
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVPg-based bidirectional synthetic mRNA circuits enable orthogonal protein regulation for high-resolution cell separation†
Zhenghua
Liang
 ,
Yaxin
Hu
,
Cheuk Yin
Li
,
Wai Laam
Yau
,
Kaixin
Tan
and
Yi
Kuang
*
,
Yaxin
Hu
,
Cheuk Yin
Li
,
Wai Laam
Yau
,
Kaixin
Tan
and
Yi
Kuang
*
Department of Chemical and Biological Engineering, Hong Kong University of Science and Technology, Room 5578, Academic Bldg, Clear Water Bay, Kowloon, Hong Kong. E-mail: kekuang@ust.hk
First published on 29th April 2024
Abstract
Synthetic mRNA circuits commonly sense input to produce binary output signals for cell separation. Based on virus-origin cap-independent translation initiation machinery and RBP-aptamer interaction, we designed smart synthetic mRNA-based circuits that sense single input molecules to bidirectionally tune output signals in an orthogonal manner, enabling high-resolution separation of cell populations.
Synthetic mRNA circuits are powerful tools for cell identification and purification.1–5 They can sense various cellular biomarker molecules (e.g., microRNA, protein, ligands) and report the information by altering the expression levels of encoded fluorescent reporter proteins.3–6 Because they promise a temporal cellular effect and no damage to genome integrity, these switches have been widely used to identify and separate cells,7,8 especially stem cell-derived cells, such as neurons and cardiomyocytes.1,2,9 However, current synthetic mRNA circuits mainly turn on or turn off output protein expression upon input-sensing, resolving cell separation in one direction. Fluorescent signal overlap between cell populations is generally observed,3,9 limiting the cell separation resolution. Using multiple one-directional circuits to sense multiple inputs only reduces but can’t solve the low-resolution issue, as each switch still produces significant cell population overlap. Inspired by the use of viral protein to initiate mRNA expression by Nakanishi and coworkers,6,10 we designed a smart strategy that changes the input-output relation simply by altering the cap structures on the synthetic mRNAs. This strategy affords circuits that sense a single input molecule to tune down and tune up different reporter expressions at the same time, bidirectionally creating orthogonal signals that substantially enhance the resolution of cell separation.
To design the circuit, we first examined the cap-independent translation initiation of viral protein genome-linked (VPg) proteins.11–13 Previous works discovered that VPg can act as an alternative to a cap structure for recruiting translation initiation factors in mammalian cells.13 Importantly, our previous works found that expressing a fusion protein of murine norovirus (MNV) VPg with RNA binding protein (RBP) in mammalian cells can efficiently locate the VPg to synthetic mRNA carrying an RBP-binding aptamer.8,14 Such localization of VPg can boost the protein production of synthetic mRNA carrying a translation-deficient cap. We hypothesize that VPg can compete with the cap for recruiting translation initiation factors to override the cap structures for translation initiation. Thus, VPg might produce opposite protein production changes to synthetic mRNAs carrying translation-efficient or deficient caps.
To evaluate this concept, we constructed an EGFP encoding synthetic mRNA carrying either a translational-deficient G(5′)ppp(5′)A RNA Cap (Acap) or a translational-efficient anti-reverse cap analogue (ARCA). An MS2 aptamer was inserted into the 5′ untranslated region (5′-UTR) of the two mRNAs (denoted as Acap-MS2-EGFP and ARCA-MS2-EGFP) for localizing a fusion protein formed by MS2 coat protein and MNV-VPg (denoted as MCP-VPg). The Acap-MS2-EGFP mRNA was transfected into HEK293 cells with gradient concentrations of MCP-VPg mRNA. As shown in Fig. 1A, a positive correlation between the EGFP expression level and the MCP-VPg mRNA concentration was observed, with the plateau of the EGFP level reached at 13.3 ng mL−1 of MCP-VPg mRNA. On the other hand, a negative correlation between EGFP expression level and MCP-VPg mRNA concentration was observed in the case of ARCA-MS2-EGFP mRNA (Fig. 1B). Such a VPg-induced differential expression pattern was also observed in a breast cancer cell line, SK-BR-3, with nearly 40 fold of expressional enhancement on Acap-mRNA and more than 60% expressional repression on ARCA-mRNA (Fig. S1A and B, ESI†). These results implied that VPg differentially affects mRNA translation, suggesting that VPg can override the cap structures for translation initiation.
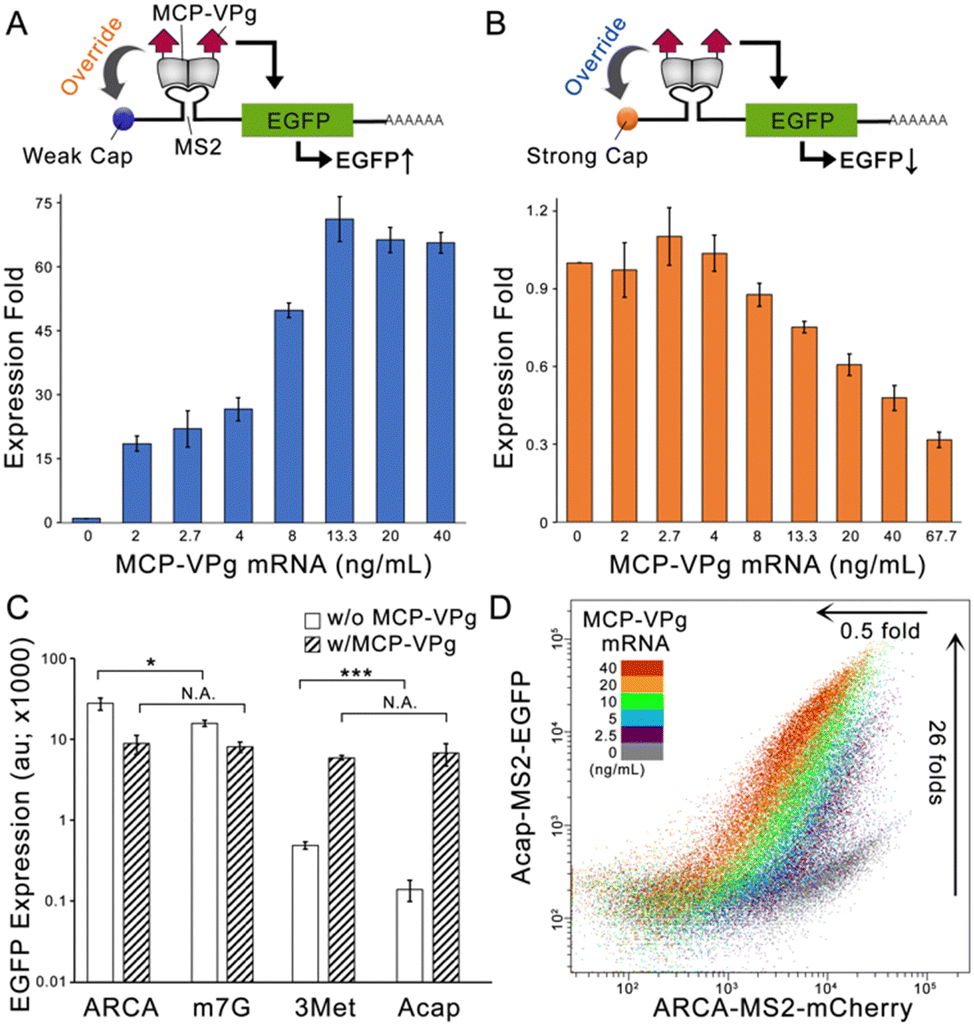 | ||
| Fig. 1 VPg overrides cap structures for translation initiation to produce opposite results on different mRNAs. (A) The scheme and the bar graph showing the MCP-VPg-induced expression enhancement on Acap-MS2-EGFP in HEK293 cells. The expression level of Acap-MS2-EGFP mRNA was set at 1. (B) The scheme and the bar graph showing the MCP-VPg-induced expression repression on ARCA-MS2-EGFP in HEK293 cells. The expression level of ARCA-MS2-EGFP mRNA was set at 1. (C) The expression levels of MS2-EGFP mRNAs carrying different cap structures with or without cotransfection of MCP-VPg mRNA in HEK293 cells. (D) The representative dot plot showing MCP-VPg mRNA-induced bidirectional output regulation in HEK293 cells. N = 3; data are presented as mean ± SD with *P < 0.05, ***P < 0.001 calculated through single-factor analysis of variance (ANOVA). | ||
To validate that the translation interference originates from the localized VPg, we cotransfected the two EGFP mRNAs with either MCP- or VPg-encoding mRNAs (Fig. S1C and D, ESI†). Agreeing with the literature, the cotransfection of MCP mRNA weakly repressed the EGFP signals on both reporter mRNAs, because the RBP-aptamer interaction can partially hinder ribosome entry.3 On the other hand, VPg mRNA cotransfection caused a minor enhancement of Acap-MS2-EGFP expression and a minor repression of ARCA-MS2-EGFP expression. This is likely due to the non-specific VPg interaction with mRNAs in mammalian cells, as demonstrated in previous findings.6 Substantial regulatory effects were only obtained via cotransfection with MCP-VPg, confirming that VPg localization to the 5′UTR is the key to expression regulation. Moreover, we also compared the EGFP expression levels of MS2-EGFP mRNAs carrying different cap structures with and without co-transfection of MCP-VPg mRNA on HEK293. As the four cap structures have different translation initiation abilities, the four MS2-EGFP mRNAs produced different levels of EGFP production (Fig. 1C). Interestingly, MCP-VPg enhanced the EGFP production of two MS2-EGFP mRNAs carrying weak caps (Acap and 3Met [m32.2.7 G(5′)ppp(5′)G]) to similar levels and also repressed the EGFP production of the mRNAs carrying strong caps (ARCA and m7G [m7G(5′)ppp(5′)G]) to similar levels. The same pattern was also observed on HeLa cells (Fig. S1E, ESI†). These results validate that the VPg can override the cap structure for translation initiation, serving as a repressor for strong caps and as an enhancer for weak caps.
To evaluate whether MCP-VPg can simultaneously and orthogonally regulate the expression of reporter mRNAs that carry different caps, we co-transfected two reporter mRNAs: Acap-MS2-EGFP and ARCA-MS2-mCherry with gradient concentrations of MCP-VPg mRNA on HEK293 cells. As shown in Fig. 1D and Fig. S2 (ESI†), with the increase of the MCP-VPg mRNA concentration, a gradual increase of the EGFP signal (maximal enhancement of 26 ± 3 fold) along with a gradient decrease of the mCherry signal (maximal repression of 0.5 ± 0.1 fold) was observed, suggesting that MCP-VPg can simultaneously and orthogonally act on the reporter mRNAs. As Fig. S2D (ESI†) validated that the co-transfection of multiple synthetic mRNAs had little influence on cell viability, these results strongly suggested that VPg-induced translation interference can orthogonally occur on reporter mRNAs with different caps to produce bidirectional output signals.
Next, we designed a microRNA-sensing MCP-VPg mRNA, following the construction of the well-established microRNA-sensing synthetic mRNA switch design.15 In the presence of the target microRNA, the expression level of the MCP-VPg is repressed, leading to a reduced influence of VPg on both reporter mRNAs, which lowers the Acap-capped reporter expression and raises the ARCA-capped reporter expression. miR-21 was chosen as the target microRNA, as it is an established cell-type marker microRNA for HepG2, and HeLa cells2,8,16 and with HEK293 being known to have low miR-21 expression.7,16 To optimize the miR-21 sensitivity, we constructed miR-21-sensing MCP-VPg mRNAs carrying native nucleotides or modified nucleotides that were used to boost the synthetic mRNA performance.8,15 To our surprise, the modified nucleotides failed to increase the effect of miR-21 mimic on the EGFP expression from both reporter mRNAs (Fig. S3A and B, ESI†). Alternatively, we increased the copy number of miR-21 binding sites on MCP-VPg mRNA. Fig. S3C and D (ESI†) show that 5 copies of binding sites could achieve the highest EGFP signal suppression on the Acap-MS2-EGFP reporter mRNAs. A further increase in copy number failed to induce a significant effect on both reporter mRNAs. Therefore, in the following experiments, native nucleotides and 5 copies of microRNA binding sites were used on all miR-sensing MCP-VPg mRNAs.
Confocal imaging (Fig. 2A and Fig. S4A, ESI†) confirmed that the miR-21 mimic can efficiently repress MCP-VPg expression from miR-21-sensing MCP-VPg mRNA (denoted as miR-21-MCP-VPg), resulting in a substantially reduced EGFP expression from Acap-MS2-EGFP (0.032 ± 0.003 fold) and a largely enhanced EGFP expression from ARCA-MS2-EGFP (1.6 ± 0.1 fold) on HEK293 cells. We compared the performance of these VPg-based one-directional circuits to the classical one-directional circuits in the literature (Fig. 2B, C and Fig. S5A, ESI†). The classical off-circuit (Fig. 2B left) directly senses miR-21 to suppress the reporter protein translation. The classical on-circuit (Fig. 2C left) senses microRNA to suppress the expression of MCP. MCP binds to an MS2 dimer to impede EGFP expression. In both comparisons, the VPg-based one-directional circuits produced similar levels of cell population overlap as the classical circuits. These results confirmed that the VPg-based one-directional circuits have comparable cell separation ability to the existing circuits.
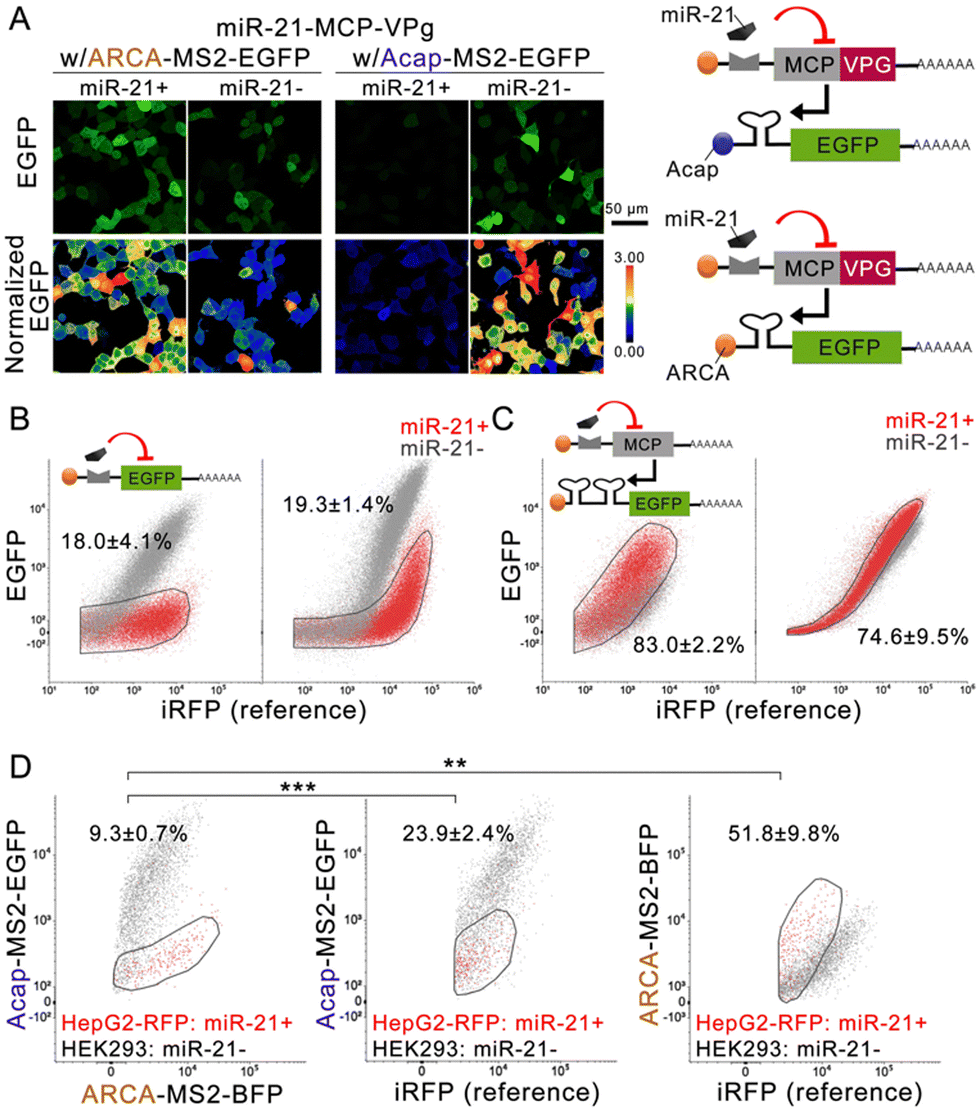 | ||
| Fig. 2 VPg-based bidirectional circuits afford high-resolution cell separation. (A) The schemes and the representative confocal images showing the miR-21 mimic-induced EGFP level change on VPg-based one-directional circuits in HEK293 cells. (B) and (C) The representative dot plots showing cell population separation by classical one-directional circuits (left) and VPg-based one-directional circuits (right) on cells with or without miR-21 mimic. The percentages represent the miR-21− cells located in the gating of the miR-21+ cell population. Cotransfection of iRFP was used as the reference. (D) The representative dot plots showing VPg-based one-directional or bidirectional separation of a mixed culture of the HepG2-RFP and HEK293 cells. The percentages represent the HEK293 cells located in the gating of the HepG2 cell population. Cotransfection of iRFP was used as the reference. N = 3; data are presented as mean ± SD with **P < 0.01, ***P < 0.001 calculated through single-factor analysis of variance (ANOVA). | ||
Next, the miR-21-MCP-VPg mRNA was co-administrated with two reporter mRNAs, carrying different caps and reporter proteins, to form an miR-sensing bidirectional circuit. The circuit was administrated to a cell mixture containing miR-21 negative HEK293 cells and miR-21 positive HepG2/RFP cells (knocked in with an RFP reporter gene) (Fig. 2D and Fig. S5B, C, ESI†). The bidirectional circuit clearly generated orthogonal reporter protein regulation, with the RFP− population producing 10.7 ± 0.8 fold of the EGFP signal and 0.56 ± 0.04 fold of the BFP signal compared with the RFP+ population. Compared with the one-directional circuits, the bidirectional circuit achieved substantially higher separation of the two cell populations (9.3 ± 0.7% overlap). The same experiment was repeated on the induced pluripotent stem cell line (201B7; miR-302 positive) and on the 201B7-derived neural progenitor cell (NPC; miR-302 negative), using a miR-302-sensing circuit. Fig. S4B (ESI†) validated that a 19-fold EGFP signal difference and 0.6-fold BFP signal difference were recorded on NPC, showing a clear separation in both signals, while 201B7 experienced little-to-no regulation by VPg. The data suggested that the circuit can be generalized to other cell-marker microRNAs to achieve cell separation.
To explore whether the VPg-based bidirectional circuit can be flexibly engineered to adapt to different needs, we first replaced the fluorescent reporter genes with two luciferase genes.17 The substantial enzyme activity change by MCP-VPg was recorded (Fig. 3A), indicating that the circuit can be adapted to different reporter genes. Next, the MCP-MS2 pair was replaced by another well-studied PP7 coat protein (PCP) and PP7 aptamer pair.18 Flow cytometry results showed that the PCP-VPg-based circuit also exhibited orthogonal and bidirectional reporter regulation (Fig. 3B and Fig. S5A, ESI†). We then engineered the circuit to sense the protein as input (Fig. 3C). Based on the design of protein-sensing mRNA circuits in the literature,3,4,15 the binding of MCP to the MS2 dimer suppresses the PCP-VPg expression. Differential EGFP expression regulations via the cotransfection of MCP encoding mRNA were recorded on the reporter mRNAs carrying different caps. Lastly, four different reporter mRNAs were employed to form a multi-output circuit. Subcellular localization tags, including the nucleal tag (NL) and the mitochondrial tag (MT), were used on the output proteins to ease signal quantification.19 The four reporter mRNAs were transfected at an equal mass into HEK293 cells with or without the co-transfection of the regulator MCP-VPg mRNA. Fig. 3E shows that the MCP-VPg regulator can simultaneously regulate all four reporter mRNAs in a bidirectional manner, suggesting that the circuit can be used for broad-spectrum protein regulation.
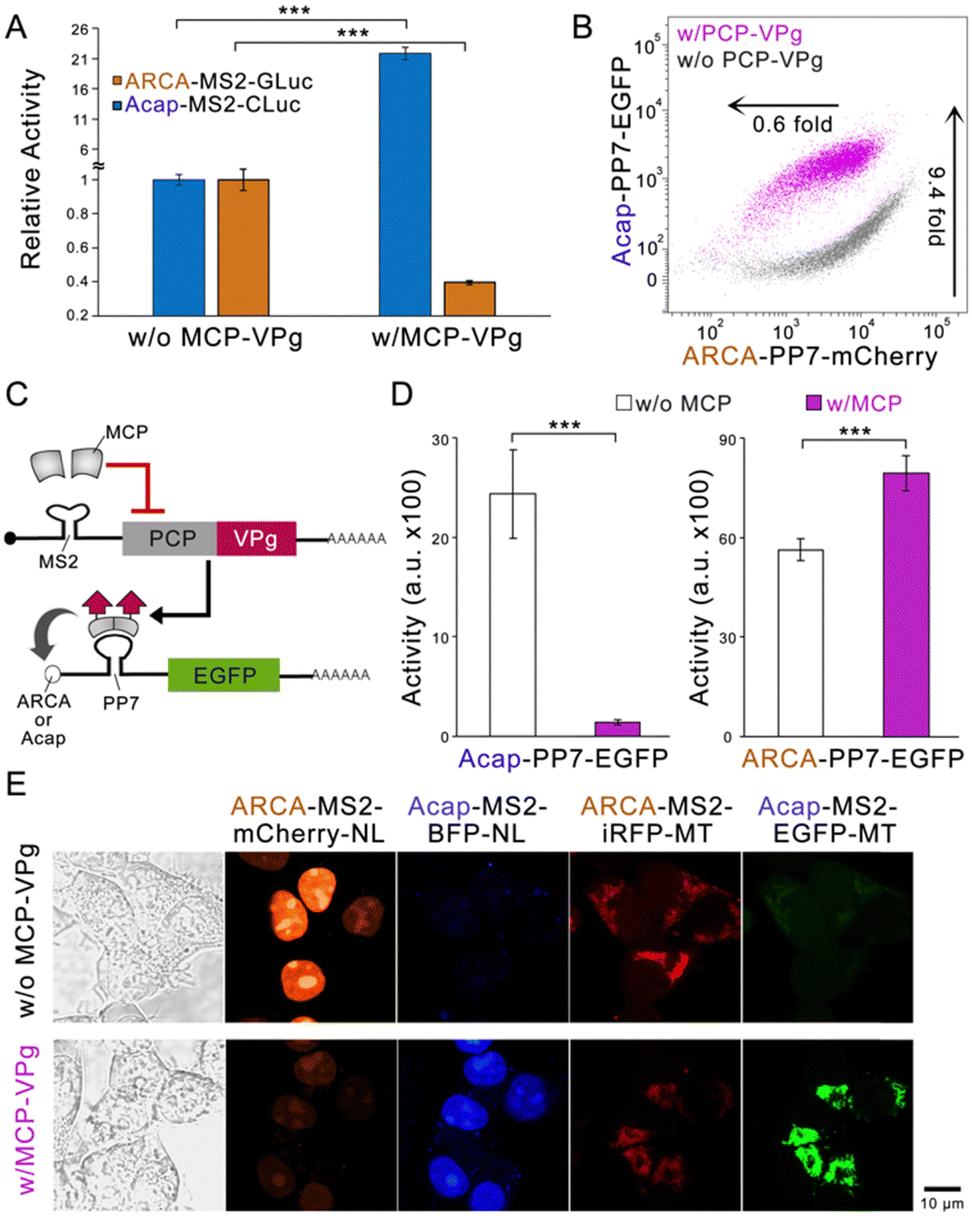 | ||
| Fig. 3 The VPg-based bidirectional circuit is flexible in design. (A) The bar graph showing the circuits bidirectionally regulates Gaussia luciferase (GLuc) and Cypridina luciferase (CLuc) in HEK293 cells. (B) The representative dot plot showing the performance of bidirectional circuits that use the PCP-PP7 pair to localize VPg in HEK293 cells. (C) and (D) The scheme and bar graphs showing the performance of the MCP-sensing circuit. (D) The EGFP regulation of the protein-controlled bidirectional circuits in HEK293 cells. (E) The representative confocal images showing the bidirectional regulation of four fluorescent proteins in HEK293 cells. NL represents the nucleal tag and MT represents the mitochondrial tag. N = 3; data are presented as mean ± SD with **P < 0.01, ***P < 0.001 calculated through single-factor analysis of variance (ANOVA). | ||
In summary, we successfully engineered an orthogonally acting bidirectional circuit based on synthetic mRNAs. Harnessing virus-origin cap-independent translation initiation machinery and RBP-aptamer interaction, the circuit conveys single input molecule information into differential output protein regulation. The merit of this design lies in that the output repression or output enhancement is controlled by the cap structure. Such a simple and effective strategy greatly increases the design flexibility for the circuit, promising further engineering for other applications, such as complex cell perturbation. The bidirectional output signals afford substantially higher resolution cell separation than the current one-directional circuits. The small fraction of the remaining overlapping cell population is likely due to the low transfection rate in these cells. This issue may be solved by engineering advanced transfection reagents for synthetic mRNAs.20–22 This work not only realizes a useful tool for accurate cell separation but also highlights a feasible approach for differential protein regulation, which sheds light on the further development of advanced synthetic mRNA-based circuits.
Yi Kuang: conceptualization, methodology, writing-original draft, visualization, supervision. Zhenghua Liang: conceptualization, methodology, validation, data curation, formal analysis, writing-original draft, visualization. Yaxin Hu: validation, methodology. Cheuk Yin Li: validation, methodology. Wai Lam Yau: validation. Kaixin Tan: validation.
We appreciate Prof. Hirohide Saito for providing the plasmids of EGFP, iRFP670, and MCP. We thank Prof. Xuhui Huang and Bio-CRF at HKUST for providing the HEK293 cell line. We thank the General Research Fund from the Research Grants Council of Hong Kong (16101722) and the Fei Chi En Education and Research Fund.
Conflicts of interest
The authors declare no conflict of interest.References
- Y. Fujita, M. Hirosawa, K. Hayashi, T. Hatani, Y. Yoshida, T. Yamamoto and H. Saito, Sci. Adv., 2022, 8, eabj1793 CrossRef CAS PubMed.
- C. Y. Li, Z. Liang, L. Liu and Y. Kuang, Angew. Chem., 2023, 135, e202306533 CrossRef.
- S. Matsuura, H. Ono, S. Kawasaki, Y. Kuang, Y. Fujita and H. Saito, Nat. Commun., 2018, 9, 4847 CrossRef PubMed.
- L. Wroblewska, T. Kitada, K. Endo, V. Siciliano, B. Stillo, H. Saito and R. Weiss, Nat. Biotechnol., 2015, 33, 839–841 CrossRef CAS PubMed.
- S. Kameda, H. Ohno and H. Saito, Nucleic Acids Res., 2023, 51, e24 CrossRef CAS PubMed.
- H. Nakanishi and H. Saito, Nat. Commun., 2020, 11, 1297 CrossRef CAS PubMed.
- Q. Lu, Y. Hu, C. Yin Li and Y. Kuang, Angew. Chem., Int. Ed., 2022, 61, e202207319 CrossRef CAS PubMed.
- Z. Liang, K. Tan, C. Yin Li and Y. Kuang, Bioorg. Chem., 2024, 144, 107081 CrossRef CAS PubMed.
- C. J. C. Parr, S. Katayama, K. Miki, Y. Kuang, Y. Yoshida, A. Morizane, J. Takahashi, S. Yamanaka and H. Saito, Sci. Rep., 2016, 6, 32532 CrossRef CAS PubMed.
- H. Nakanishi, H. Saito and K. Itaka, ACS Synth. Biol., 2022, 11, 1077–1085 CrossRef CAS PubMed.
- E. N. Leen, F. Sorgeloos, S. Correia, Y. Chaudhry, F. Cannac, C. Pastore, Y. Xu, S. C. Graham, S. J. Matthews, I. G. Goodfellow and S. Curry, PLoS Pathog., 2016, 12, e1005379 CrossRef PubMed.
- L. Chung, D. Bailey, E. N. Leen, E. P. Emmott, Y. Chaudhry, L. O. Roberts, S. Curry, N. Locker and I. G. Goodfellow, J. Biol. Chem., 2014, 289, 21738–21750 CrossRef PubMed.
- E. Royall and N. Locker, Viruses, 2016, 8, 104 CrossRef PubMed.
- K. Tan, Y. Hu, Z. Liang, C. Y. Li, W. L. Yau and Y. Kuang, ACS Synth. Biol., 2023, 12, 2516–2523 CrossRef CAS PubMed.
- C. J. C. Parr, S. Wada, K. Kotake, S. Kameda, S. Matsuura, S. Sakashita, S. Park, H. Sugiyama, Y. Kuang and H. Saito, Nucleic Acids Res., 2020, 48, e35 CrossRef CAS PubMed.
- W. Tian, X. Dong, X. Liu, G. Wang, Z. Dong, W. Shen, G. Zheng, J. Lu, J. Chen, Y. Wang, Z. Wu and X. Wu, PLoS One, 2012, 7, e29551 CrossRef CAS PubMed.
- C. Wu, C. Suzuki-Ogoh and Y. Ohmiya, Biotechniques, 2007, 42, 290–292 CrossRef CAS PubMed.
- M. Jazurek, A. Ciesiolka, J. Starega-Roslan, K. Bilinska and W. J. Krzyzosiak, Nucleic Acids Res., 2016, gkw803 Search PubMed.
- Y. Hu, C. Y. Li, Q. Lu and Y. Kuang, Cell Chem. Biol., 2024, 31, 150–162 CrossRef CAS PubMed.
- K. McCormick, J. Moreno Herrero, H. Haas, S. Fattah, A. Heise, F. J. O’Brien and S.-A. Cryan, Mol. Pharmaceutics, 2024, 21, 1662–1676 CrossRef CAS PubMed.
- J. R. Melamed, S. S. Yerneni, M. L. Arral, S. T. LoPresti, N. Chaudhary, A. Sehrawat, H. Muramatsu, M.-G. Alameh, N. Pardi, D. Weissman, G. K. Gittes and K. A. Whitehead, Sci. Adv., 2023, 9, eade1444 CrossRef CAS PubMed.
- Y. Zeng, M. Estapé Senti, M. C. I. Labonia, P. Papadopoulou, M. A. D. Brans, I. Dokter, M. H. Fens, A. Van Mil, J. P. G. Sluijter, R. M. Schiffelers, P. Vader and A. Kros, ACS Nano, 2023, 17, 23466–23477 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc01725k |
| This journal is © The Royal Society of Chemistry 2024 |