
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards tailoring hydrophobic interaction with uranyl(VI) oxygen for C–H activation†
Satoru
Tsushima
 *ab,
Jérôme
Kretzschmar
a,
Hideo
Doi
c,
Koji
Okuwaki
c,
Masashi
Kaneko
d,
Yuji
Mochizuki
de and
Koichiro
Takao
f
*ab,
Jérôme
Kretzschmar
a,
Hideo
Doi
c,
Koji
Okuwaki
c,
Masashi
Kaneko
d,
Yuji
Mochizuki
de and
Koichiro
Takao
f
aInstitute of Resource Ecology, Helmholtz–Zentrum Dresden–Rossendorf (HZDR), Dresden, 01328, Germany. E-mail: s.tsushima@hzdr.de
bInternational Research Frontiers Initiative (IRFI), Institute of Innovative Research, Tokyo Institute of Technology, Tokyo, 152-8550, Japan
cDepartment of Chemistry and Research Center for Smart Molecules, Rikkyo University, Tokyo, 171–8501, Japan
dDepartment of Chemistry, Osaka University, Osaka, 560-0043, Japan
eInstitute of Industrial Science, The University of Tokyo, Tokyo, 153-8505, Japan
fLaboratory for Zero-Carbon Energy, Institute of Innovative Research, Tokyo Institute of Technology, Tokyo, 152-8550, Japan
First published on 25th March 2024
Abstract
Bovine serum albumin (BSA) has a uranyl(VI) binding hotspot where uranium is tightly bound by three carboxylates. Uranyl oxygen is “soaked” into the hydrophobic core of BSA. Isopropyl hydrogen of Val is trapped near UO22+ and upon photoexcitation, C–H bond cleavage is initiated. A unique hydrophobic contact with “yl”-oxygen, as observed here, can be used to induce C–H activation.
The excited state of uranyl(VI) has a high oxidizing power of E° = +2.6 ± 0.1 V vs. standard hydrogen electrode (SHE), which is almost as strong as the oxidizing power of F2.1,2 This allows UO22+ to exhibit photocatalytic behavior toward C(sp3)–H bonds of aliphatic compounds, which have a bond dissociation energy of nearly 100 kcal mol−1.3 A well-known example of uranyl(VI) photochemistry is the photodegradation of organic compounds in the presence of uranyl(VI) under extremely mild conditions. As such, uranium has the potential to be a sustainable catalyst. Upon photoexcitation, the “yl”-oxygen of UO22+ becomes highly reactive to induce either H-abstraction from aliphatic carbon, C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond cleavage,4,5 or Ligand–Metal Charge Transfer (LMCT).6 Uranyl(VI) photolysis can be utilized in two different ways; either to transform U(VI) into U(IV) for U recovery, or to reoxidize U(V) with air and introduce U into a catalytic cycle.7,8 While photochemical reduction and functionalization of uranyl(VI) has often been reported in “exotic” systems, here we show that H-abstraction can be performed in a well-controlled manner in mild systems containing only ubiquitous biological substances, such as amino acids and proteins, to which direct C–H to C–C,3,9 C–O,10,11 or C–F12,13 conversion can be applied.
C bond cleavage,4,5 or Ligand–Metal Charge Transfer (LMCT).6 Uranyl(VI) photolysis can be utilized in two different ways; either to transform U(VI) into U(IV) for U recovery, or to reoxidize U(V) with air and introduce U into a catalytic cycle.7,8 While photochemical reduction and functionalization of uranyl(VI) has often been reported in “exotic” systems, here we show that H-abstraction can be performed in a well-controlled manner in mild systems containing only ubiquitous biological substances, such as amino acids and proteins, to which direct C–H to C–C,3,9 C–O,10,11 or C–F12,13 conversion can be applied.
First, we test our hypothesis that the excited uranyl(VI) aquo ion is a stronger oxidant compared to its coordination complexes, as is indicated by the fact that methanol is readily oxidized by [UO22+]*14 while acetic acid remains intact.15 For a uranyl-methanol mixture, a previous theoretical study suggested H-abstraction from –CH3 of methanol by Oyl of [UO22+]*.16 The same mechanism does not take place in the case of uranyl-acetate mixture.15 Ligand coordination apparently downgrades the oxidizing power of [UO22+]*. On the other hand, several coordination complexes of uranyl are reported to be photoreactive, such as nitrates,9,17,18 [UO2(NO3)2(Ph2phen)],19 and [UO2(OPMe3)4]2+.20 We have also recently discovered that photoexcited [UO2(CO3)3]4−, which has significantly weaker oxidizing power than photoexcited [UO2(H2O)5]2+, is capable of abstracting H from BH4−.21 Peptides22,23 and proteins24,25 are also known to be degraded by [UO22+]*. We first used quantum chemical calculations to theoretically estimate the reduction potentials of uranyl(VI) coordination complexes with the aim of confirming our hypothesis that ligand coordination reduces the oxidizing power of [UO22+]*. We followed a protocol of previous studies26–28 for calculating reduction potentials of U(VI)/U(V) pairs and extended the calculations to include excited states. Details of the calculations are given in the ESI.† The CAM-B3LYP functional with the aug-cc-PVTZ basis set on H, C, and O as well as the small-core effective core potential (ECP) on U29 and the CPCM solvation model in combination with Pauling's ionic radius were found to give reasonable agreement with experimental values after including spin–orbit corrections. The reduction potentials were estimated to be −0.082 and +2.478 V for [UO2(H2O)5]2+/+ and [UO2(H2O)5]2+*/+ couples, respectively. The corresponding experimental values are +0.08830 and +2.60 V,2 respectively. Using the same level of theory, we extended the calculation to the acetate (Ac) complexes [UO2(Ac)(H2O)3]0/−1 as well as to [UO2(Ac)(H2O)3]0*/−1 couples and obtained reduction potentials of −0.397 and +2.228 V, respectively. The latter value is significantly lower than that of an aquo complex redox couple. This suggests that coordination of the carboxyl group does indeed reduce the oxidizing power of UO22+ and that excited uranyl(VI) becomes a milder oxidant when coordinated by carboxyl groups. On the other hand, there are clear examples of peptides22,23 and proteins24,25 being degraded by [UO22+]*. Therefore our first question is whether amino acids alone have the ability to photochemically reduce uranyl(VI) carboxylate. Using a 365 nm light source, we illuminated aqueous uranyl(VI) solutions containing excess Gly, Val, or Phe, and measured time-dependent UV-Vis-NIR spectra (Fig. 1).
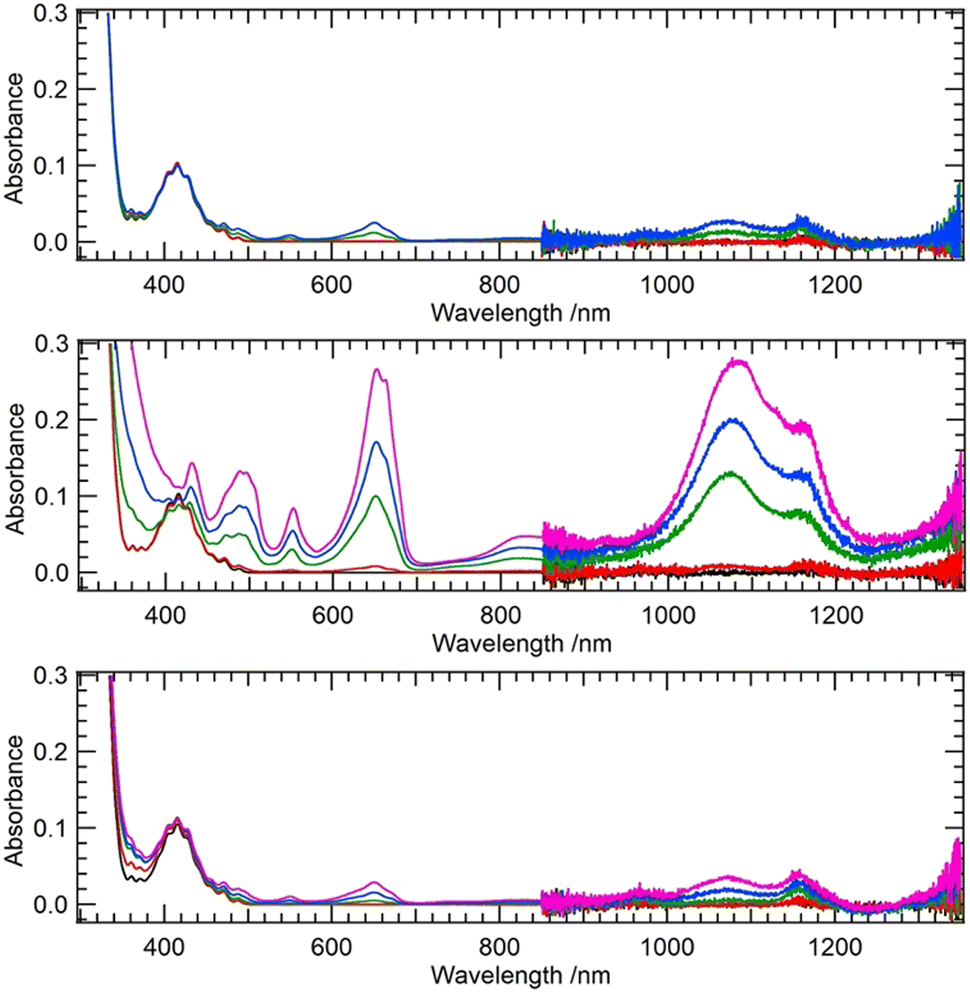 | ||
| Fig. 1 Time-dependent UV-Vis-NIR spectra of 10 mM uranyl(VI) mixtures with 90.4 mM Gly (top), Val (middle), and Phe (bottom) under illumination of 365 nm light. Before illumination (black), 60 s (red), 360 s (green), 600 s (blue), and 900 s (pink) after illumination. | ||
Among the three amino acids studied, Val undergoes the fastest photodegradation with U(VI) reduction to U(IV), while Gly and Phe are only slightly photoreactive. Gaseous by-products of the photodegradation of the U(VI)–Val system were CO2 and H2 (Shimadzu, GC-2014), although we could not quantify their yields. Nevertheless, it strongly suggests an H-abstraction mechanism involving aliphatic CH3. The aliphatic amino acid Val seems to act as an effective reductant, while CH2 from the backbone may also partially contribute to H-abstraction. However, the latter mechanism involving the backbone is likely to be difficult in the case of a peptide or protein due to steric hindrance. Therefore, we believe that the latter mechanism can only occur with amino acids and not with proteins and peptides. On the other hand, H-abstraction from protein side chains by [UO22+]* is likely to occur provided that Oyl is in a hydrophobic environment within the protein. UO22+ is known to interact with metalloproteins,24,31,32 but it is usually bound to carboxyl groups of Asp or Glu, which are in a hydrophilic environment, and U-protein remains photoinactive.
Bovine serum albumin (BSA) has a high affinity UO22+ binding site which undergoes site-specific photocleavage.24 To obtain a clue about its photoreactivity and its relation to molecular conformation, classical molecular dynamics (MD) simulations of UO22+-bound BSA were performed, and the MD trajectory was used to analyze the mechanism leading to site-specific photocleavage. For uranyl(VI) MD simulations, although novel 12-6-4 force fields have recently been developed by Guilbaud et al.34 and have been shown to have excellent performance, we used the conventional 12-6 FF (see ESI† for details) for consistency with our previous MD-FMO work on uranyl(VI).33 The validity of 12-6 FF in biological systems has been tested in several recent studies.35–37 Since previous studies suggested that photocleavage of UO22+-bound BSA occurs at Val314–Cys315,24 the MD simulation was performed by first aligning UO22+ in the vicinity of these residues. The simulation converged to a conformation in which uranyl(VI) is coordinated by three carboxyl groups from Asp307, Asp311, Asp313, and additionally with a water (Fig. 2). In addition, Oyl is H-bonded to the side chains of Phe308, Val314, Phe325, and occasionally to Asn317. Ultimately, this Oyl is soaked into the hydrophobic core of the protein. On the other hand, Oyl at the opposite end remains in a hydrophilic environment and in loose contact with bulk water, as is generally the case for Oyl in water.27 The formation of an H-bond between Oyl and Val314 confirms the experimental finding that illumination of uranyl(VI)-bound BSA leads to cleavage of the protein at Val314–Cys315.24 Presumably, the photoexcited state of uranyl(VI) abstracts hydrogen from the isopropyl side chain of Val314, leading to protein cleavage at Val314–Cys315. Although Val is not one of the most easily oxidized amino acids,38 it apparently acts as an effective reductant as long as its aliphatic side chain is readily accessible to Oyl.
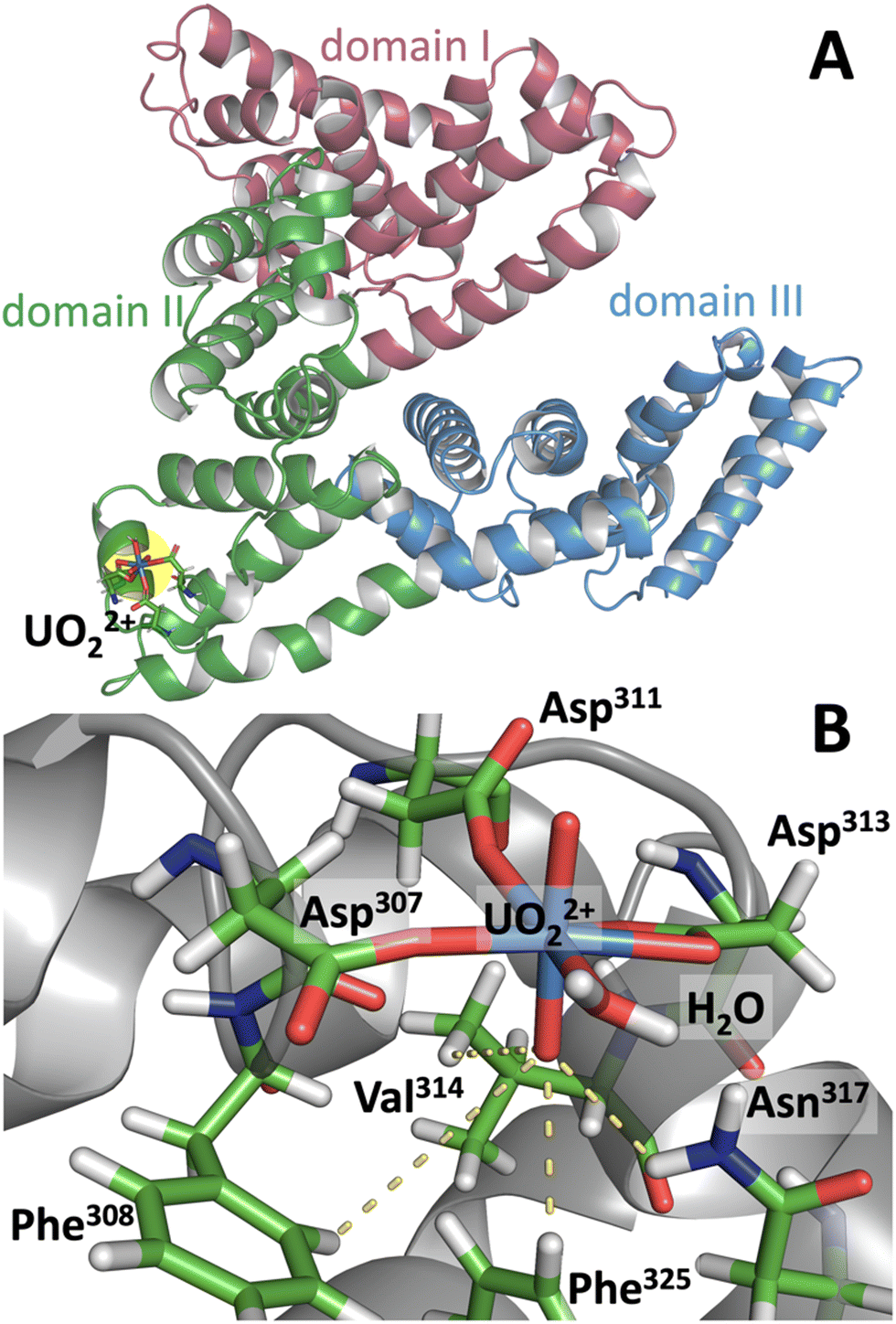 | ||
| Fig. 2 Representative snapshot from the MD trajectory of UO22+-bound BSA. Upper panel A shows the global conformation of the protein and its uranyl binding hotspot (in yellow). The lower panel B highlights the uranyl and its associated residues. Yellow dots represent hydrogen bonds involving Oyl. | ||
In order to investigate this point further and to have an extended view on the interactions acting between uranyl and protein residues, the structures obtained by MD simulations were additionally analyzed by the Fragment Molecular Orbital (FMO) method.39–41 In FMO, the molecular system of interest is divided into smaller fragments and each fragment as well as fragment pair is subjected to self-consistent field calculations and to successive correlation energy calculations. The electronic structure of the whole system is then reconstructed. This procedure drastically reduces the computational cost and allows MP2 or even MP3 calculations of gigantic biomolecules such as fully hydrated proteins42 or uranium–bound DNA.33 By combining MD with FMO, we are able to compensate for some of the problems associated with the use of classical MD and can more accurately assess energetics involving weak interactions such as H-bonds and dispersion interactions. The inter-fragment interaction energy (IFIE) obtained by FMO has been used to evaluate interactions with uranyl(VI).43–45 Although FMO is based on a full quantum chemical description, the input structures are obtained by classical MD. Calculating energy at the quantum chemical level using MD-based structures can be controversial as these structures are not necessarily at the energy minima. Therefore, this approach was further strengthened by taking 100 snapshots of MD trajectories (each 1 ns from the total 100 ns simulation), all of which were subjected to FMO calculations. We used FMO in a scaled third-order Møller–Plesset perturbation scheme (MP2.5) as reported elsewhere.46,47 The average IFIE was calculated using 100 MD structures. Note that the IFIE is not a binding energy and refers to the interaction energy between two individual fragments (residues), thus giving us an indication of which residues are actively interacting with each other.
In Fig. 3, representative structures of BSA with and without UO22+ from MD simulations are superimposed. The two structures overlap well with each other, and there is little conformational change upon uranyl binding as far as the vicinity of the uranyl-biding site is concerned. Three Asp side chains also remain essentially in the same position and are only slightly tilted to adapt to uranyl binding. This implies that BSA can bind uranyl with little energetic cost to protein stability. This is also evident from the fact that the secondary structure of BSA remains essentially the same even after uranyl(VI) binding. On the other hand, analysis of the IFIE from FMO calculations (Table 1) shows that uranyl(VI) is not only captured by three Asp, but that there are also attractive interactions between uranyl(VI) and other side chains. These residues include Leu304, Thr305, Ala306, Phe308, and Val314, most of which have hydrophobic side chains. For Val314, although the average IFIE is a positive value (i.e., the interaction is repulsive), the actual value at each time step varies from −20.9 to +20.2 kcal mol−1, reflecting the dynamic fluctuation of the UO22+–Val distance from 2.23 to 4.61 Å. Only when the UO22+–Val distance is close does the interaction becomes attractive. We observed so-called “Asn flipping” during the simulation, which resulted in a large standard deviation of the IFIE for Asn317. However, such behavior at atomic resolution is well known48 and is thought to be a computational artifact. Therefore, we do not take the interaction with Asn seriously.
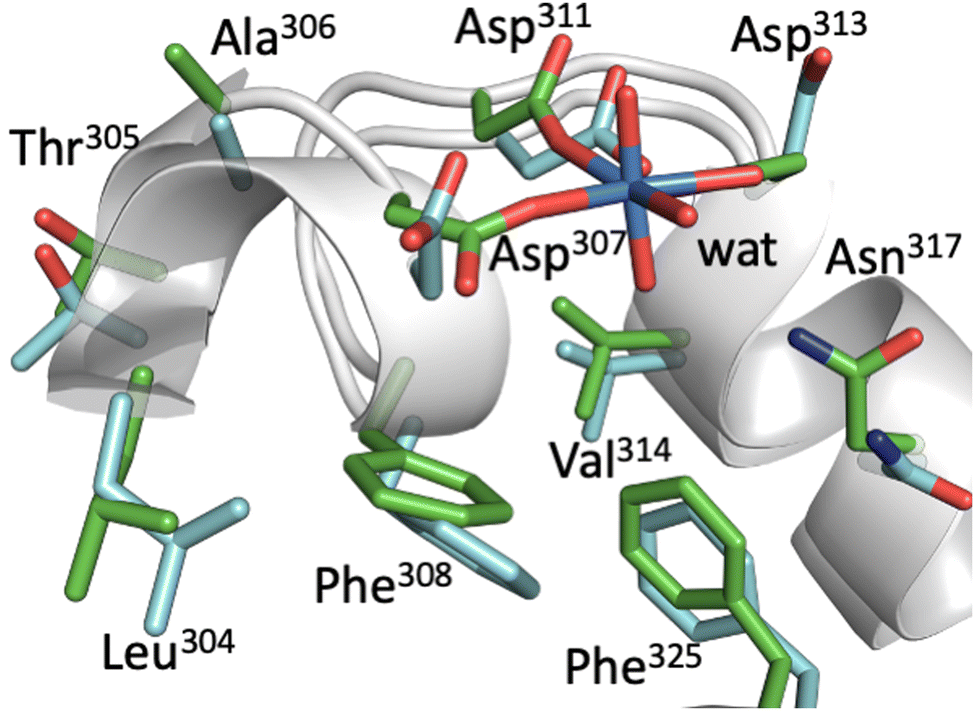 | ||
| Fig. 3 Overlay of representative structures of BSA with (light green) and without (light blue) uranyl(VI). The side chains of the uranyl-interacting residues are highlighted as sticks, while the secondary structures of the protein are shown as ribbons. | ||
| Distance (Å) | IFIE (kcal mol−1) | |||
|---|---|---|---|---|
| Mean | s.d. | Mean | s.d. | |
| Leu304 | 8.79 | 0.73 | −9.6 | 1.4 |
| Thr305 | 7.82 | 0.50 | −10.8 | 2.1 |
| Ala306 | 7.65 | 0.29 | −10.2 | 1.7 |
| Asp307 | 2.19 | 0.04 | −350.1 | 9.9 |
| Phe308 | 2.79 | 0.19 | −26.0 | 13.2 |
| Asp311 | 2.20 | 0.05 | −327.3 | 12.9 |
| Asp313 | 2.24 | 0.06 | −350.3 | 25.0 |
| Val314 | 3.26 | 0.49 | +4.8 | 8.6 |
| Asn317 | 3.61 | 0.77 | −8.6 | 42.0 |
| Phe325 | 3.29 | 0.53 | −6.5 | 1.7 |
In summary, BSA has a binding hot spot where uranyl(VI) ions can be accommodated with high affinity. At this binding site, UO22+ is coordinated by three Asp in its equatorial shell and additionally captured by hydrophobic side chains. Upon photoexcitation of UO22+, Val314 residing in its vicinity is oxidized by [UO22+]* and BSA is cleaved (proteolysis). Apparently, such a binding pattern is peculiar to UO22+–BSA, because such site-specific photocleavage has not been reported in any other uranyl(VI) protein system. Such a unique binding pattern may be adopted in uranyl photocatalysis for C–H activation. In this regard, we have recently designed a new uranyl(VI) binding peptide that also has hydrophobic interaction with Oyl.49 We are also interested in further exploring the importance and relevance of the hydrophobicity of uranyl(VI) oxygen and how it relates to the environmental impact of uranium, including possible U(VI) reduction to U(V) and its stabilization. This will be the subject of our future investigation.
This work was supported by Japan Society for the Promotion of Science (ID: JP20KK0119). MD calculations were performed at the Center for Information Services and High-Performance Computing at Technische Universität Dresden, Germany. FMO calculations were performed on the Fugaku supercomputer provided by the RIKEN Center of Computational Science, Japan, as part of the HPCI System Research Project ID hp220229. YM was supported by Rikkyo University SFR. We thank Ellen Adams (HZDR/TU Dresden) for proofreading the manuscript.
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. E. Cowie, J. M. Purkis, J. Austin, J. B. Love and P. L. Arnold, Chem. Rev., 2019, 119, 10595 CrossRef CAS PubMed.
- N. Behera and S. Sethi, Eur. J. Inorg. Chem., 2021, 95 CrossRef CAS.
- B. Maity, S. Dutta and L. Cavallo, Chem. Soc. Rev., 2023, 52, 5373 RSC.
- S. Tsushima, V. Brendler and K. Fahmy, Dalton Trans., 2010, 39, 10953 RSC.
- S.-B. Tang, S.-Y. Zhang, W.-J. Li, Y.-X. Jiang, Z.-X. Wang, B. Long and J. Su, Org. Chem. Front., 2023, 10, 5130 RSC.
- S. Tsushima, C. Götz and K. Fahmy, Chem. – Eur. J., 2010, 16, 8029 CrossRef CAS PubMed.
- Z. Wang, B. Li, H. Shang, X. Dong, L. Huang, Q. Qing, C. Xu, J. Chen, H. Liu, X. Wang, X.-G. Xiong and Y. Lu, Green Chem., 2022, 24, 7092 RSC.
- I. M. DiMucci, H. D. Root, Z. R. Jones, S. A. Kozimor, M. M. Maclnnes, J. L. Miller, V. Mocko, W. J. Oldham and B. W. Stein, Chem. Commun., 2022, 58, 10961 RSC.
- L. Capaldo, D. Merli, M. Fagnoni and D. Ravelli, ACS Catal., 2019, 9, 3054 CrossRef CAS.
- D. Hu and X. Jiang, Green Chem., 2022, 24, 124 RSC.
- S.-Y. Zhang, S.-B. Tang, Y.-X. Jiang, R.-Y. Zhu, Z.-X. Wang, B. Long and J. Su, Inorg. Chem., 2024, 63, 2418 CrossRef CAS PubMed.
- L. Wu, X. Cao, X. Chen, W. Fang and M. Dolg, Angew. Chem., Int. Ed., 2018, 57, 11812 CrossRef CAS PubMed.
- C.-L. Chen, H.-Y. Wang, Z.-Z. Weng, L.-S. Long, L.-S. Zheng and X.-J. Kong, Inorg. Chem., 2023, 62, 17041 CrossRef CAS PubMed.
- H. D. Burrows and S. J. Formosinho, J. Chem. Soc., Faraday Trans., 1977, 73, 201 RSC.
- K. Müller, et al. , ACS Omega, 2019, 4, 8167 CrossRef PubMed.
- S. Tsuhsima, Inorg. Chem., 2009, 48, 4856 CrossRef PubMed.
- J. G. West, T. A. Bedell and E. J. Sorensen, Angew. Chem., Int. Ed., 2016, 55, 8923–8927 CrossRef CAS PubMed.
- S. Lv, Q. Li, J.-W. Sang, J. Wang and W.-D. Zhang, RSC Adv., 2023, 13, 11929 RSC.
- P. L. Arnold, J. M. Purkis, R. Rutkauskaite, D. Kovacs, J. B. Love and J. Austin, ChemCatChem, 2019, 11, 3786 CrossRef CAS.
- T. Mashita, S. Tsushima and K. Takao, ACS Omega, 2019, 4, 7194 CrossRef CAS.
- K. Takao and S. Tsushima, Dalton Trans., 2018, 47, 5149 RSC.
- L. H. Kristensen, P. E. Nielsen, C. I. Jørgensen, B. B. Kragelund and N. E. Møllegaard, ChemBioChem, 2008, 9, 2377 CrossRef CAS PubMed.
- R. L. B. Elnegaard, N. E. Møllegaard, Q. Zhang, F. Kjeldsen and T. J. D. Jørgensen, ChemBioChem, 2017, 18, 1117 CrossRef CAS PubMed.
- M. R. Duff and C. V. Kumar, Angew. Chem., Int. Ed., 2006, 45, 137 CrossRef CAS PubMed.
- T. Feng, Y. Yuan, S. Zhao, L. Feng, B. Yan, M. Cao, J. Zhang, W. Sun, K. Lin and N. Wang, Angew. Chem., Int. Ed., 2022, 61, e202115886 ( Angew. Chem. , 2022 , 134 , e202115886 ) CrossRef CAS PubMed.
- G. A. Shamov and G. Schreckenbach, J. Phys. Chem. A, 2005, 109, 10961 CrossRef CAS PubMed.
- S. Tsushima, U. Wahlgren and I. Grenthe, J. Phys. Chem. A, 2006, 110, 9175 CrossRef CAS PubMed.
- K. E. Gutowski and D. A. Dixon, J. Phys. Chem. A, 2006, 110, 8840 CrossRef CAS PubMed.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuß, Mol. Phys., 1993, 80, 1431 CrossRef CAS.
- I. Grenthe, X. Gaona, A. V. Plyasunov, L. Rao, W. H. Runde, B. Grambow, R. J. M. Konings, A. L. Smith and E. E. Moore, Second Update on the Chemical Thermodynamics of Uranium, Neptunium, Plutonium, Americium and Technetium, OECD Nuclear Energy Agency Data Bank, OECD Publications, Paris, France, 2020.
- G. Montavon, C. Apostolidis, F. Bruchertseifer, U. Repinc and A. Morgenstern, J. Inorg. Biochem., 2009, 103, 1609 CrossRef CAS PubMed.
- C. Basset, O. Averseng, P.-J. Ferron, N. Richaud, A. Hagege, O. Pible and C. Vidaud, Chem. Res. Toxicol., 2013, 26, 645 Search PubMed.
- A. Rossberg, T. Abe, K. Okuwaki, A. Barkleit, K. Fukuzawa, T. Nakano, Y. Mochizuki and S. Tsushima, Chem. Commun., 2019, 55, 2015 RSC.
- D. M. Martinez, D. Guillaumont and P. Guilbaud, J. Chem. Inf. Model., 2022, 62, 2432 CrossRef PubMed.
- S. O. Odoh, G. D. Bondarevsky, J. Karpus, Q. Cui, C. He, R. Spezia and L. Gagliardi, J. Am. Chem. Soc., 2014, 50, 17484–17494 CrossRef PubMed.
- L. Li, W. Ma, S. Shen, H. Huang, Y. Bai and H. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 31032–31041 CrossRef CAS PubMed.
- T. Lan, H. Wang, J. Liao, Y. Yang, Z. Chai, N. Liu and D. Wang, Environ. Sci. Technol., 2016, 50, 11121–11128 CrossRef CAS PubMed.
- J. R. Milligan, J. A. Aguilera, A. Ly, N. Q. Tran, O. Hoang and J. F. Ward, Nucleic Acids Res., 2003, 31, 6258 CrossRef CAS PubMed.
- D. G. Fedorov, T. Nagata and K. Kitaura, Phys. Chem. Chem. Phys., 2012, 14, 7562 RSC.
- S. Tanaka, Y. Mochuzuki, Y. Komeiji, Y. Okiyama and K. Fukuzawa, Phys. Chem. Chem. Phys., 2014, 16, 10310–10344 RSC.
- K. Fukuzawa and S. Tanaka, Curr. Opin. Struct. Biol., 2022, 72, 127 CrossRef CAS PubMed.
- B. Drobot, M. Schmidt, Y. Mochizuki, T. Abe, K. Okuwaki, F. Brulfert, S. Falke, S. A. Samsonov, Y. Komeiji, C. Betzel, T. Stumpf, J. Raff and S. Tsushima, Phys. Chem. Chem. Phys., 2019, 21, 21213 RSC.
- T. Tokiwa, S. Nakano, Y. Yamamoto, T. Ishikawa, S. Ito, V. Sladek, K. Fukuzawa, Y. Mochizuki, H. Tokiwa, F. Misaizu and Y. Shigeta, J. Chem. Inf. Model., 2019, 59, 25 CrossRef CAS PubMed.
- K. Takaba, C. Watanabe, A. Tokuhisa, Y. Akinaga, B. Ma, R. Kanada, M. Araki, Y. Okuno, Y. Kawashima, H. Moriwaki, N. Kawashita, T. Honma, K. Fukuzawa and S. Tanaka, J. Comput. Chem., 2022, 43, 1362 CrossRef CAS PubMed.
- S. Matsuoka, K. Sakakura, Y. Akinaga, K. Akisawa, K. Okuwaki, H. Doi and Y. Mochizuki, J. Comput. Chem., 2024, 45, 898 CrossRef CAS PubMed.
- M. Pitonak, P. Neogrady, J. Cerny, S. Grimme and P. Hobza, Chem. Phys. Chem., 2009, 10, 282 CrossRef CAS PubMed.
- K. Akisawa, R. Hatada, K. Okuwaki, Y. Mochizuki, K. Fukuzawa, Y. Komeiji and S. Tanaka, RSC Adv., 2021, 11, 3272 RSC.
- W. G. Touw, R. P. Joosten and G. Vriend, J. Mol. Biol., 2016, 428, 1375 CrossRef CAS PubMed.
- S. Tsushima and K. Takao, Phys. Chem. Chem. Phys., 2022, 24, 4455 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc01030b |
| This journal is © The Royal Society of Chemistry 2024 |