
Cyano disubstituted tetrabenzoindeno[2,1-a]fluorene: open-shell or closed-shell?†
Priyank Kumar
Sharma
 a,
Palash
Jana
b,
Subhajit
Bandyopadhyay
b and
Soumyajit
Das
*a
a,
Palash
Jana
b,
Subhajit
Bandyopadhyay
b and
Soumyajit
Das
*a
aDepartment of Chemistry, Indian Institute of Technology Ropar, Rupnagar 140001, Punjab, India. E-mail: chmsdas@iitrpr.ac.in
bDepartment of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur 741246, West Bengal, India
First published on 7th May 2024
Abstract
Organic diradicaloids have lately emerged as potential spintronic materials. We report the unprecedented synthesis of a near-IR absorbing indeno[2,1-a]fluorene derivative that displays remarkably low LUMO (−4.15 eV) and a small HOMO–LUMO gap (0.85 eV). NMR/EPR studies indicated its open-shell diradical property, which was supported by DFT calculations while suggesting a 30% diradical character and a small singlet (S)–triplet (T) gap (−2.52 kcal mol−1). A large bond length alternation of the as-indacene core for its single-crystals indicated a quinoidal contribution with greater antiaromaticity, which is in line with the small diradical character despite showing a small S–T gap.
Exploration of polycyclic hydrocarbons (PHs) with antiaromatic and diradical properties has attracted immense interest in recent years,1 as tuning such properties may provide paths toward new organic semiconductor materials with tunable band gaps. Formally antiaromatic indeno[1,2-b]fluorene21 and indeno[2,1-a]fluorene32 regioisomers are stable closed-shell (CS) molecules that display moderate antiaromaticity for the central s-indacene and as-indacene units,4 respectively (Fig. 1a). Unsymmetrical disubstitution of [2,1-a]IF also resulted in a stable CS molecule 3,5 but benzo-extension of the central as-indacene unit of [2,1-a]IF afforded an open-shell (OS) molecule benzo-2 with a large diradical character index (y0 = 0.63), likely causing instability (half-life 77 min) and 8% synthetic yield.6
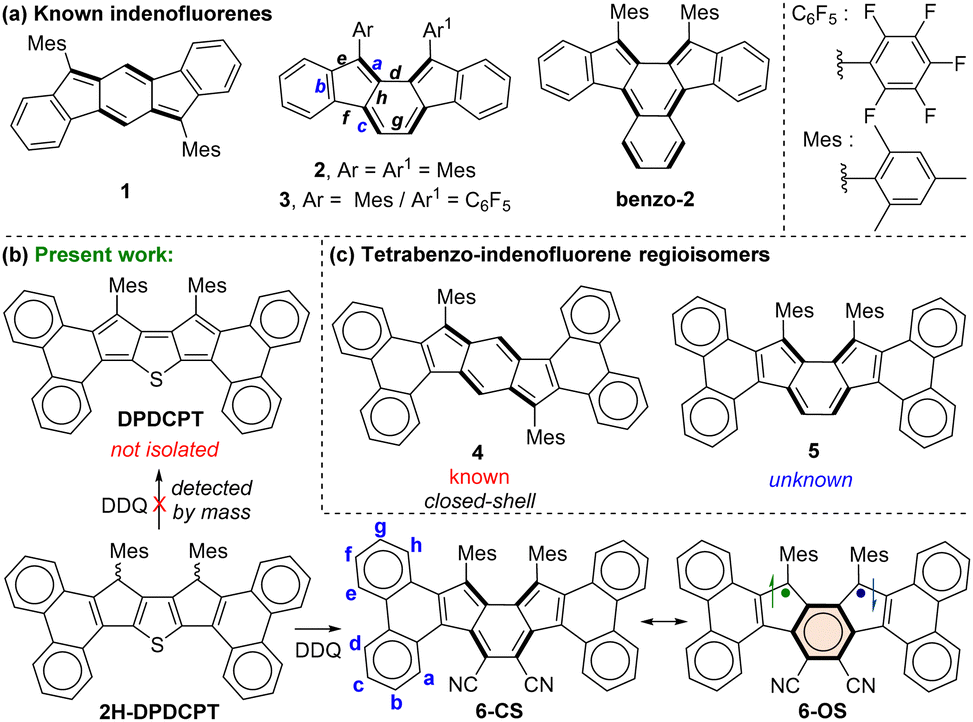 | ||
| Fig. 1 (a) Indeno[1,2-b]fluorene 1, indeno[2,1-a]fluorenes 2 & 3, and benzo-fused [2,1-a]IF benzo-2. (b) Targeted diphenanthro-DCPT, and the synthesized tetrabenzoindeno[2,1-a]fluorene 6 and its resonance forms. (c) Isomeric tetrabenzoindeno[1,2-b]fluorene 4 and tetrabenzoindeno[2,1-a]fluorene 5 derivatives. | ||
In continuation of efforts7 to synthesize the elusive arene-fused dicyclopenta[b,d]thiophene (DCPT),7a,b a diphenanthro-DCPT (DPDCPT) derivative was targeted (Fig. 1b). While attempting its synthesis by a DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) mediated dehydrogenation from dihydro precursor 2H-DPDCPT, we failed to isolate it presumably due to the high reactivity (electron-rich, large y0 = 0.51, and a small singlet–triplet energy gap (ΔES–T) = −0.64 kcal mol−1; Table S2, ESI†), as analysed by density functional theory (DFT) calculation. Instead, we isolated a cyano disubstituted tetrabenzoindeno[2,1-a]fluorene 6 (Fig. 1b) as a degradation product, and it was confirmed by single-crystal X-ray diffraction (SCXRD) analysis (vide infra). Isolation of 6 was interesting since there is no straightforward synthetic approach known thus far to construct 5, which is a constitutional isomer of known tetrabenzoindeno[1,2-b]fluorene 4,8 which displays a greater degree of antiaromaticity for the s-indacene unit than that of 1. Our DFT optimization of 5 indicated its CS ground state, like 4, and its OS singlet state was found to be isoenergetic with the CS state with a small ΔES–T = −3.46 kcal mol−1 (Table S2, ESI†). A lowering of the triplet state for 5, despite showing a negligible diradical character (y0 = 0.01), is in line with a recent report9 by Ottosson and Solà et al. explaining that diphenanthro-fused antiaromatics (pentalene, s-indacene) may display low-lying triplet states. Despite the lowering of the triplet excited state, 5 didn’t show an OS ground state, theoretically.
We envisaged that the cyano (–CN) substituents for 6 may result in a smaller HOMO–LUMO energy spacing than that of 5, seemingly due to the greater stabilization effect exerted by electron-withdrawing –CN groups (negative resonance effect), which are attached to as-indacene carbons containing large LUMO coefficients (Fig. S22, ESI†).10 A small HOMO–LUMO energy gap is crucial to show an OS ground state, as evident from CS heptazethrene-triisopropylsilylethynyl (HZ-TIPS)11vs. OS heptazethrene-dicarboximide (HZ-DI),12 though the number of Clar sextet recoveries remains the same for them. Therefore, in this work, compound 6 was thoroughly studied by various analytical techniques and DFT calculations to conclude its electronic ground state, while reporting its unprecedented synthesis.
Suzuki coupling between commercially available 7 and 2,5-dibromothiophene-3,4-dicarbaldehyde 8,13 as depicted in Scheme 1, afforded dialdehyde derivative 9. Treatment of 9 with 2-mesitylmagnesiumbromide afforded a diol intermediate, which was subsequently treated with BF3·Et2O to give 2H-DPDCPT, which is the dihydro precursor of DPDCPT. DDQ-mediated oxidative dehydrogenation of 2H-DPDCPT in dichloromethane (DCM) remained partially complete, even after refluxing or increasing the reaction time, as monitored by thin layer chromatography. To ensure complete conversion, 4 equiv. DDQ was added, and the reactant was consumed within 30 minutes at room temperature (rt). The formation of the targeted DPDCPT could be detected from the crude reaction mixture by direct mass spectrometric (MS) analysis (Fig. S25, ESI†), but unfortunately, no desired product trace was obtained after traditional silica gel column chromatography; instead, DPDCPT-DDQ was isolated as a 10+2 cycloaddition adduct,14 likely due to the reaction of DPDCPT and DDQ on the silica surface (Fig. S26, ESI†).
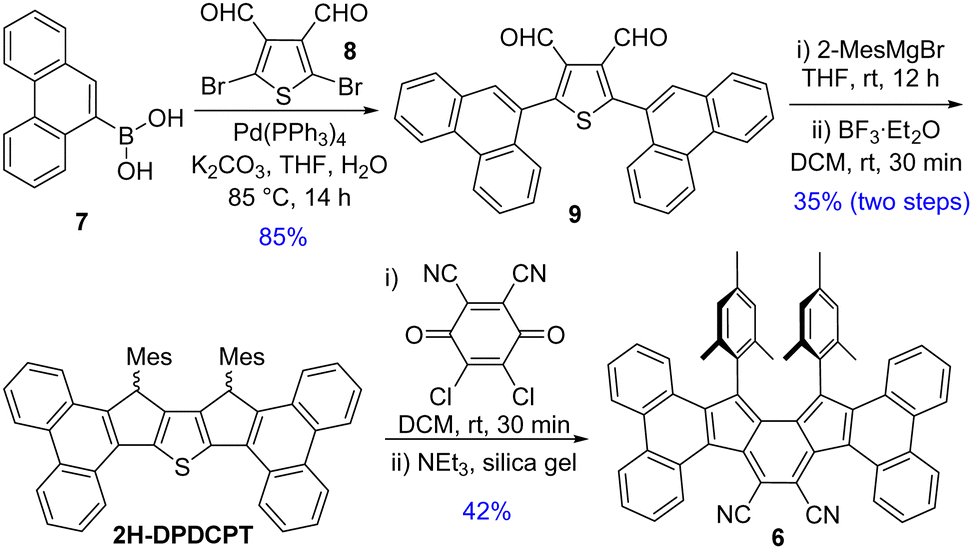 | ||
| Scheme 1 Synthesis of 6. | ||
Treatment of silica gel with triethylamine, prior to loading the slurry of the crude reaction mixture containing DDQ, afforded a deep purple solid in 42% yield. However, the high-resolution mass (m/z = 739.3099) didn’t match the desired mass of DPDCPT. It was found to be an unexpected product 6 after SCXRD analysis of the purple solid (Fig. 2), plausibly formed by the loss of a sulfur atom and ClC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO)C(Cl)CO unit from the DPDCPT-DDQ adduct on neutral silica gel or an alumina column (Fig. S26, ESI†).14b,c Nonetheless, 6 is the first diphenanthro-as-indacene derivative with an ortho-quinoidal backbone, which is a structural isomer of 4.
CO)C(Cl)CO unit from the DPDCPT-DDQ adduct on neutral silica gel or an alumina column (Fig. S26, ESI†).14b,c Nonetheless, 6 is the first diphenanthro-as-indacene derivative with an ortho-quinoidal backbone, which is a structural isomer of 4.
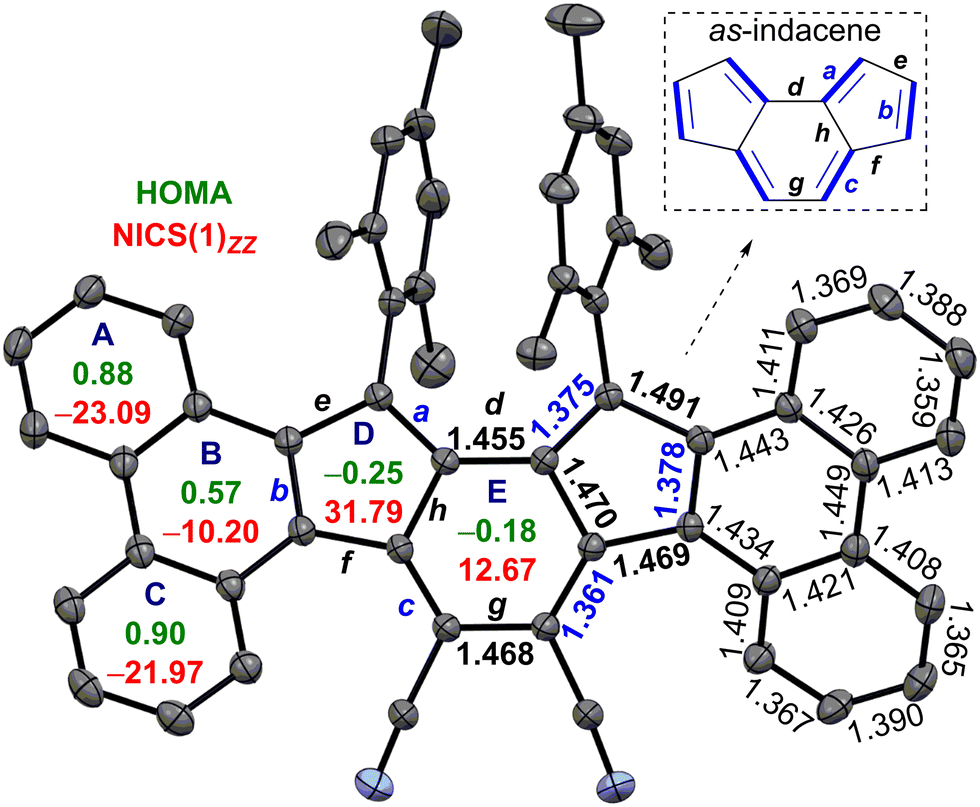 | ||
| Fig. 2 ORTEP drawing of 6 with thermal ellipsoids at the 30% probability level (hydrogens and disordered solvent omitted), including the mean bond lengths (in angstroms), NICS(1)zz (red) and HOMA (green) indices. | ||
The single crystals of 6 were grown in a DCM/acetonitrile mixture (Fig. 2). SCXRD analysis of 6 revealed a contorted π-backbone (Fig. S13a, ESI†) due to the steric congestion between the –CN group and ring-C hydrogen in the cove region and repulsive interaction between the bulky mesityl groups, which are orthogonal to the IF backbone. The terminal phenanthrene units were twisted by an avg. torsional angle of 30.1°, as measured from the mean planes between central ring E and outer phenanthrene units (Fig. S13a, ESI†). The C–C double bond b linking as-indacene and the phenanthrene unit and the exo-methylene bond a for 6 are 0.032 Å and 0.016 Å, respectively, shorter than those of 2 (Fig. 2 and Table 1). The C–C single bonds d–h for 6 possess more single bond character (1.455 to 1.491 Å), with e and g bonds 0.016 and 0.037 Å longer than those of 2. The degree of bond length alternation (BLA) in the as-indacene unit has significantly enhanced for 6 in comparison to 2, suggesting a reduced extent of π-delocalization for the as-indacene core in 6, indicating a greater degree of antiaromaticity for 6 than that of 2. On the other hand, the homogeneous bond length distribution for rings A (avg. 1.394 Å) and C (avg. 1.393 Å) in 6 suggested a greater degree of benzene-like aromaticity than that of ring B (avg. 1.425 Å).
| a | b | c | d | e | f | g | h | |
|---|---|---|---|---|---|---|---|---|
| 6 | 1.375 | 1.378 | 1.361 | 1.455 | 1.491 | 1.469 | 1.468 | 1.470 |
| 2 | 1.391 | 1.410 | 1.359 | 1.454 | 1.475 | 1.463 | 1.431 | 1.480 |
The harmonic oscillator model of aromaticity (HOMA)15 analyses of the optimized structure 6 (at BHandHLYP/6-31G*) supported the large BLA for rings D (−0.25) and E (−0.18), whereas small (A = 0.88, C = 0.90) to moderate (B = 0.57) BLA for phenanthrene indicated its aromaticity (Fig. 2). Nuclear independent chemical shift (NICS)16 calculation showed larger positive NICS(1)zz values for rings D (31.79) and E (12.67) for 6 than those of 2, suggesting enhanced antiaromaticity of the as-indacene unit for 6. The negative NICS values of rings A (−23.09), B (−10.20), and C (−21.97) for 6 are in line with the HOMA values, supporting phenanthrene ring aromaticity.
The observation of proton NMR (nuclear magnetic resonance) line broadening for 6 at rt (Fig. S5, ESI†) prompted us to study its variable temperature (VT) NMR behaviour. In CDCl3, 6 exhibited broad NMR signals for some of the aromatic protons (d, c, b protons in Fig. 3a, see Fig. 1b for proton labels) at rt, while other ring protons were also broadened upon heating (Fig. S9, ESI†). These broad NMR signals became sharper upon gradual cooling and became reasonably sharper at −60 °C (213 K). This is a typical observation for OS diradicaloid PHs with small ΔES–T,11,17 due to which the excited triplet state can be thermally populated at rt or by slight heating. The observation was also supported by VT-EPR (electron paramagnetic resonance), exhibiting a featureless broad signal (ge = 2.0041) at rt, while the signal intensity decreased as the temperature was lowered from 260 K to 150 K (Fig. 3b), which is a consequence of reduced population of magnetically active triplet species at lower temperatures.18 A careful fitting of the VT-EPR data using the Bleaney–Bowers equation19 gave a ΔES–T = −0.97 kcal mol−1 for 6 (Fig. S11a, ESI†), suggesting singlet OS ground state, which is in line with VT-EPR in solution (ΔES–T = −1.73 kcal mol−1, Fig. S11b and c, ESI†).
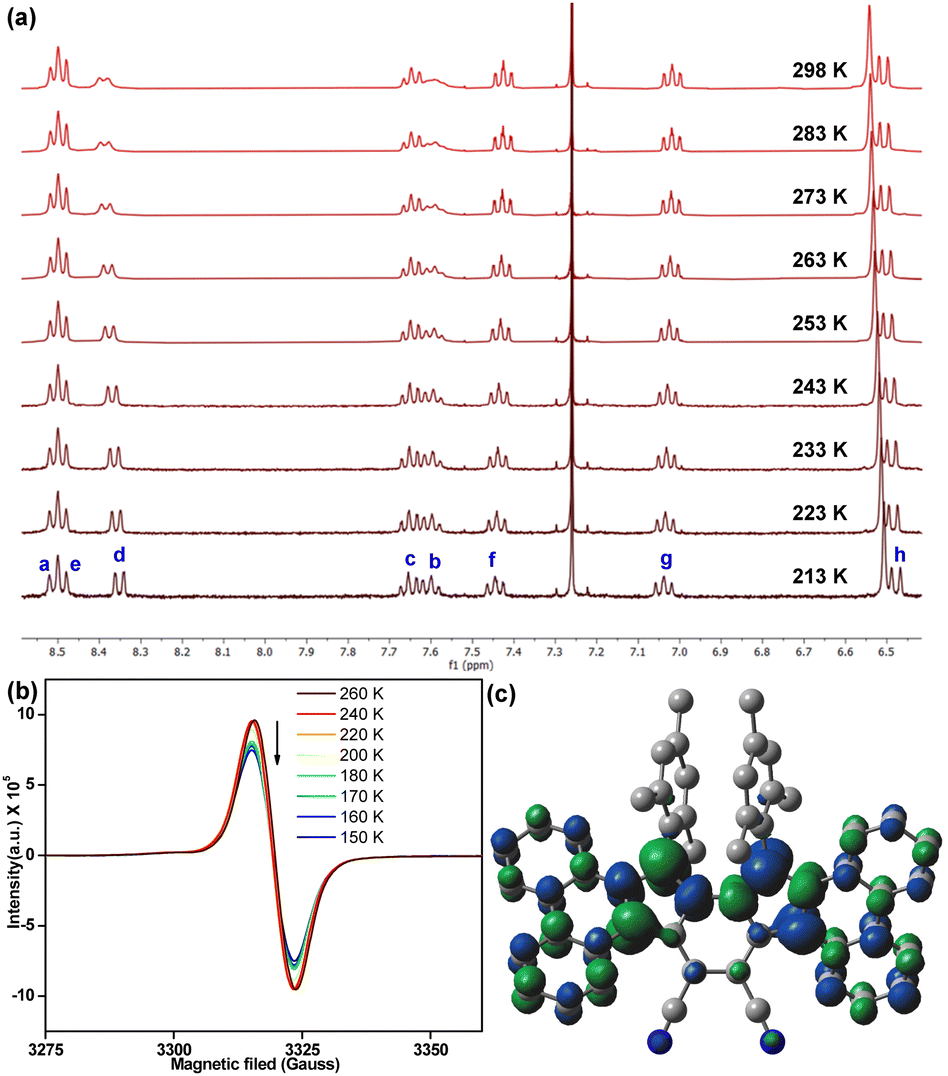 | ||
| Fig. 3 (a) Partial VT-NMR of 6 in CDCl3 showing aromatic protons (see Fig. 1b for proton labels); (b) VT-EPR spectra of solid sample 6; (c) spin density distribution of singlet OS 6. | ||
DFT calculation of 6 at the BHandHLYP/6-31G* level of theory20 confirmed that the energy of an OS singlet state was 1.96 kcal mol−1 and 2.52 kcal mol−1 lower than the energy of the CS singlet (quinoidal) and triplet (biradical) states, respectively (Table S2, ESI†). The y0 for 6 was estimated to be 0.30, based on the natural orbital occupation numbers using the broken symmetry formalism. The frontier molecular orbital profiles for α- and β-spins (Fig. S21, ESI†) displayed the characteristic disjointed nature in the ground state singlet diradical form. The spin densities were observed to be uniformly dispersed across the entire π-conjugated framework (Fig. 3c), giving thermodynamic stability.
The UV-vis-NIR (UV-visible-near infrared) spectrum (Fig. 4a) of 6 exhibited intense absorption peaks at λmax = 312 nm (ε = 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) and λmax = 365 nm (ε = 19000 M−1 cm−1) in the UV region and a moderate absorption in the visible region λmax = 554 nm (ε = 11000 M−1 cm−1), which is associated with a weak absorption band in the lowest energy region stretching from 750 to 1500 nm in the NIR region with the absorption maximum at λmax = 1000 nm (ε = 650 M−1 cm−1). Time dependent-DFT (TD-DFT) calculations of 6 in toluene (Fig. S19, ESI†) suggested that the absorption in the visible region is dominated by the HOMO−1 → LUMO transition (λmax = 476 nm, oscillator strength (f) = 0.55, Table S3, ESI†), while the weaker lowest energy absorption tail has originated from a forbidden HOMO → LUMO transition (λmax = 877 nm, f = 0.028). Such a long wavelength absorption tail could originate due to the admixing of the doubly excited electronic configuration (H,H → L,L) for 6 owing to its OS ground state, as observed for Kubo's diphenaleno-DCPT.14a Compound 6 displayed a very small optical HOMO–LUMO energy gap of 0.85 eV, as roughly estimated from the lowest energy absorption onset (1240/λonset); yet, a good photostability under ambient conditions with a half-life of 9 days was observed for 6 (Fig. S10, ESI†).
000 M−1 cm−1) and λmax = 365 nm (ε = 19000 M−1 cm−1) in the UV region and a moderate absorption in the visible region λmax = 554 nm (ε = 11000 M−1 cm−1), which is associated with a weak absorption band in the lowest energy region stretching from 750 to 1500 nm in the NIR region with the absorption maximum at λmax = 1000 nm (ε = 650 M−1 cm−1). Time dependent-DFT (TD-DFT) calculations of 6 in toluene (Fig. S19, ESI†) suggested that the absorption in the visible region is dominated by the HOMO−1 → LUMO transition (λmax = 476 nm, oscillator strength (f) = 0.55, Table S3, ESI†), while the weaker lowest energy absorption tail has originated from a forbidden HOMO → LUMO transition (λmax = 877 nm, f = 0.028). Such a long wavelength absorption tail could originate due to the admixing of the doubly excited electronic configuration (H,H → L,L) for 6 owing to its OS ground state, as observed for Kubo's diphenaleno-DCPT.14a Compound 6 displayed a very small optical HOMO–LUMO energy gap of 0.85 eV, as roughly estimated from the lowest energy absorption onset (1240/λonset); yet, a good photostability under ambient conditions with a half-life of 9 days was observed for 6 (Fig. S10, ESI†).
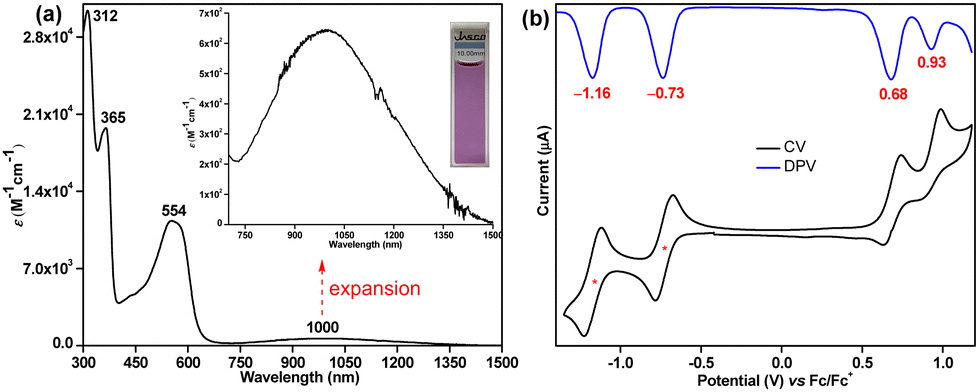 | ||
| Fig. 4 (a) UV-vis-NIR absorption spectrum of 6 in toluene (715–1500 nm expansion shown in inset); (b) CV and DPV of 6. | ||
Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) analyses (Fig. 4b) suggested that 6 could be easily reversibly reduced at Ered11/2 = −0.73 V and at Ered21/2 = −1.16 V (vs. ferrocene/ferrocenium (Fc/Fc+) couple). It can also be oxidized to its radical cation and dication species at Eox1peak = 0.68 V and Eox2peak = 0.93 V, respectively. The HOMO and LUMO energy levels were −5.40 eV and −4.15 eV, as estimated from the onset potentials of the oxidation and reduction waves, respectively, giving a small electrochemical energy gap of 1.25 eV. The LUMO energy level of 6 is remarkably stabilized (low-lying) due to the electron-withdrawing cyano substituents, as hypothesized.
The greater stabilization of the LUMO basically reduced the HOMO–LUMO energy gap, and the observation is comparable to HZ-TIPSvs.HZ-DI, where HZ-DI with a small HOMO–LUMO gap displayed an OS diradicaloid ground state despite one Clar sextet being recovered for both molecules in their OS forms.11,12 The smaller ΔES–T for 6 than that of 5 could be caused by the narrower HOMO–LUMO gap of 6 pushing its triplet excited state to locate closer to the singlet ground state.21 As a result, the excited triplet state could be easily thermally populated at rt or by slight heating, despite showing a small y0. Usually, the greater the y0, the smaller the ΔES–T;22 with a few noteworthy exceptions23 (including benzo-2)6 showing a larger ΔES–T although exhibiting a larger y0. To the best of our knowledge, compound 6 is a unique example that exhibits a small y0 despite showing a small ΔES–T.
Typically, an OS PH with large y0 displays a large energy difference between its CS singlet and OS singlet states, and a small energy difference between its OS singlet state and first excited triplet state.24 Our experimental and theoretical studies confirmed that 6 exists as an OS diradicaloid in the singlet ground state with its CS quinoidal state lying in between the OS ground state and triplet excited state, which is unlike the known diradicaloid PHs17,24 with large y0 displaying CS quinoidal states lying vertically at a much higher energy level above the triplet state. A significant BLA for the central as-indacene unit of 6 in its crystalline form indicates a dominant quinoidal structure presumably due to the rt accessible low-lying CS state, which is in line with the small y0. This observation suggested that both CS and OS resonance forms for 6 may contribute to the singlet ground state.
In summary, a formally antiaromatic diphenanthro-as-indacene derivative 6 was synthesized, that exhibits diradical-like and antiaromatic properties in its OS singlet ground state as a result of a low-lying CS state. Though the formation of 6 was unexpected, its detailed characterization gave newer insights in diradical chemistry as our study suggested that substitution driven HOMO–LUMO energy gap modulation can affect the ground state properties for antiaromatic PHs. The small y0 (30%) for 6 could likely be attributed to the dominant as-indacene antiaromaticity owing to the low-lying quinoidal CS form, as supported by SCXRD analysis and NICS(1)zz calculation. The VT-NMR and VT-EPR experiments indicated its singlet diradicaloid ground state, which is the consequence of a small HOMO–LUMO energy gap. Qualitative correlations between the structural features and the properties of different diradicaloid systems usually indicate an increased y0 as the energy separation between the singlet and triplet states decreased. However, compound 6 is an example of a rare diradicaloid exhibiting a small y0 despite showing a small ΔES–T, implying potential for applications in organic spintronics and photonics (non-linear optics and singlet-fission) studies.25
S. D. thanks SERB, India (CRG/2022/003012) for funding. P. K. S. and P. J. thank CSIR for the senior research fellowship. S. B. thanks SERB for a research grant (CRG/2022/006776). The authors thank Kamlesh Satpute (IIT Ropar) for SCXRD data of 6.
Conflicts of interest
There are no conflicts to declare.Notes and references
-
(a) A. Konishi, Y. Okada, M. Nakano, K. Sugisaki, K. Sato, T. Takui and M. Yasuda, J. Am. Chem. Soc., 2017, 139, 15284 CrossRef CAS PubMed
; (b) T. Xu, X. Hou, Y. Han, H. Wei, Z. Li and C. Chi, Angew. Chem., Int. Ed., 2023, 62, e202304937 CrossRef CAS PubMed
- D. T. Chase, A. G. Fix, S. J. Kang, B. D. Rose, C. D. Weber, Y. Zhong, L. N. Zakharov, M. C. Lonergan, C. Nuckolls and M. M. Haley, J. Am. Chem. Soc., 2012, 134, 10349 CrossRef CAS PubMed
- A. Shimizu and Y. Tobe, Angew. Chem., Int. Ed., 2011, 50, 6906 CrossRef CAS PubMed
- Y. Tobe, Top. Curr. Chem., 2018, 376, 12 CrossRef PubMed
- H. Sharma, N. Bhardwaj and S. Das, Org. Biomol. Chem., 2022, 20, 8071 RSC
- H. Miyoshi, S. Nobusue, A. Shimizu, I. Hisaki, M. Miyata and Y. Tobe, Chem. Sci., 2014, 5, 163 RSC
-
(a) X. Shi and C. Chi, Top. Curr. Chem., 2017, 375, 68 CrossRef PubMed
- C. K. Frederickson, L. N. Zakharov and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 16827 CrossRef CAS PubMed
- R. Ayub, O. El Bakouri, K. Jorner, M. Solà and H. Ottosson, J. Org. Chem., 2017, 82, 6327 CrossRef CAS PubMed
- A. Casey, S. D. Dimitrov, P. Shakya-Tuladhar, Z. Fei, M. Nguyen, Y. Han, T. D. Anthopoulos, J. R. Durrant and M. Heeney, Chem. Mater., 2016, 28, 5110 CrossRef CAS
- Y. Li, W.-K. Heng, B. S. Lee, N. Aratani, J. L. Zafra, N. Bao, R. Lee, Y. M. Sung, Z. Sun, K.-W. Huang, R. D. Webster, J. T. López Navarrete, D. Kim, A. Osuka, J. Casado, J. Ding and J. Wu, J. Am. Chem. Soc., 2012, 134, 14913 CrossRef CAS PubMed
- Z. Sun, K.-W. Huang and J. Wu, J. Am. Chem. Soc., 2011, 133, 11896 CrossRef CAS PubMed
- P. K. Sharma, D. Mallick, H. Sharma and S. Das, Org. Lett., 2023, 25, 2201 CrossRef CAS PubMed
-
(a) T. Kubo, M. Sakamoto, M. Akabane, Y. Fujiwara, K. Yamamoto, M. Akita, K. Inoue, T. Takui and K. Nakasuji, Angew. Chem., Int. Ed., 2004, 43, 6474 CrossRef CAS PubMed
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed
- H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Org. Lett., 2006, 8, 863 CrossRef CAS PubMed
-
(a) S. Das, S. Lee, M. Son, X. Zhu, W. Zhang, B. Zheng, P. Hu, Z. Zeng, Z. Sun, W. Zeng, R.-W. Li, K.-W. Huang, J. Ding, D. Kim and J. Wu, Chem. – Eur. J., 2014, 20, 11410 CrossRef CAS PubMed
- P. Jana, S. Koppayithodi, S. Mahato, S. Molla and S. Bandyopadhyay, J. Phys. Chem. Lett., 2023, 14, 7433 CrossRef CAS PubMed
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451 CAS
- S. Lehtola, M. Dimitrova, H. Fliegl and D. Sundholm, J. Chem. Theory Comput., 2021, 17, 1457 CrossRef CAS PubMed
- W. Zeng, Q. Qi and J. Wu, Eur. J. Org. Chem., 2018, 2018, 7 CrossRef CAS
- J. J. Dressler and M. M. Haley, J. Phys. Org. Chem., 2020, 33, e4114 CrossRef CAS
-
(a) J. J. Dressler, M. Teraoka, G. L. Espejo, R. Kishi, S. Takamuku, C. J. Gómez-García, L. N. Zakharov, M. Nakano, J. Casado and M. M. Haley, Nat. Chem., 2018, 10, 1134 CrossRef CAS PubMed
- S. S. K. Boominathan, K.-H. Chang, Y.-C. Liu, C.-S. Wang, C.-F. Wu, M.-H. Chiang, P.-T. Chou and Y.-T. Wu, Chem. – Eur. J., 2019, 25, 7280 CrossRef CAS PubMed
-
(a) S. Datta, Y. Cai, I. Yudhistira, Z. Zeng, Y.-W. Zhang, H. Zhang, S. Adam, J. Wu and K. P. Loh, Nat. Commun., 2017, 8, 677 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Detailed syntheses and characterization data for 6; X-ray crystallographic data; DFT calculations; NMR spectra; direct mass spectrum of DPDCPT; and plausible mechanism for formation of 6. CCDC 2309430. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc00683f |
| This journal is © The Royal Society of Chemistry 2024 |