
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of sterically congested double helicene by alkyne cycloisomerization†
Junichiro
Hirano
a,
Sayaka
Miyoshi
a,
Eiji
Yashima
 a,
Tomoyuki
Ikai
ab,
Hiroshi
Shinokubo
ac and
Norihito
Fukui
*ab
a,
Tomoyuki
Ikai
ab,
Hiroshi
Shinokubo
ac and
Norihito
Fukui
*ab
aDepartment of Molecular and Macromolecular Chemistry, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8603, Japan. E-mail: fukui@chembio.nagoya-u.ac.jp
bPRESTO, Japan Science and Technology Agency (JST), Kawaguchi, Saitama 332-0012, Japan
cIntegrated Research Consortium on Chemical Science (IRCCS), Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8603, Japan
First published on 17th May 2024
Abstract
Alkyne cycloisomerization of 2,7,10,15-tetra(ortho-alkynylphenyl)benzo[g,p]chrysene containing bulky 4-alkoxy-2,6-dimethylphenyl groups at the alkyne terminals selectively proceeded at the sterically crowded bay-region. The obtained double helicene adopts a distorted structure with a high racemization barrier due to the intramolecular steric repulsion.
Helicene represents a non-planar polycyclic aromatic hydrocarbon composed of ortho-fused benzene rings, which exhibits a distinctive chiral structure resulting from intramolecular steric repulsion between helically arranged fused benzene rings.1–16 This chirality endows helicene with potential applications in chiroptical materials, asymmetric catalysis, supramolecular chemistry, and liquid crystals. To further improve these applications, a rational strategy should be established for finely tuning the structures of parent helicene cores. One representative strategy is the incorporation of substituents at the sterically hindered inner rim, thereby inducing structural distortion and increasing the racemization barrier.17–26 However, the introduction of such steric repulsion is synthetically challenging, heavily depending on photodehydrocyclization, radical cyclization, and Diels–Alder reactions. Even in these cases, the introduced substituents at the inner rim remain relatively small (i.e., methyl and methoxy groups). Furthermore, the introduction of bulky substituents at the inner rims of multiple helicenes, which represent active targets in modern organic synthesis,27–36 remains unexplored.
Alkyne cycloisomerization37–44 is a rational strategy to expand the fused benzene rings in polycyclic aromatic hydrocarbons, which has also been applied to the synthesis of helicenes.45,46 Nonetheless, this methodology necessitates the careful design of precursors to avert the formation of undesired regioisomers and undesirable rearrangements.47 On the other hand, Ikai, Yashima, and co-workers have recently demonstrated that a bulky 4-alkoxy-2,6-dimethylphenyl group at the alkyne terminal circumvents such rearrangements, enabling the precise synthesis of ladder polymers.48 Herein, we describe the applicability of this approach to the synthesis of sterically congested double helicene 1 (Fig. 1). The core structure of 1: benzo[m]naphtho[1,2-c]diphenanthro[1,2-f:1′,2′-s]picene, which represents a π-extended analogue of double [5]helicene 2,29 has not been previously reported.
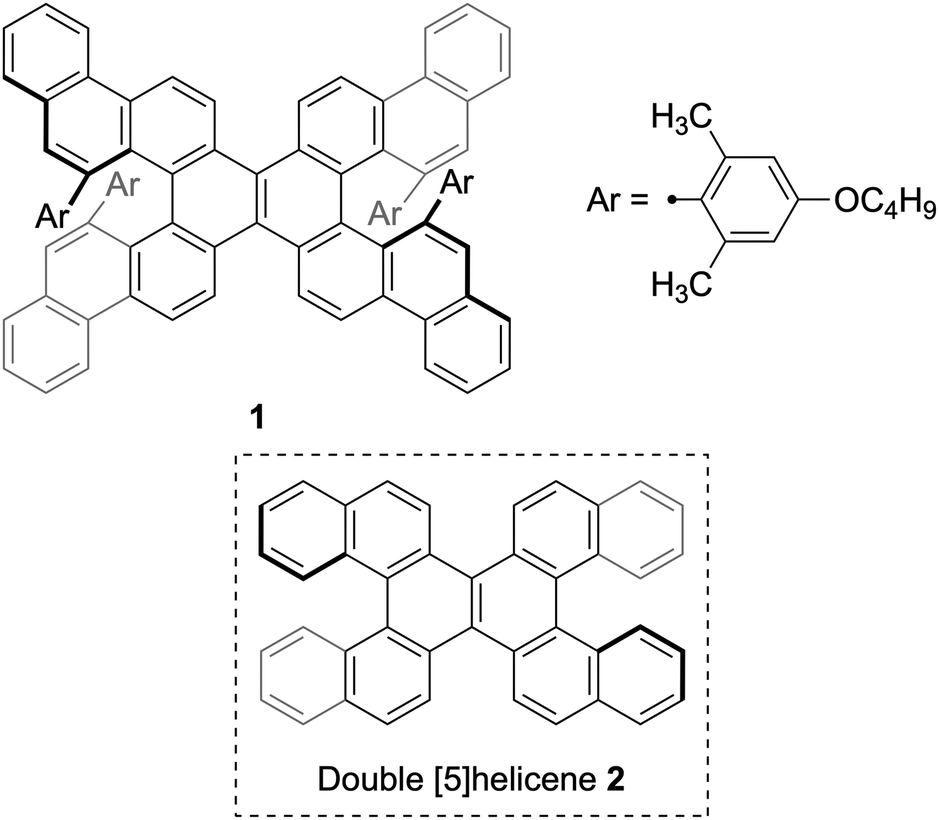 | ||
| Fig. 1 Structures of sterically congested double helicene 1 and double [5]helicene 2. | ||
The synthesis of compound 1 is shown in Scheme 1. The Suzuki–Miyaura cross-coupling reaction of 2,7,10,15-tetrabromodibenzo[g,p]chrysene 349 with ortho-alkynylphenylboronic acid afforded the precursor 4 in 35% yield. Treatment of 4 with trifluoroacetic acid (TFA) furnished the desired double helicene 1 in 31% yield. The 1H NMR analysis of the obtained crude mixture after the cycloisomerization indicates compound 1 is the major product along with trace amounts of side products (Fig. S5, ESI†). Considering that the formation of 1 requires four-fold cycloisomerization, each step proceeds with approximately 75% efficiency. This result indicates that the cycloisomerization reaction selectively proceeded at the 1,8,9,16-positions (bay regions) of the dibenzo[g,p]chrysene unit without undesirable rearrangement. The observed regioselectivity in cycloisomerization can be explained by the distribution of the highest occupied molecular orbital (HOMO) of 4. The MO coefficient of 4′, in which the alkynyl groups are replaced with hydrogens to simplify the calculations, is substantially large at the bay region, while the HOMO has a node at 3,6,11,14-positions (Fig. S13, ESI†). These results suggest that the bay region in 4 is more nucleophilic than 3,6,11,14-positions. A similar trend was noted in phenanthrene derivatives.50
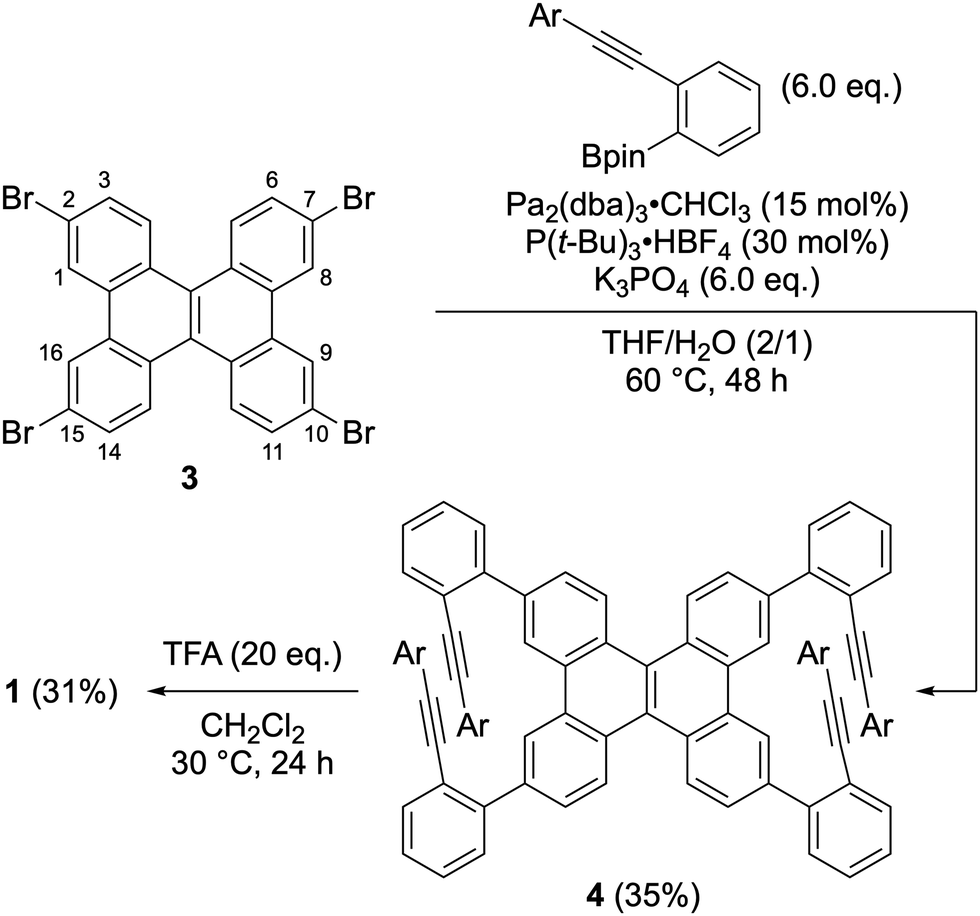 | ||
| Scheme 1 Synthesis of sterically congested double helicene 1. | ||
The molecular structure of compound 1 was confirmed via single-crystal X-ray diffraction analysis (Fig. 2). The space group is P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , wherein the asymmetric unit comprises one molecule, leading to a pair of enantiomers within the unit cell. In the crystal packing, molecules with the same configuration form one-dimensional slipped-stacking array. The vertical distances (d), defined as the centroid of rings A (or E) to the average plane of rings E (or A), range from 3.04 to 3.28 Å. These distances exceed those observed in double [5]helicene 2 (1.80–2.35 Å).29 Moreover, the angles (θ) between rings A and E in compound 1 range from 58 to 66°, which are also larger than those in compound 2 (53–59°). These structural characteristics indicate that the 4-alkoxy-2,6-dimethylphenyl groups of compound 1 increase structural distortion within the double [5]helicene core due to steric congestion.
, wherein the asymmetric unit comprises one molecule, leading to a pair of enantiomers within the unit cell. In the crystal packing, molecules with the same configuration form one-dimensional slipped-stacking array. The vertical distances (d), defined as the centroid of rings A (or E) to the average plane of rings E (or A), range from 3.04 to 3.28 Å. These distances exceed those observed in double [5]helicene 2 (1.80–2.35 Å).29 Moreover, the angles (θ) between rings A and E in compound 1 range from 58 to 66°, which are also larger than those in compound 2 (53–59°). These structural characteristics indicate that the 4-alkoxy-2,6-dimethylphenyl groups of compound 1 increase structural distortion within the double [5]helicene core due to steric congestion.
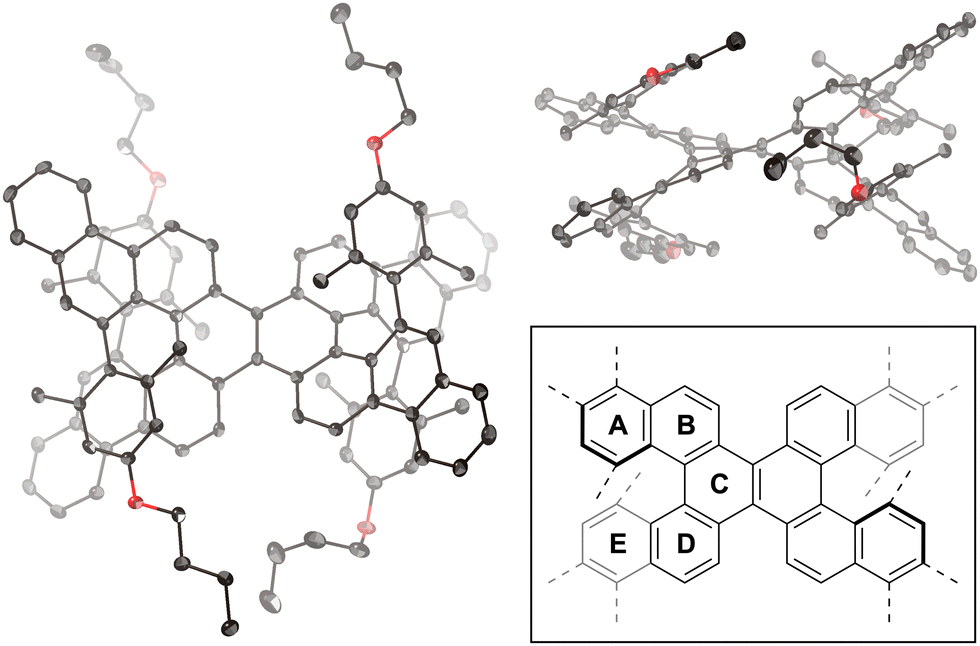 | ||
| Fig. 2 X-ray crystal structure of 1 with thermal ellipsoids at 50% probability; all hydrogen atoms are omitted for clarity. Inset: definition of rings A–E. | ||
The cyclic voltammogram and differential pulse voltammogram of 1 were measured in CH2Cl2 using 0.1 M Bu4NPF6 as the supporting electrolyte and Ag/AgNO3 as the reference electrode (Fig. S10, ESI†). The ferrocene/ferrocenium couple (Fc/Fc+) was used as an external reference. While the reduction wave was not observed under the current conditions, compound 1 exhibited one oxidation wave at 0.56 V.
The enantiomers of 1 were separated using high-performance liquid chromatography (HPLC) equipped with the DAICEL CHIRALPAK IA-3 eluted with 2-propanol/hexane (1/9). The first and second fractions have been assigned to be (P,P)- and (M,M)-enantiomers, respectively, by time-dependent density functional theory (TD-DFT) calculations (vide infra). Heating the enantiomer (M,M)-1 at 180 °C in 1,2,4-trichlorobenzene for 25 h did not cause racemization (Fig. S9, ESI†). The Eyring equation estimates that the racemization barrier of 1 is higher than 40 kcal mol−1, which is larger than the DFT-simulated racemization barrier of double [5]helicene 2 (32 kcal mol−1).29 The high racemization barrier of 1 is attributable to the steric congestion by 4-alkoxy-2,6-dimethylphenyl groups.
The UV/vis absorption and emission spectra of sterically congested double helicene 1 and its precursor 4 are depicted in Fig. 3a. Compound 1 exhibits an intense absorption band at 384 nm with a broad shoulder peak extending to approximately 490 nm. Its fluorescence is observed at 506 nm with a quantum yield of 5.2% and a lifetime of 7.4 ns. These parameters yield radiative and nonradiative decay constants of 7.0 × 106 s−1 and 1.3 × 108 s−1, respectively. The observed weak absorption tail and small radiative decay constant suggest that the S0–S1 transition of 1 is forbidden. Moreover, the absorption spectrum of 1 is red-shifted compared to its precursor 4 and double [5]helicene 229 by approximately 3400 and 1800 cm−1, respectively. These results indicate the effective π-extension in the benzo[m]naphtho[1,2-c]diphenanthro[1,2-f:1′,2′-s]picene core of 1.
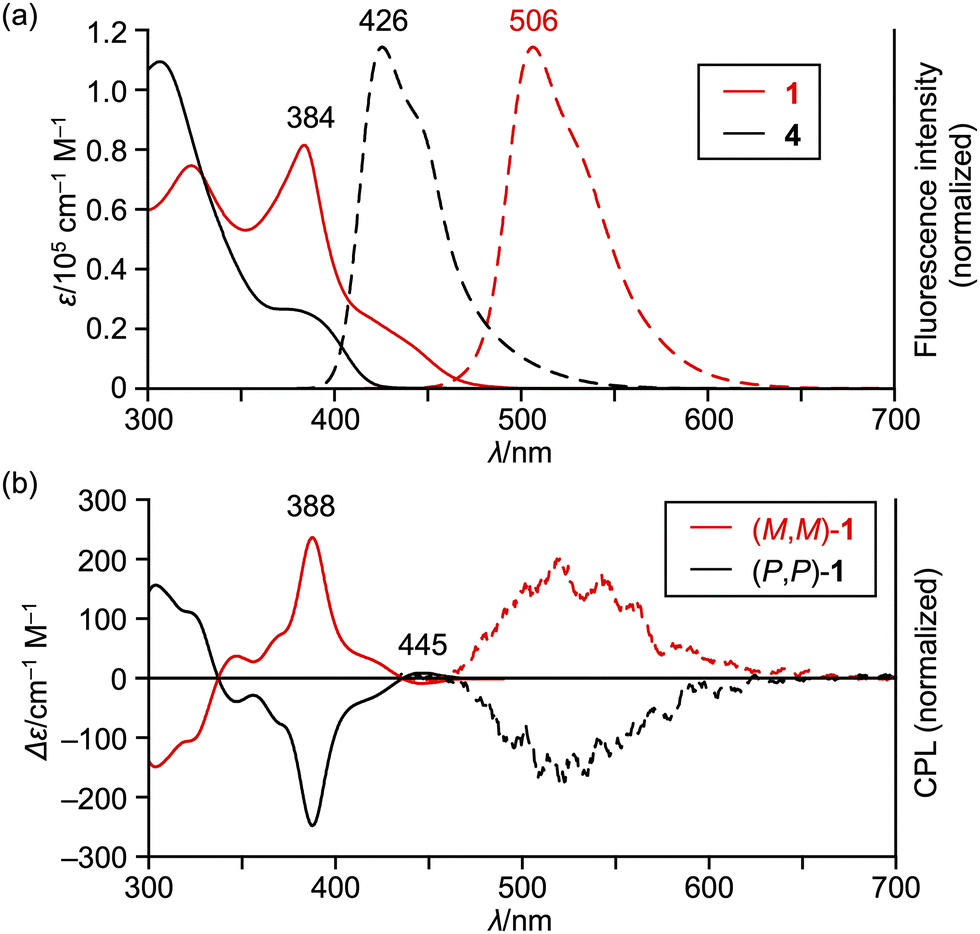 | ||
| Fig. 3 (a) UV/vis absorption (solid lines) and emission (dashed lines) spectra of 1 and 4 in CH2Cl2. (b) CD (solid lines) and CPL (dashed lines) spectra of (M,M)-1 and (P,P)-1 in CH2Cl2. λ = wavelength; ε = extinction coefficient. | ||
The circular dichroism (CD) and circularly polarized luminescence (CPL) of the enantiomers of 1 are shown in Fig. 3b. The CD spectra consist of intense peaks at 388 nm, whose Δε exceeds 200 cm−1 M−1. The first Cotton peaks appear in 430–470 nm, which are positive and negative for the first and second fractions given by HPLC resolution, respectively. The dissymmetry factors |gCD| are 3.1–3.3 × 10−3 at 388 nm and 6.6–7.1 × 10−4 at 445 nm. The CPL spectra of the enantiomers of 1 range from 460 to 650 nm in a reciprocal mirror image. The dissymmetry factors |gCPL| at 506 nm are 6–8 × 10−4, which is comparable to those of previously reported double helicenes.27
To get more insight into the effect of 4-alkoxy-2,6-dimethylphenyl groups in 1, DFT calculations were conducted at the B3LYP/6-31G(d) level using the Gaussian 16 software package (Fig. 4). The optimized structure of 1, whose initial geometry was generated from its crystal structure, exhibits a vertical distance d of 3.18 Å and a dihedral angle θ of 63°, which nicely accord with those of the crystal structure. Then, we conducted the structural optimization for the reference compound 1′, in which the 4-alkoxy-2,6-dimethylphenyl groups are replaced with hydrogen atoms. The vertical distance d and dihedral angle θ of 1′ are 2.31 Å and 51°, respectively. The replacement of 4-alkoxy-2,6-dimethylphenyl groups of 1 with less bulky phenyl groups, namely compound 1′′, affords a vertical distance d of 2.83 Å and a dihedral angle θ of 43°. These parameters are sufficiently smaller than those of 1, indicating that the 4-alkoxy-2,6-dimethylphenyl groups increase the structural distortion. The calculated total energy of (M,M)-1 is smaller than that of the diastereomer: meso-form (P,M)-1, by 7.7 kcal mol−1.
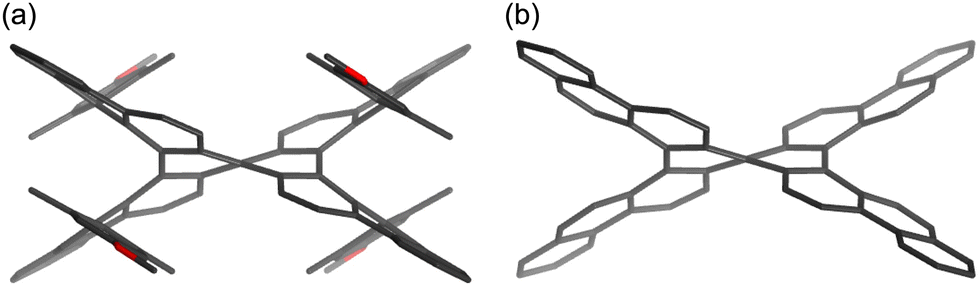 | ||
| Fig. 4 Optimized structures of (a) 1 and (b) 1′. Calculation level: B3LYP/6-31G(d). | ||
We also conducted the TD-DFT calculations for 1 at the B3LYP/6-31G(d) level, which nicely reproduced the absorption and CD spectra (Fig. S14, ESI†). The calculations indicate that the S0–S1 transition of 1 originates from the configuration interaction between [HOMO−1 → LUMO] and [HOMO → LUMO+1] transitions. The S0–S1 transition is forbidden with an oscillator strength of 0.0023, which is consistent with the observed results. Another counterpart given by the configuration interaction is assigned to the intense absorption band at 384 nm, which is an allowed transition with an oscillator strength of 0.4497. The broad peak in 400–460 nm is assigned as the HOMO–LUMO transition, which is slightly blue-shifted compared to the S0–S1 transition by approximately 190 cm−1. The simulated CD spectrum of (M,M)-1 exhibits a weak and negative first Cotton peak, which corresponds to the experimental CD signal of the second fraction given by the HPLC resolution. The calculated g value at the first Cotton peak is −2.9 × 10−3. The angle between the transition electric dipole moment μ and transition magnetic dipole moment m is 180° due to the D2-symmetric structure, which is ideal for affording a high g value.51 However, |μ| value (47 × 10−20 esu cm) is much larger than |m| value (0.034 × 10−20 erg G−1), which should be the reason for the relatively small g value.
In conclusion, we synthesized a sterically congested double helicene 1 through alkyne cycloisomerization of 2,7,10,15-tetra(ortho-alkynylphenyl)benzo[g,p]chrysene 4. This cycloisomerization proceeded with high selectivity at the sterically crowded bay region despite the presence of bulky 4-alkoxy-2,6-dimethylphenyl groups at the alkyne terminals. The 4-alkoxy-2,6-dimethylphenyl groups increase the distortion of the double helicene core, as evidenced by both experimental and theoretical examinations. The large steric repulsion results in the increase of racemization barrier. The core structure of 1: benzo[m]naphtho[1,2-c]diphenanthro[1,2-f:1′,2′-s]picene, represents a new π-extended double [5]helicene derivative, whose physical, photophysical, and chiroptical properties have been systematically explored. The current work expands the scope of alkyne cycloisomerization for the synthesis of distorted helicenes, thus offering important insights for the design of advanced organic materials.
The manuscript was written through the contributions of all authors. All authors have approved of the final version of the manuscript. N. F. designed and conducted the project, wrote the original draft, and finalized the manuscript. J. H. and S. M. carried out all the experiments including the synthesis and characterization. T. I. and E. Y. supervised the alkyne cycloisomerization. H. S. supported the finalization of the manuscript.
This work was supported by JSPS KAKENHI grants JP20H05863, JP20H05867, and JP22K14663 as well as JST, PRESTO grant JPMJPR21Q7 (Japan).
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Gingras, Chem. Soc. Rev., 2013, 42, 968 RSC.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 1007 RSC.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 1051 RSC.
- Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463 CrossRef CAS PubMed.
- W.-L. Zhao, M. Li, H.-Y. Lu and C.-F. Chen, Chem. Commun., 2019, 55, 13793 RSC.
- M. S. Newman and R. M. Wise, J. Am. Chem. Soc., 1956, 78, 450 CrossRef CAS.
- F. Morita, Y. Kishida, Y. Sato, H. Sugiyama, M. Abekura, J. Nogami, N. Toriumi, Y. Nagashima, T. Kinoshita, G. Fukuhara, M. Uchiyama, H. Uekusa and K. Tanaka, Nat. Synth., 2024 DOI:10.1038/s44160-024-00527-3.
- Y. Nakakuki, T. Hirose, H. Sotome, M. Gao, D. Shimizu, R. Li, J.-Y. Hasegawa, H. Miyasaka and K. Matsuda, Nat. Commun., 2022, 13, 1475 CrossRef CAS PubMed.
- Y.-J. Shen, N.-T. Yao, L.-N. Diao, Y. Yang, X.-L. Chen and H.-Y. Gong, Angew. Chem., Int. Ed., 2023, 62, e202300840 CrossRef CAS PubMed.
- G. R. Kiel, H. M. Bergman, A. E. Samkian, N. J. Schuster, R. C. Handford, A. J. Rothenberger, R. Gomez-Bombarelli, C. Nuckolls and T. D. Tilley, J. Am. Chem. Soc., 2022, 144, 23421 CrossRef CAS PubMed.
- S. T. Bao, S. Louie, H. Jiang, Q. Jiang, S. Sun, M. L. Steigerwald, C. Nuckolls and Z. Jin, J. Am. Chem. Soc., 2024, 146, 51 CrossRef CAS PubMed.
- K. Mori, T. Murase and M. Fujita, Angew. Chem., Int. Ed., 2015, 54, 6847 CrossRef CAS PubMed.
- Z. Qiu, C.-W. Ju, L. Frédéric, Y. Hu, D. Schollmeyer, G. Pieters, K. Müllen and A. Narita, J. Am. Chem. Soc., 2021, 143, 4661 CrossRef CAS PubMed.
- S. Oda, B. Kawakami, Y. Yamasaki, R. Matsumoto, M. Yoshioka, D. Fukushima, S. Nakatsuka and T. Hatakeyama, J. Am. Chem. Soc., 2022, 144, 106 CrossRef CAS PubMed.
- J.-K. Li, X.-Y. Chen, Y.-L. Guo, X.-C. Wang, A. C.-H. Sue, X.-Y. Cao and X.-Y. Wang, J. Am. Chem. Soc., 2021, 143, 17958 CrossRef CAS PubMed.
- N. J. Schuster, L. A. Joyce, D. W. Paley, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2020, 142, 7066 CrossRef CAS PubMed.
- M. Nakazaki, K. Yamamoto, T. Ikeda, T. Kitsuki and Y. Okamoto, J. Chem. Soc., Chem. Commun., 1983, 787 RSC.
- R. H. Janke, G. Haufe, E.-U. Würthwein and J. H. Borkent, J. Am. Chem. Soc., 1996, 118, 6031 CrossRef CAS.
- L. Liu, B. Yang, T. J. Katz and M. K. Poindexter, J. Org. Chem., 1991, 56, 3769 CrossRef CAS.
- L. Minuti, A. Taticchi, A. Marrocchi, E. Gacs-Baitz and R. Galeazzi, Eur. J. Org. Chem., 1999, 3155 CrossRef CAS.
- D. C. Harrowven, M. I. T. Nunn and D. R. Fenwick, Tetrahedron Lett., 2002, 43, 3189 CrossRef CAS.
- D. C. Harrowven, M. I. T. Nunn and D. R. Fenwick, Tetrahedron Lett., 2002, 43, 7345 CrossRef CAS.
- D. C. Harrowven, I. L. Guy and L. Nanson, Angew. Chem., Int. Ed., 2006, 45, 2242 CrossRef CAS PubMed.
- Y. Zhang, J. L. Petersen and K. K. Wang, Org. Lett., 2007, 9, 1025 CrossRef CAS PubMed.
- J. Storch, J. Sýkora, J. Čermák, J. Karban, I. Císařová and A. Růžička, J. Org. Chem., 2009, 74, 3090 CrossRef CAS PubMed.
- T. Hartung, R. Machleid, M. Simon, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2020, 132, 5709 CrossRef.
- T. Mori, Chem. Rev., 2021, 121, 2373 CrossRef CAS PubMed.
- C. Li, Y. Yang and Q. Miao, Chem. – Asian J., 2018, 13, 884 CrossRef CAS PubMed.
- H. Kashihara, T. Asada and K. Kamikawa, Chem. – Eur. J., 2015, 21, 6523 CrossRef CAS PubMed.
- T. Fujikawa, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 7763 CrossRef CAS PubMed.
- T. Katayama, S. Nakatsuka, H. Hirai, N. Yasuda, J. Kumar, T. Kawai and T. Hatakeyama, J. Am. Chem. Soc., 2016, 138, 5210 CrossRef CAS PubMed.
- Y. Hu, G. M. Paternò, X.-Y. Wang, X.-C. Wang, M. Guizzardi, Q. Chen, D. Schollmeyer, X.-Y. Cao, G. Cerullo, F. Scotognella, K. Müllen and A. Narita, J. Am. Chem. Soc., 2019, 141, 12797 CrossRef CAS PubMed.
- K. Nakamura, S. Furumi, M. Takeuchi, T. Shibuya and K. Tanaka, J. Am. Chem. Soc., 2014, 136, 5555 CrossRef CAS PubMed.
- Y. Chen, C. Lin, Z. Luo, Z. Yin, H. Shi, Y. Zhu and J. Wang, Angew. Chem., Int. Ed., 2021, 60, 7796 CrossRef CAS PubMed.
- J. Tan, X. Xu, J. Liu, S. Vasylevskyi, Z. Lin, R. Kabe, Y. Zou, K. Müllen, A. Narita and Y. Hu, Angew. Chem., Int. Ed., 2023, 62, e202218494 CrossRef CAS PubMed.
- J. Luo, X. Xu, R. Mao and Q. Miao, J. Am. Chem. Soc., 2012, 134, 13796 CrossRef CAS PubMed.
- M. B. Goldfinger and T. M. Swager, J. Am. Chem. Soc., 1994, 116, 7895–7896 CrossRef CAS.
- M. B. Goldfinger, K. B. Crawford and T. M. Swager, J. Am. Chem. Soc., 1997, 119, 4578–4593 CrossRef CAS.
- H. V. Anderson, N. D. Gois and W. A. Chalifoux, Org. Chem. Front., 2023, 10, 4167 RSC.
- J. Labella, G. Durán-Sampedro, M. V. Martínez-Díaz and T. Torres, Chem. Sci., 2020, 11, 10778 RSC.
- J. Labella, G. Durán-Sampedro, S. Krishna, M. V. Martínez-Díaz, D. M. Guldi and T. Torres, Angew. Chem., Int. Ed., 2023, 62, e202214543 CrossRef CAS PubMed.
- J. Carreras, G. Gopakumar, L. Gu, A. Gimeno, P. Linowski, J. Petuškova, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2013, 135, 18815 CrossRef CAS PubMed.
- W. Yang, A. Lucotti, M. Tommasini and W. A. Chalifoux, J. Am. Chem. Soc., 2016, 138, 9137 CrossRef CAS PubMed.
- W. Yang, G. Longhi, S. Abbate, A. Lucotti, M. Tommasini, C. Villani, V. J. Catalano, A. O. Lykhin, S. A. Vaganov and W. A. Chalifoux, J. Am. Chem. Soc., 2017, 139, 13102 CrossRef CAS PubMed.
- V. Mamane, P. Hannen and A. Fürstner, Chem. – Eur. J., 2004, 10, 4556 CrossRef CAS PubMed.
- K. Fujise, E. Tsurumaki, G. Fukuhara, N. Hara, Y. Imai and S. Toyota, Chem. – Asian J., 2020, 15, 2456 CrossRef CAS PubMed.
- Y. Yu, L. Wang, C. Wang, F. Liu, H. Ling and J. Liu, Small Sci., 2023, 3, 2300040 CrossRef CAS.
- W. Zhang, T. Ikai and E. Yashima, Angew. Chem., Int. Ed., 2021, 60, 11294 CrossRef PubMed.
- M. M. Hossain, M. S. Mirzaei, S. V. Lindeman, S. Mirzaei and R. Rathore, Org. Chem. Front., 2021, 8, 2393 RSC.
- P. Redero, T. Hartung, J. Zhang, L. D. M. Nicholls, G. Zichen, M. Simon, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2020, 59, 23527 CrossRef CAS PubMed.
- H. Kubo, D. Shimizu, T. Hirose and K. Matsuda, Org. Lett., 2020, 22, 9276 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2345868. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc01573h |
| This journal is © The Royal Society of Chemistry 2024 |