
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simple generation of cleavable labels for multiplexed imaging†
Vincent
Van Deuren
 *a,
Silke
Denis
a,
Robin
Van den Eynde
a,
Jonathan Sai-Hong
Chui
bc,
Francesca
Bosisio
bc,
Frederik
De Smet
cd,
Wim
Dehaen
e,
Wim
Vandenberg
f and
Peter
Dedecker
ag
*a,
Silke
Denis
a,
Robin
Van den Eynde
a,
Jonathan Sai-Hong
Chui
bc,
Francesca
Bosisio
bc,
Frederik
De Smet
cd,
Wim
Dehaen
e,
Wim
Vandenberg
f and
Peter
Dedecker
ag
aLaboratory for Nanobiology, Department of Chemistry, KU Leuven, Leuven, Belgium. E-mail: vincent.vandeuren@kuleuven.be
bTranslational Cell and Tissue Research Unit, Department of Imaging and Pathology, KU Leuven, Leuven, Belgium
cThe Leuven Institute for Single-cell Omics (LISCO), KU Leuven, Leuven, Belgium
dThe Laboratory for Precision Cancer Medicine, Translational Cell and Tissue Research Unit, Department of Imaging and Pathology, KU Leuven, Leuven, Belgium
eLaboratory for Organic Synthesis, Department of Chemistry, KU Leuven, Leuven, Belgium
fLaboratory for Bioimaging, Department of Chemistry, KU Leuven, Leuven, Belgium
gUniversité de Lille, LASIRE CNRS, Lille, France
First published on 13th November 2024
Abstract
The most common methods for multiplexed immunohistochemistry rely on cyclic procedures, whereby cells or tissues are repeatedly stained, imaged, and regenerated. Here, we present a simple and inexpensive approach for amine-targeted labeling of antibodies using a linker that can be easily cleaved by a mild reducing agent. This method requires only inexpensive and readily-available reagents, and can be carried out without synthetic experience in a simple one-pot reaction. We demonstrate the applicability of this approach by performing repeated staining-imaging-removal cycles on isolated cells and tissue sections, finding that over 94% of the labels can be removed within 30 minutes using only the gentle application of reducing agent, increasing up to 99% by extending the incubation duration to 1 hour. By providing a convenient way to introduce cleavable linkers, our method simplifies methodologies such as high-content imaging or multiplexed immunohistochemistry.
Fluorescence imaging is a key method in the life sciences, although the amount of information that can be captured is limited by the number of distinct fluorophores that can be visualized. One way to mitigate this is to distinguish fluorophores not just by their colors, but also based on other properties such as their lifetimes1 or photochemistry,2,3 although these approaches require additional instrumentation or specific probes, and even then the number of fluorophores that can be imaged remains limited.
An alternative approach is to stain and image the same sample multiple times, each time staining with different labels and removing the fluorescence after each imaging step. In principle, this approach allows for the visualization of an unlimited number of structures provided that the sample has been fixed and permeabilized.4 A key challenge lies in the removal of the fluorescence after each imaging round, since on the one hand the stains should bind tightly to the corresponding structure, yet on the other hand the removal should be gentle and not perturb the sample structuring.
Several methods to perform this removal have been proposed, including the removal of the entire stain (e.g. stripping of antibody complexes including the connected fluorophore), inactivation of the fluorophores so that they are no longer fluorescent, or removal of only the fluorophores. The first approach usually requires harsher treatment due to the tight binding of the stain to the target structure. Typical strategies include elevated temperatures, detergents, changes in pH, and denaturing agents.5–7 These procedures are often slow and pose high risks of sample degradation. The second approach can occur via light-induced photodestruction, which is slow, can be applied only to a small region of the sample, and may only yield partial removal of the fluorescence.8 An alternative possibility is the use of reactive molecules such as hydrogen peroxide to degrade the fluorophores, which is typically harsh and works only for a limited number of dyes.9,10
The least perturbing approach is to introduce a chemically-labile link between the fluorophore and targeting moiety, such that it can be cleaved using comparatively mild chemistry. One method is to simply use a non-covalent interaction such as the spontaneous and reversible association of two matching DNA strands. This offers comparatively straightforward and gentle multiplexing, though at the cost of an increase in materials and the complexity associated with keeping track of the proper DNA sequences.11–13 Alternatively, covalent bonds susceptible to cleavage can be engineered into the linker connecting the fluorophore and targeting moiety. One study introduced an azide-based linker that could be cleaved with the mild reducing agent TCEP, although this required high temperatures that can induce sample degradation.14 The synthesis of these linkers was also laborious, low yield, and requires expertise in synthetic chemistry. Another approach made use of a simpler synthesis procedure based on click chemistry, and requires much milder cleavage conditions, but likewise required extensive expertise in synthetic chemistry to develop.15 More recent developments focused on removal kinetics and efficiency, but still required extensive organic synthesis steps16–21 (Table S1, ESI†).
We reasoned that easily-cleavable fluorophore labeling could be achieved in a much simpler way by combining click chemistry with the use of disulfide (S–S) bonds that can be cleaved using mild denaturing agents.22 In this study, we describe a very simple method for the labeling of amine-containing molecules with azide-containing fluorophores such that these are connected with a linker containing a disulfide bond. The method itself requires just a single-pot reaction using inexpensive and commercially-available reagents, thereby omitting the need for expertise in synthetic chemistry. Furthermore, no purification beyond a simple buffer exchange is required. We show that this procedure can be used to obtain fluorescently-labeled antibodies that can be cleaved via the addition of a mild reducing agent and incubation for minutes. Overall, our approach facilitates the development and application of highly-multiplexed fluorescence imaging.
The overall labeling strategy is schematically depicted in Fig. 1. Briefly, the antibody (or another molecule containing amine groups) is mixed with a commercially-available dibenzocyclooctyne-SS-N-hydroxysuccinimidyl ester and an azide-containing fluorophore (or another molecule containing an azide group). The dibenzocyclooctyne (DBCO) moiety selectively reacts with azides via a strain-promoted alkyne-azide cycloaddition, which is fast, copper-free, highly specific, and makes use of stable reagents.23 The N-hydroxysuccinimidyl (NHS) ester is highly reactive towards amine groups and is widely used for the labeling of molecules such as proteins.
 | ||
| Fig. 1 Basic principle of labeling and destaining. Chemical reaction mechanism of antibody labeling and DTT-destaining using a DBCO-SS-NHS linker and azide-fluorophore. | ||
Because the reactions involved are highly specific, the full labeling procedure can be carried out within a single reaction vessel in an overnight incubation step at 4 °C. Unreacted dyes and linkers can then be removed using simple size exclusion chromatography carried out on the bench. Overall, this procedure offers several important advantages: simple synthesis using off-the-shelf reagents, straightforward purification, and a physically small linker that can reduce linkage errors in fields such as super-resolution imaging. A downside, however, is that molecules such as antibodies contain many amine groups, potentially leading to multiple labeling events or labeling events that may interfere with the antibody target recognition.24
To test this approach, we first labeled an antibody against α-Tubulin using both our strategy and a commercial fluorophore-labeling kit that also makes use of NHS-mediated chemistry but does not introduce a cleavable linker. The resulting antibodies were used to stain fixed and permeabilized U2OS cells, revealing the expected cell structuring when visualized using a fluorescence microscope (Fig. 2). We then exposed both samples to 50 mM of the reducing agent dithiothreitol (DTT) in order to cleave the disulfide bond present in the linker, resulting in a fast decrease of the fluorescence in cells stained with antibodies labeled using our approach (Fig. 2a), but not in cells stained with the antibodies labeled using the commercial kit (Fig. 2b). Approximately 94% of the fluorescence could be removed in this way, with about 6% of the fluorescence remaining post-removal. This could be enhanced to about 99% by increasing the incubation time to 1 hour, although complete removal was unattainable with the conditions used here. This limitation also reflects the difficulty assessing this removal in the presence of unavoidable background emission, but may also result from incomplete accessibility of the denaturing agent to the cleavable linkers, reflecting the crowded cellular environment and tight antibody-target interaction. Lower concentrations of DTT resulted in lower fluorescence losses (Fig. S1, ESI†). We then performed a three-cycle staining experiment in which anti α-Tubulin, anti-Vimentin and anti β-Actin antibodies were labeled with AF555 using our protocol (Fig. 3). The cells were first stained with the Vimentin antibody containing the cleavable linker and imaged, followed by treatment with 50 mM DTT and on-stage washout. The staining/destaining procedure was then repeated with anti β-Actin and α-Tubulin respectively. The resulting images faithfully recapitulated the features observed in single-label imaging and did not depend on the order in which the staining was performed (Fig. S2, ESI†). Overall, this approach readily shows the feasibility of staining samples with multiple cycles.
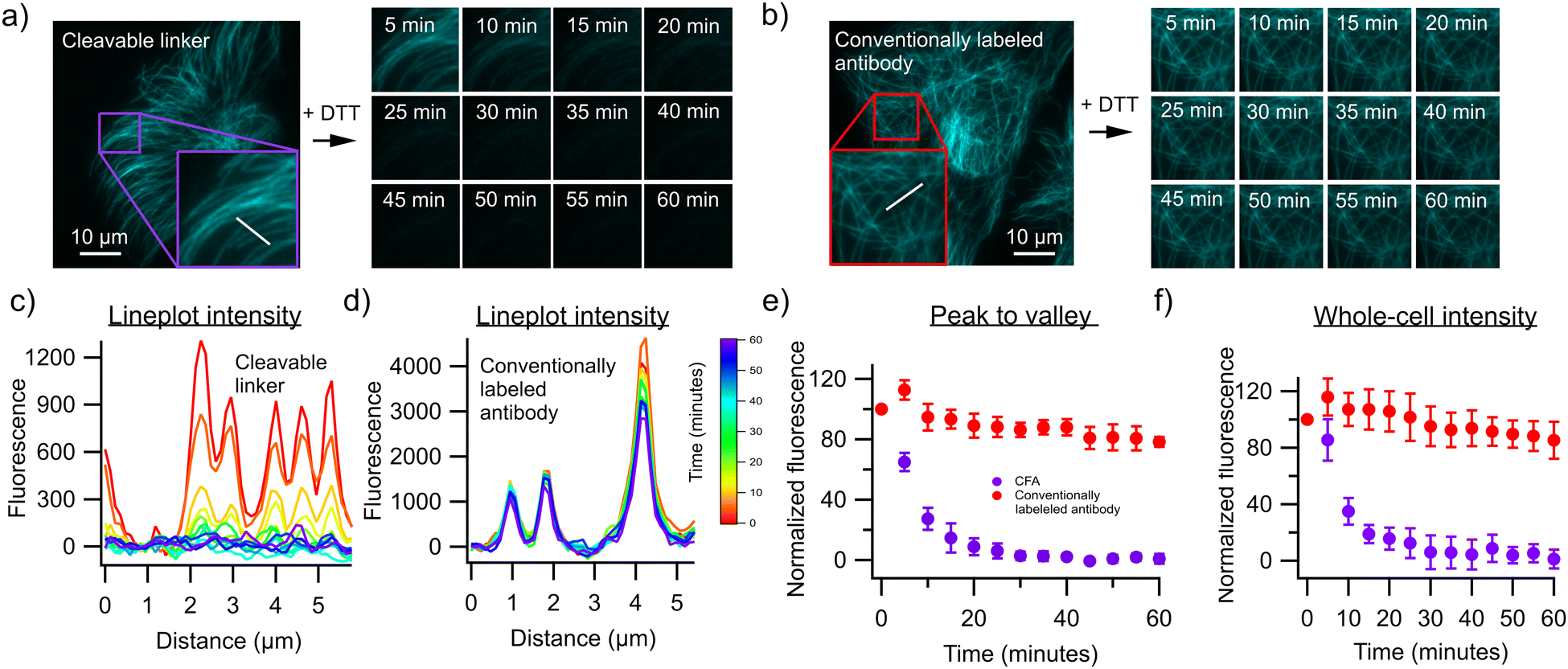 | ||
| Fig. 2 Destaining kinetics and efficiency. (a) Timelapse of tubulin fibers stained with antibodies containing the cleavable linker after DTT treatment. Insets are 9 μm × 9 μm. (b) Timelapse of tubulin fibers stained with conventionally labeled antibodies after DTT treatment. Insets are 13 μm × 13 μm. (c) Lineplots through different tubulin fibers in U2OS cells stained with cleavable fluorescent antibodies. (d) Line plots through different tubulin fibers in U2OS cells stained with conventionally labeled antibodies. (e) Timetraces of peak to valley fluorescence calculated from lineplots of U2OS cells stained with α-Tubulin antibodies containing the cleavable linker and conventionally labeled antibody (n = 5 for each condition, data points on graph are mean values together with the standard deviation). (f) Timetraces of average fluorescence intensity of U2OS cells stained with α-Tubulin antibodies containing the cleavable linker and conventionally labeled antibody (n = 10 for each condition, data points on graph are mean values together with the standard deviation). | ||
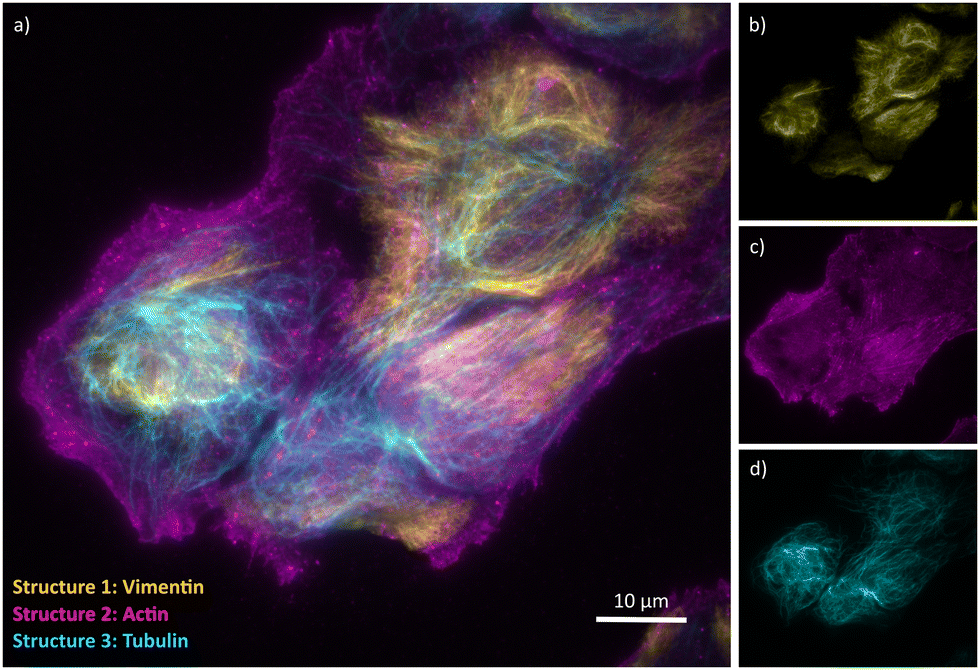 | ||
| Fig. 3 Cyclic staining proof-of-concept. (a) Sequential staining of U2OS cells with three AF555-labeled antibodies using the cyclic immunostaining protocol. Yellow: Vimentin, magenta: β-Actin, cyan: α-Tubulin. (b)–(d) Monochrome panels for each structure. | ||
Next, we expanded the antibody repertoire with two additional markers (KI-67 and CD44) commonly used in tissue immunohistochemistry. These antibodies were validated together with the α-Tubulin antibody on HeLa cells (Fig. S3, ESI†). We then performed a live cell staining/destaining experiment with the CD44 antibody, showing that these can also be used to target the plasma membrane of live cells and cleaved using DTT (Fig. S4, ESI†). The anti KI-67 and CD44 antibodies and the α-Tubulin antibody were then used together with our cyclic staining protocol to stain formalin-fixed paraffin-embedded (FFPE) human glioblastoma sections. The tissue was first stained with the KI-67 antibody containing the cleavable linker and imaged, followed by treatment with 50 mM DTT. The staining/destaining procedure was then repeated with anti α-Tubulin and CD44 respectively (Fig. 4). The resulting image faithfully represents the targeted structures and shows no bleedthrough of signal one cycle from another. In line with expectations, α-Tubulin stained cytoskeletal structures in every cell, KI-67 mostly indicated cycling tumor cells, while CD44 was present in glioblastoma tumor cells and infiltrating macrophages.25
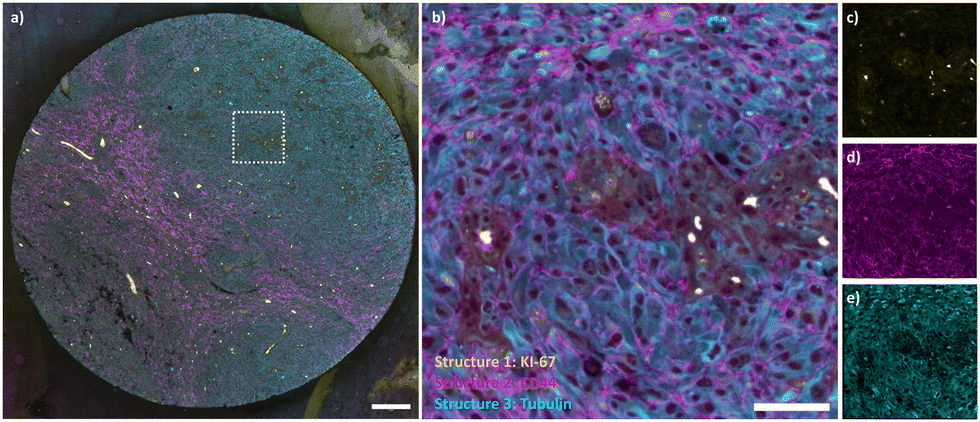 | ||
| Fig. 4 Cyclic tissue staining. (a) FFPE glioblastoma sections stained with three AF555-labeled antibodies using the cyclic immunostaining protocol. Yellow: KI-67, magenta: CD44, cyan: α-Tubulin. Scalebar is 200 μm. (b)–(e) Inset and monochrome panels for each structure. Scalebar is 50 μm. | ||
Lastly, we also verified whether the addition of 50 mM DTT induced additional perturbations of the cellular structure during washout. To do so, we stained actin filaments with a phalloidin-Atto488 conjugate that should not be cleavable with DTT and performed 10 consecutive destaining-imaging cycles with and without 50 mM DTT (Fig. S5, ESI†). The sample structuring was persistent over the imaging cycles, demonstrating that the addition of DTT did not introduce appreciable additional sample degradation.
In summary, we have presented a straightforward and inexpensive approach to label an amine-containing molecule such as an antibody or protein with an azide-containing molecule in such a way that the linker between them can be cleaved with a mild reducing agent. Performing this labeling requires only a one-pot reaction overnight followed by a single purification step such as benchtop size-exclusion chromatography. Antibodies labeled using our method resulted in comparable staining with respect to conventional antibody labeling using non-cleavable linkers, but allowed nearly complete fluorophore removal within minutes upon addition of 50 mM DTT. By providing a straightforward approach for the repeated staining-imaging-stripping cycles inherent in highly multiplexed imaging, our method considerably expands the ease of use and applicability of methods such as high-content imaging and spatial transcriptomics.
VVD: investigation; writing – original draft. SD: investigation; writing – review and editing. RVdE: investigation; visualisation. JSHC: investigation. FB: writing – review & editing. FDS: writing – review & editing; conceptualization (supporting). WD: conceptualization (supporting). WV: writing – review & editing. PD: conceptualization (lead); writing – review & editing; supervision.
VVD thanks the KU Leuven for postdoctoral fellowship PDMt1/22/006. VVD, SD and RVdE thank the Research Foundation-Flanders (FWO) for the fellowships. This work was supported by KU Leuven grant IDN/20/021 OrganADVANCE.
Data availability
Data for this article, including images are available at Zenodo at https://doi.org/10.5281/zenodo.13866826.Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Niehörster, A. Löschberger, I. Gregor, B. Krämer, H.-J. Rahn, M. Patting, F. Koberling, J. Enderlein and M. Sauer, Nat. Methods, 2016, 13, 257–262 CrossRef PubMed.
- T. Roebroek, W. Vandenberg, F. Sipieter, S. Hugelier, C. Stove, J. Zhang and P. Dedecker, Nat. Commun., 2021, 12, 2005 CrossRef PubMed.
- J. Quérard, R. Zhang, Z. Kelemen, M.-A. Plamont, X. Xie, R. Chouket, I. Roemgens, Y. Korepina, S. Albright, E. Ipendey, M. Volovitch, H. L. Sladitschek, P. Neveu, L. Gissot, A. Gautier, J.-D. Faure, V. Croquette, T. Le Saux and L. Jullien, Nat. Commun., 2017, 8, 969 CrossRef PubMed.
- J. W. Hickey, E. K. Neumann, A. J. Radtke, J. M. Camarillo, R. T. Beuschel, A. Albanese, E. McDonough, J. Hatler, A. E. Wiblin, J. Fisher, J. Croteau, E. C. Small, A. Sood, R. M. Caprioli, R. M. Angelo, G. P. Nolan, K. Chung, S. M. Hewitt, R. N. Germain, J. M. Spraggins, E. Lundberg, M. P. Snyder, N. L. Kelleher and S. K. Saka, Nat. Methods, 2022, 19, 284–295 CrossRef PubMed.
- T. Tsujikawa, S. Kumar, R. N. Borkar, V. Azimi, G. Thibault, Y. H. Chang, A. Balter, R. Kawashima, G. Choe, D. Sauer, E. El Rassi, D. R. Clayburgh, M. F. Kulesz-Martin, E. R. Lutz, L. Zheng, E. M. Jaffee, P. Leyshock, A. A. Margolin, M. Mori, J. W. Gray, P. W. Flint and L. M. Coussens, Cell Rep., 2017, 19, 203–217 CrossRef PubMed.
- Y.-G. Park, C. H. Sohn, R. Chen, M. McCue, D. H. Yun, G. T. Drummond, T. Ku, N. B. Evans, H. C. Oak, W. Trieu, H. Choi, X. Jin, V. Lilascharoen, J. Wang, M. C. Truttmann, H. W. Qi, H. L. Ploegh, T. R. Golub, S.-C. Chen, M. P. Frosch, H. J. Kulik, B. K. Lim and K. Chung, Nat. Biotechnol., 2019, 37, 73–83 CrossRef.
- E. Murray, J. H. Cho, D. Goodwin, T. Ku, J. Swaney, S.-Y. Kim, H. Choi, Y.-G. Park, J.-Y. Park, A. Hubbert, M. McCue, S. Vassallo, N. Bakh, M. P. Frosch, V. J. Wedeen, H. S. Seung and K. Chung, Cell, 2015, 163, 1500–1514 CrossRef PubMed.
- K. Holzwarth, R. Köhler, L. Philipsen, K. Tokoyoda, V. Ladyhina, C. Wählby, R. A. Niesner and A. E. Hauser, Cytometry A, 2018, 93, 876–888 CrossRef.
- J.-R. Lin, B. Izar, S. Wang, C. Yapp, S. Mei, P. M. Shah, S. Santagata and P. K. Sorger, eLife, 2018, 7, e31657 CrossRef PubMed.
- M. J. Gerdes, C. J. Sevinsky, A. Sood, S. Adak, M. O. Bello, A. Bordwell, A. Can, A. Corwin, S. Dinn, R. J. Filkins, D. Hollman, V. Kamath, S. Kaanumalle, K. Kenny, M. Larsen, M. Lazare, Q. Li, C. Lowes, C. C. McCulloch, E. McDonough, M. C. Montalto, Z. Pang, J. Rittscher, A. Santamaria-Pang, B. D. Sarachan, M. L. Seel, A. Seppo, K. Shaikh, Y. Sui, J. Zhang and F. Ginty, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11982–11987 CrossRef PubMed.
- F. Schueder, M. T. Strauss, D. Hoerl, J. Schnitzbauer, T. Schlichthaerle, S. Strauss, P. Yin, H. Harz, H. Leonhardt and R. Jungmann, Angew. Chem., Int. Ed., 2017, 56, 4052–4055 CrossRef.
- R. Jungmann, M. S. Avendaño, J. B. Woehrstein, M. Dai, W. M. Shih and P. Yin, Nat. Methods, 2014, 11, 313–318 CrossRef.
- S. Pallikkuth, C. Martin, F. Farzam, J. S. Edwards, M. R. Lakin, D. S. Lidke and K. A. Lidke, PLoS One, 2018, 13, e0203291 CrossRef.
- J. Guo, N. Xu, Z. Li, S. Zhang, J. Wu, D. H. Kim, M. Sano Marma, Q. Meng, H. Cao, X. Li, S. Shi, L. Yu, S. Kalachikov, J. J. Russo, N. J. Turro and J. Ju, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9145–9150 CrossRef.
- J. Ko, M. Wilkovitsch, J. Oh, R. H. Kohler, E. Bolli, M. J. Pittet, C. Vinegoni, D. B. Sykes, H. Mikula, R. Weissleder and J. C. T. Carlson, Nat. Biotechnol., 2022, 1–9 Search PubMed.
- T. Reiber, C. Dose and D. A. Yushchenko, RSC Chem. Biol., 2024, 5, 684–690 RSC.
- Y. Goltsev, N. Samusik, J. Kennedy-Darling, S. Bhate, M. Hale, G. Vazquez, S. Black and G. P. Nolan, Cell, 2018, 174, 968–981.e15 CrossRef.
- J. Ko, M. Wilkovitsch, J. Oh, R. H. Kohler, E. Bolli, M. J. Pittet, C. Vinegoni, D. B. Sykes, H. Mikula, R. Weissleder and J. C. T. Carlson, Nat. Biotechnol., 2022, 40, 1654–1662 CrossRef.
- M. Wilkovitsch, M. Haider, B. Sohr, B. Herrmann, J. Klubnick, R. Weissleder, J. C. T. Carlson and H. Mikula, J. Am. Chem. Soc., 2020, 142, 19132–19141 CrossRef PubMed.
- N. Mougios, E. R. Cotroneo, N. Imse, J. Setzke, S. Rizzoli, N. A. Simeth, R. Tsukanov and F. Opazo, Nat. Commun., 2024, 15, 8771 CrossRef PubMed.
- E. A. Halabi and R. Weissleder, J. Am. Chem. Soc., 2023, 145, 8455–8463 CrossRef.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef.
- E. Kim and H. Koo, Chem. Sci., 2019, 10, 7835–7851 RSC.
- S. Vira, E. Mekhedov, G. Humphrey and P. S. Blank, Anal. Biochem., 2010, 402, 146–150 CrossRef.
- A. R. Pombo Antunes, I. Scheyltjens, F. Lodi, J. Messiaen, A. Antoranz, J. Duerinck, D. Kancheva, L. Martens, K. De Vlaminck, H. Van Hove, S. S. Kjølner Hansen, F. M. Bosisio, K. Van der Borght, S. De Vleeschouwer, R. Sciot, L. Bouwens, M. Verfaillie, N. Vandamme, R. E. Vandenbroucke, O. De Wever, Y. Saeys, M. Guilliams, C. Gysemans, B. Neyns, F. De Smet, D. Lambrechts, J. A. Van Ginderachter and K. Movahedi, Nat. Neurosci., 2021, 24, 595–610 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc01909a |
| This journal is © The Royal Society of Chemistry 2024 |