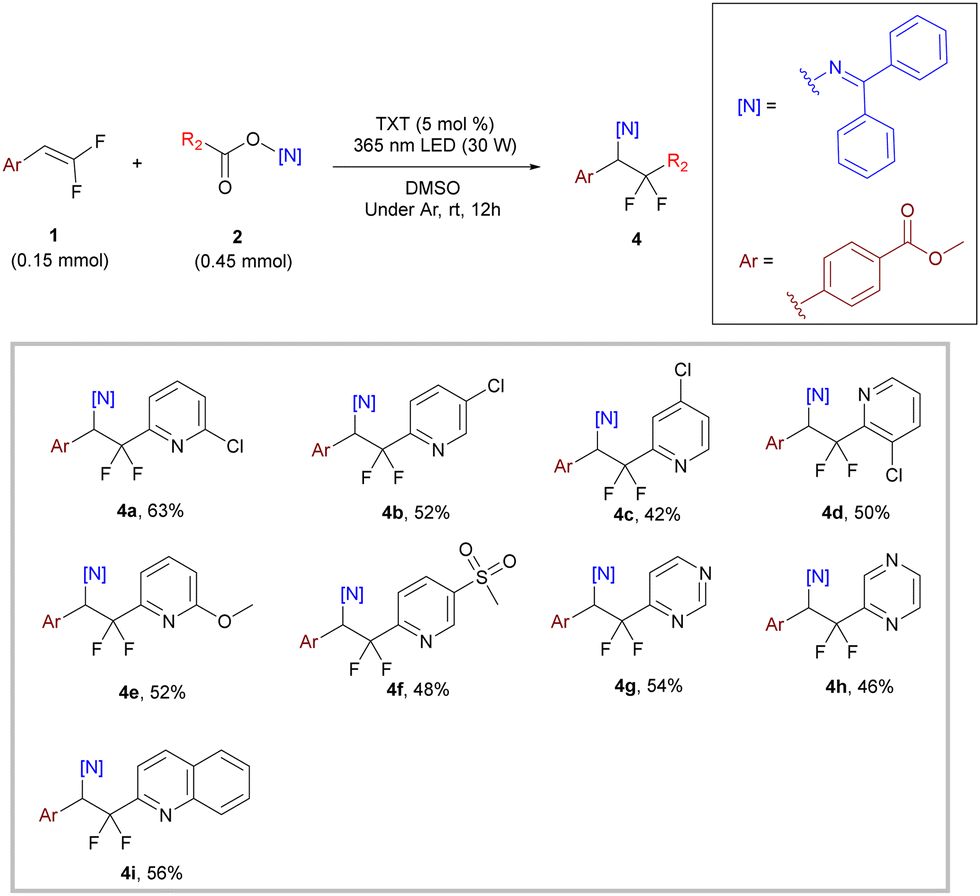
After determining the optimal reaction conditions, we explored the generality of this difunctionalization reaction using gem-difluoroalkenes with different substituents on the benzene ring. As shown in Scheme 2, within the tested scope, both substrates with electron-withdrawing and electron-donating substituents reacted smoothly to afford the target compounds in moderate to good yields (3a–3j, 56–76%), and changing the substitution position also gave good yield for meta-substitution (3k, 75%) and acceptable yield for ortho-substitution (3l, 52%). A slightly decreased yield of 3l suggested that the reaction may be influenced by the steric effect of the substituents. It is worth noting that even with some reactive substituents (–Br, –CN, and –COOMe), the yield was not significantly affected, indicating the mildness of this transformation (3h–3l, 52–75% yield). Subsequently, we further expanded the substrate scope to fused aryl and heteroaryl gem-difluoroalkenes, which also reacted well with moderate to good yields (3m–3p, 30–80%).
 | ||
| Scheme 2 Substrate scope of gem-difluoroalkenes.a,b a Reaction conditions: 1 (0.15 mmol, 1.0 equiv.), 2a (0.45 mmol, 3 equiv.), thioxanthone (5 mol%), 365 nm LED (30 W), argon atmosphere, at room temperature in DMSO (0.75 mL, 0.2 M) for 12 h. b Isolated yield. | ||
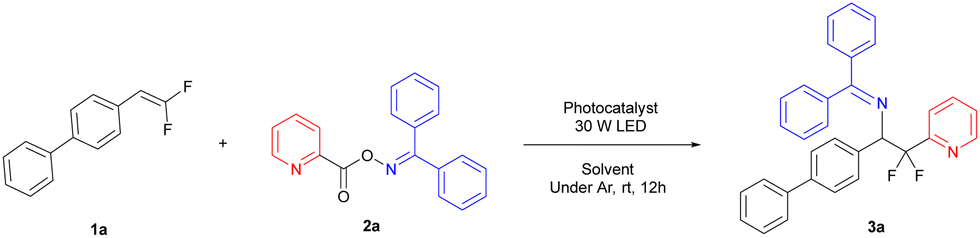
For the oxime ester part, we also explored the compatibility of the reaction using different substituted pyridine carboxylate oxime esters. As shown in Scheme 3, these reactions proceeded smoothly with yields of 42–63% as expected (4a–4f). To our delight, expanding the substrate to other electron-deficient six-member heterocycles such as pyrimidine (4g, 54% yield), pyrazine (4h, 46% yield) and quinoline (4i, 56% yield) also gave the desired products. Meanwhile, we also used some electron-rich heterocycles (thiophene and furan) and electron-withdrawing substituted benzene rings (–COOMe and –CN), but unfortunately, the target products were not obtained due to the difficulty of decarboxylation.
 | ||
| Scheme 3 Substrate scope of the (hetero)aryl radical precursor.a,b a Reaction conditions: 1 (0.15 mmol, 1.0 equiv.), 2 (0.45 mmol, 3 equiv.), thioxanthone (5 mol%), 365 nm LED (30 W), argon atmosphere, at room temperature in DMSO (0.75 mL, 0.2 M) for 12 h. b Isolated yield. | ||
To evaluate the scalability of this protocol, the reaction was successfully scaled up to ten-fold (Schemes 4a, 1.5 mmol, 60% yield). Considering the potential application of the heteroaryl difluoroethylamine structures in the field of medicinal chemistry, we further applied the reaction to the structural modification of natural products or drugs to evaluate the practicality of this difunctionalization reaction. As shown in Scheme 4b, structural modifications were carried out on both asaraldehyde (natural product, COX-2 inhibitor) and picolinafen (herbicide) intermediates with yields of 61% and 54% (yields given for the last step), respectively. It is well known that imine structures can be rapidly hydrolyzed under acidic conditions, and product 3a was deprotected to give the corresponding amine with a yield of 95% (see the ESI,† S2.6).
 | ||
| Scheme 4 Scale-up reaction and late-stage modification. Isolated yield. | ||
We next turned our attention to the reaction mechanism with a series of experiments. The radical crossover study proved the existence of the aryl radical and N-centered imine radical, respectively, as the crossover products were detected by mass spectrometry (Scheme 5a). Moreover, the generation of these two types of radicals was further confirmed via radical scavenger trapping experiments with (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) or azobis-(isobutyronitrile) (AIBN) (Scheme 5b).
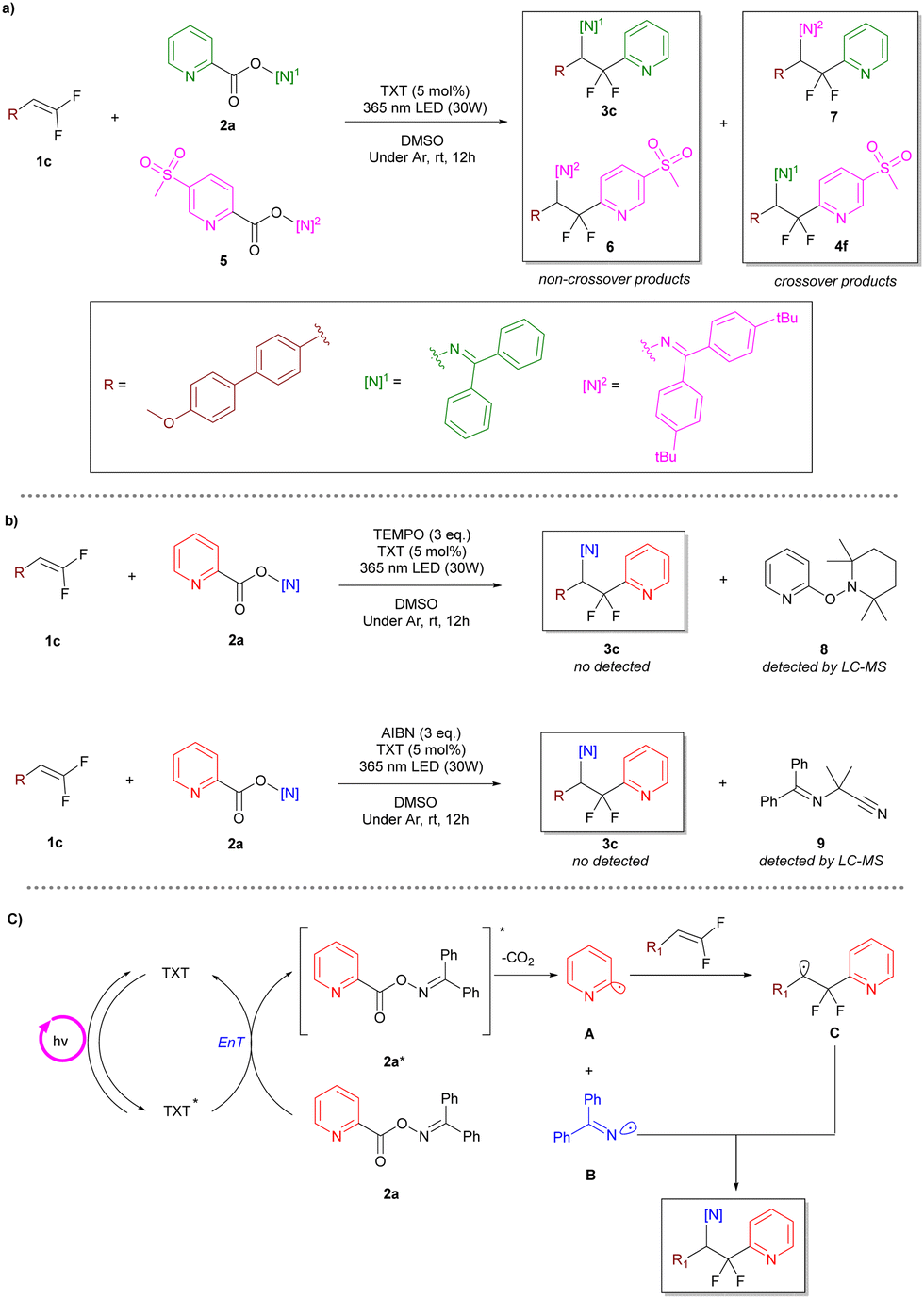 | ||
| Scheme 5 Mechanistic studies. (a) Crossover study. (b) Trapping experiments. (c) Proposed mechanism. | ||
Based on the experimental facts mentioned above and previous reports,23,27,31 we proposed a possible reaction mechanism as shown in Scheme 5. First, under the irradiation of suitable wavelength light, thioxanthone (TXT) is excited to a triplet excited state. Subsequently, a triplet–triplet energy-transfer process between TXT and oxime ester leads to the sensitization of the oxime ester and homolysis of the N–O bond, forming an imine radical (B) and a carboxyl radical (A). The carboxyl radical further undergoes decarboxylation to form an aryl radical (C), which is then captured by a gem-difluoroalkene to form a carbon-centered radical. Finally, a radical–radical cross-coupling between the carbon-centered radical and the imine radical produces the target product.
In summary, we have developed a metal-free photocatalytic method for the amination and heteroarylation of gem-difluoroalkenes. This method features good functional group compatibility and mild and metal-free reaction conditions, making it environmentally friendly. Above all, this benign atom-economical method can be used for the rapid construction of heteroaryl difluoroethylamine structures, and at the same time, we expect that this method can be a good complement to the arylation of gem-difluoroalkenes.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Acena, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed
.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed
- C. S. Burgey, K. A. Robinson, T. A. Lyle, P. E. Sanderson, S. D. Lewis, B. J. Lucas, J. A. Krueger, R. Singh, C. Miller-Stein, R. B. White, B. Wong, E. A. Lyle, P. D. Williams, C. A. Coburn, B. D. Dorsey, J. C. Barrow, M. T. Stranieri, M. A. Holahan, G. R. Sitko, J. J. Cook, D. R. McMasters, C. M. McDonough, W. M. Sanders, A. A. Wallace, F. C. Clayton, D. Bohn, Y. M. Leonard, T. J. Detwiler, Jr., J. J. Lynch Jr., Y. Yan, Z. Chen, L. Kuo, S. J. Gardell, J. A. Shafer and J. P. Vacca, J. Med. Chem., 2003, 46, 461–473 CrossRef CAS PubMed
-
G. Deng and J. Peng, CN Pat., CN117069693A, 2023 Search PubMed
- F. Xue, H. Li, S. L. Delker, J. Fang, P. Martasek, L. J. Roman, T. L. Poulos and R. B. Silverman, J. Am. Chem. Soc., 2010, 132, 14229–14238 CrossRef CAS PubMed
- I. Kers, I. Macsari, G. Csjernyik, M. Nylof, K. Skogholm, L. Sandberg, A. Minidis, T. Bueters, J. Malmborg, A. B. Eriksson, P. E. Lund, E. Venyike, L. Luo, J. E. Nystrom and Y. Besidski, Bioorg. Med. Chem. Lett., 2012, 22, 6108–6115 CrossRef CAS PubMed
- M.-Z. Lu, J. Goh, M. Maraswami, Z. Jia, J.-S. Tian and T.-P. Loh, Chem. Rev., 2022, 122, 17479–17646 CrossRef CAS PubMed
- J. P. Sorrentino and R. A. Altman, Synthesis, 2021, 3935–3950 CAS
- X. Zhang and S. Cao, Tetrahedron Lett., 2017, 58, 375–392 CrossRef CAS
- X. Chen, Z. Zhang, W. Y. Shi, Y. N. Ding, Y. Y. Luan, Y. C. Huang, Q. Wang, X. Y. Liu and Y. M. Liang, Org. Lett., 2023, 25, 4456–4461 CrossRef CAS PubMed
- F. Liu, Z. Zhuang, Q. Qian, X. Zhang and C. Yang, J. Org. Chem., 2022, 87, 2730–2739 CrossRef CAS PubMed
- X. Y. Wang, M. Yang, Y. Zhou, J. Zhou and Y. J. Hao, Org. Biomol. Chem., 2023, 21, 1033–1037 RSC
- R. M. Herrick, M. K. Abd El-Gaber, G. Coy and R. A. Altman, Chem. Commun., 2023, 59, 5623–5626 RSC
- X. Han, X. Liu, C. Len, L. Liu, D. Wang, Y. Zhang, X. H. Duan and M. Hu, J. Org. Chem., 2023, 88, 12744–12754 CrossRef CAS PubMed
- J. P. Sorrentino, R. M. Herrick, M. K. Abd El-Gaber, A. Z. Abdelazem, A. Kumar and R. A. Altman, J. Org. Chem., 2022, 87, 16676–16690 CrossRef CAS PubMed
- C. Liu, C. Zhu, Y. Cai and H. Jiang, Angew. Chem., Int. Ed., 2021, 60, 12038–12045 CrossRef CAS PubMed
- W. Zhu, H. Xi, W. Jiao, L. Huang, L. Wang and J. Wu, Org. Lett., 2022, 24, 720–725 CrossRef CAS PubMed
- X. Yu, A. Maity and A. Studer, Angew. Chem., Int. Ed., 2023, 62, e202310288 CrossRef CAS PubMed
- Y. Cai, H. Jiang and C. Zhu, Adv. Synth. Catal., 2023, 365, 342–354 CrossRef CAS
- B. Zhang and X. Zhang, Chem. Commun., 2016, 52, 1238–1241 RSC
- K. Yuan, T. Feoktistova, P. H. Cheong and R. A. Altman, Chem. Sci., 2021, 12, 1363–1367 RSC
- T. Patra, S. Mukherjee, J. Ma, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10514–10520 CrossRef CAS PubMed
- V. K. Soni, S. Lee, J. Kang, Y. K. Moon, H. S. Hwang, Y. You and E. J. Cho, ACS Catal., 2019, 9, 10454–10463 CrossRef CAS
- J. Kang, H. S. Hwang, V. K. Soni and E. J. Cho, Org. Lett., 2020, 22, 6112–6116 CrossRef CAS PubMed
- Z. D. Chen, X. Zhou, J. T. Yi, H. J. Diao, Q. L. Chen, G. Lu and J. Weng, Org. Lett., 2022, 24, 2474–2478 CrossRef CAS PubMed
- Z. Zhuang, Y. Sun, Y. Zhong, Q. He, X. Zhang and C. Yang, Org. Lett., 2024, 26, 713–718 CrossRef CAS PubMed
- Y. Zheng, Z. J. Wang, Z. P. Ye, K. Tang, Z. Z. Xie, J. A. Xiao, H. Y. Xiang, K. Chen, X. Q. Chen and H. Yang, Angew. Chem., Int. Ed., 2022, 61, e202212292 CrossRef CAS PubMed
- S. Kim, H. Oh, W. Dong, J. Majhi, M. Sharique, B. Matsuo, S. Keess and G. A. Molander, ACS Catal., 2023, 13, 9542–9549 CrossRef CAS
- C. P. Yuan, Y. Zheng, Z. Z. Xie, K. Y. Deng, H. B. Chen, H. Y. Xiang, K. Chen and H. Yang, Org. Lett., 2023, 25, 1782–1786 CrossRef CAS PubMed
- X. K. Qi, M. J. Zheng, C. Yang, Y. Zhao, L. Guo and W. Xia, J. Am. Chem. Soc., 2023, 145, 16630–16641 CrossRef CAS PubMed
- J. E. Erchinger, R. Hoogesteger, R. Laskar, S. Dutta, C. Humpel, D. Rana, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2023, 145, 2364–2374 CrossRef CAS PubMed
- S. S. Li, Y. S. Jiang, X. L. Luo, C. X. Pan and P. J. Xia, Org. Lett., 2023, 25, 1595–1599 CrossRef CAS PubMed
- J. Majhi, R. K. Dhungana, A. Renteria-Gomez, M. Sharique, L. Li, W. Dong, O. Gutierrez and G. A. Molander, J. Am. Chem. Soc., 2022, 144, 15871–15878 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc00528g |
| ‡ These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2024 |