
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aminoarylation of alkynes using diarylanilines†
Zi
Liu
and
Michael F.
Greaney
 *
*
Dept of Chemistry, University of Manchester, Oxford Rd, Manchester M13 9PL, UK. E-mail: michael.greaney@manchester.ac.uk
First published on 22nd May 2024
Abstract
Intermolecular aminoarylation of alkynes is described, via addition of diarylanilines to alkynes and Smiles–Truce rearrangement. The transformation manipulates the C–N bond of anilines directly, with no requirement for organometallic reagents or transition metal catalysis. The enaminoate products are versatile building blocks for different classes of heterocycles.
The Smiles–Truce rearrangement (STR) is a powerful approach to C–C bond formation that enables arylation under simple, sustainable conditions (Scheme 1A).1 By exchanging an aryl C-heteroatom bond for a C–C bond, functionalised arene and hetero-arene structures can be built efficiently from simple starting materials, with no requirement for precious metal catalysis. The STR gains substantial utility if it is set up as a domino or multi-component coupling process, whereby an initial intermolecular bond formation creates the key reactive intermediate for arene transfer, which can undergo rearrangement to the desired arene in one operation. Some recent examples are shown in Scheme 1, which illustrate different domino STR design approaches in the anionic and radical regimes.2
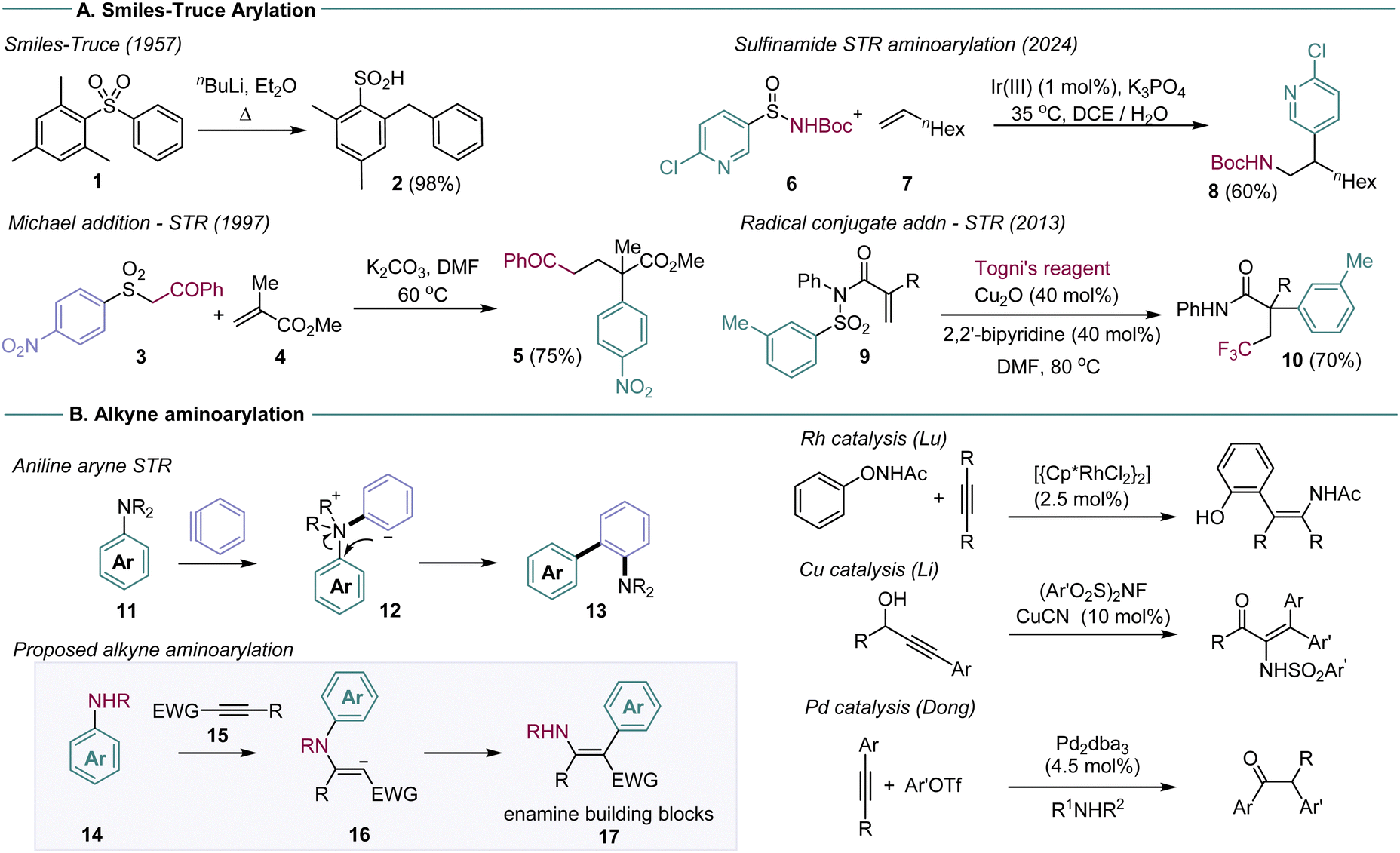 | ||
| Scheme 1 (A). The Smiles–Truce rearrangement (STR) and domino examples. (B) Proposed aminoarylation. | ||
Early work in this area was defined by the sulfonamide functional group, used as the key linkage in the vast majority of domino STR reactions.3 Sulfonamides are easy to prepare, enable versatile aminoarylations in both anionic and radical reaction regimes, and drive the actual rearrangement through irreversible loss of SO2. The weak nucleophilicity of sulfonamides, however, can be problematic for domino reactions that rely on C–N bond formation as the first step. Recent work from the Stephenson and Nevado groups, for example, showed that stereogenic sulfinamides were superior to their sulfonamide analogs for radical alkene aminoarylation.4 Domino STR processes have also been described for sulfones,2a,k,p,ab amides,2w ureas,2e and sulfonates.2z,ac The exploration of alternative STR linkages is in general a productive direction to develop new classes of arylating agents.
We have recently reported that dialkylanilines can undergo domino STR with the reactive intermediate benzyne, to form aminobiaryl compounds 13 (Scheme 1B).5 The reaction enables anilines to be used as arylating agents at the site of the C–N bond, a difficult manipulation outside of high energy diazonium chemistry, with few methods available.6 We were interested in developing this reaction for general alkyne aminoarylation, through reaction with ground state alkynes. Alkyne aminoarylation is a potentially high value transformation, affording versatile enamine products (17), but is restricted by a paucity of viable synthetic methods. Very few examples have been reported in the intermolecular mode: Lu described enamide synthesis using Rh catalysis and N-phenoxyacetamides, and Li has reported the Cu-catalysed addition of NFSI derivatives to make enesulfonamides. The Dong laboratory used Pd catalysis for in situ aminoarylation with amines and aryl halides, affording α-arylketones on work-up.7 Work from our laboratory described a STR approach using metal-free addition of arylsulfonamides to make enaminoates.8 New methods for alkyne aminoarylation are thus required, particularly ones that exploit readily available starting materials (e.g.14 and 15).
A challenge in the planned aminoarylation concerns the four-membered transition state intrinsic to the aniline STR. In our previous aryne system, the exceptional reactivity of benzyne enables capture by tertiary anilines and a subsequent charge-quenching STR from intermediate 12 (Scheme 1B). This substrate design is unlikely to work for ground state alkynes, which are typically unreactive with electron-poor tertiary anilines. We were encouraged, however, by reports of domino sulfonamide SN2/STRs through four-membered transition states, providing some precedent for the idea.9 We set out to investigate secondary anilines that would be nucleophilic enough to undergo conjugate alkyne addition, but containing an electrophilic arene ring that could support the intramolecular SNAr character of the STR.
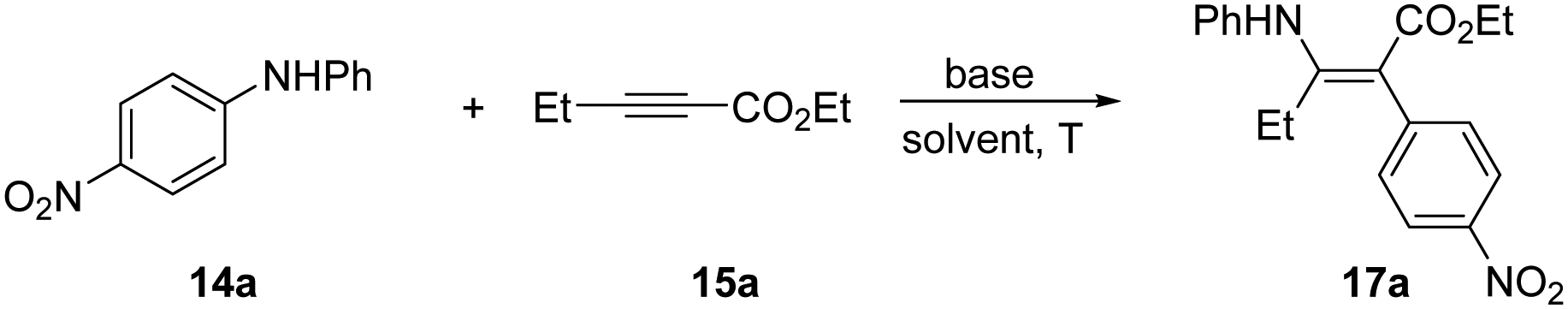
We initially screened a series of secondary N-alkyl anilines with propargylate substrates, and did not observe any reactivity. Moving to diarylanilines, however, did result in a successful STR with N,N-(4-nitrophenyl)phenylaniline (14a) reacting with ethyl pent-2-ynoate (15a) in low conversion. Under conditions of mild base, Cs2CO3, in MeCN at 70 °C we isolated the aminoarylated product 17a as the Z-isomer in 14% (Table 1, entry 1). X-Ray analysis confirmed the Z-geometry,10 in line with the selectivity we have previously observed with sulfonamide nucleophiles.8 The resonance-assisted H-bond11 (δH = 11.3 ppm) present in the Z, but not the E, geometrical isomer likely drives isomerisation in situ.12 A solvent screen established DMA as a better solvent choice delivering 17a in 56% yield (Table 1, entry 3). Further modifications to temperature, stoichiometry and base choice did not advance reaction efficiency, with stronger bases in particular being poor for the reaction. We were pleased to find that conducting the reaction under inert atmosphere supplied the corresponding product 17a in 73% overall yield (Table 1, entry 13).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Ratio (14a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15a:base) :15a:base) |
Solvent | T (°C) | Yield (%) |
| a The reaction was performed under N2. | |||||
| 1 | Cs2CO3 | 1.2:1.0:1.5 |
MeCN | 70 | 14 |
| 2 | Cs2CO3 | 1.2:1.0:1.5 |
DMSO | 70 | 19 |
| 3 | Cs2CO3 | 1.2:1.0:1.5 |
DMA | 70 | 56 |
| 4 | Cs2CO3 | 1.2:1.0:1.5 |
DMA | 90 | 24 |
| 5 | Cs2CO3 | 1.2:1.0:1.5 |
DMA | 50 | 19 |
| 6 | Cs2CO3 | 1.5:1.0:1.5 |
DMA | 70 | 11 |
| 7 | Cs2CO3 | 1.0:1.0:1.5 |
DMA | 70 | 18 |
| 8 | Cs2CO3 | 1.0:1.2:1.5 |
DMA | 70 | 27 |
| 9 | K2CO3 | 1.2:1.0:1.5 |
DMA | 70 | ND |
| 10 | KOH | 1.2:1.0:1.5 |
DMA | 70 | 34 |
| 11 | t BuOK | 1.2:1.0:1.5 |
DMA | 70 | 19 |
| 12 | NaH | 1.2:1.0:1.5 |
DMA | 70 | 13 |
| 13a | Cs2CO3 | 1.2:1.0:1.5 |
DMA | 70 | 73 |
With optimal conditions in hand, we screened a variety of differentially substituted diarylanilines reacting with 15a (Scheme 2). A broad range of electron-deficient or electron-rich substituents on the phenyl ring at different positions (meta- or para-) were all tolerated under the mild reaction conditions, furnishing the corresponding rearrangement products 17 with yields up to 87%. Substrates encompassing strongly electron-withdrawing groups (17b–17g), including nitro, nitrile, acetyl, ester, trifluoromethyl and pyridyl, could be successfully incorporated in this process.
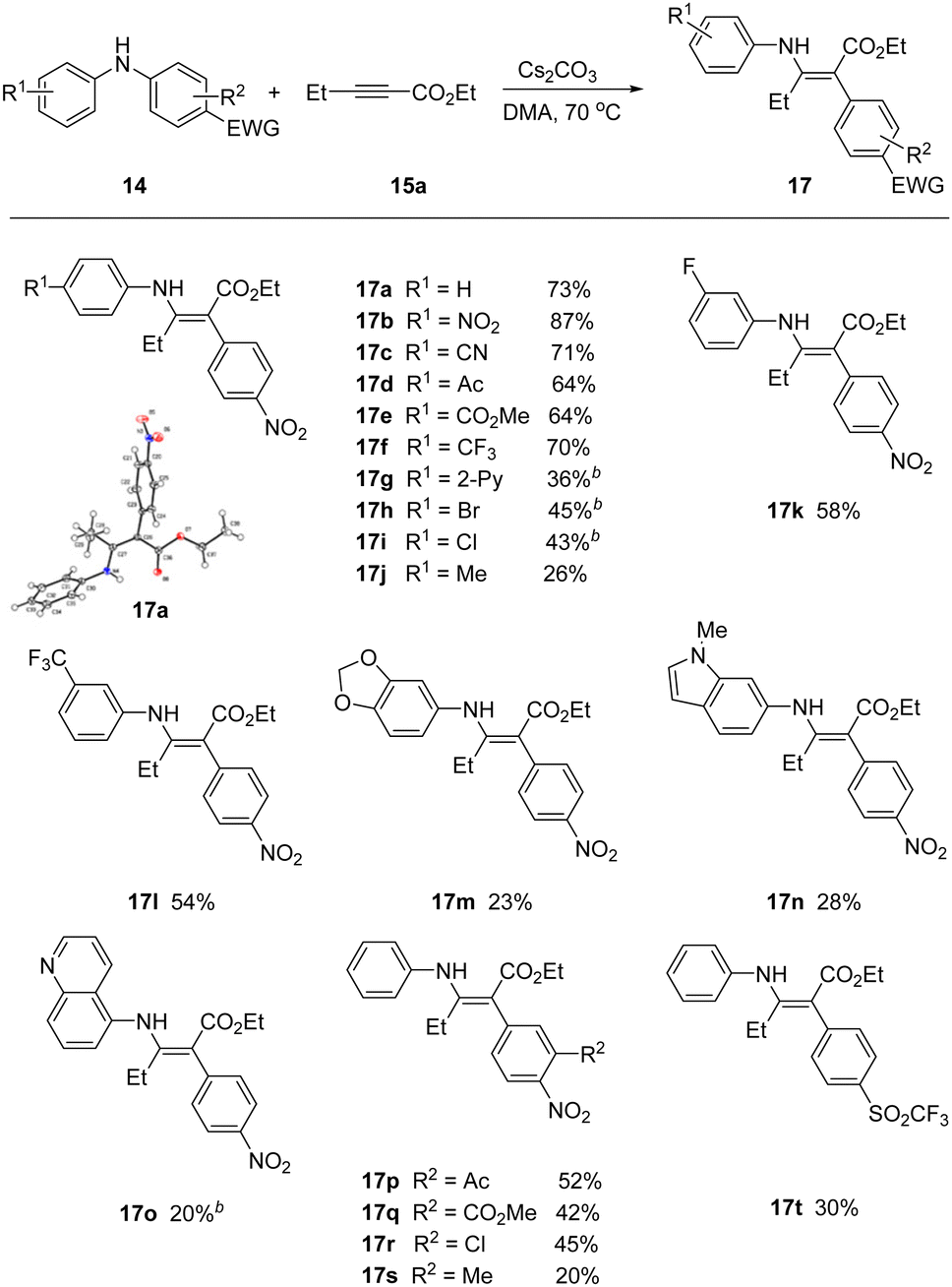 | ||
| Scheme 2 Scope of anilines. a14 (1.2 equiv.), 15a (1.0 equiv.) and Cs2CO3 (1.5 equiv.) in DMA at 70 °C; busing KOH as base. | ||
The rearrangement products 17h, 17i and 17r with halogen groups, providing the opportunity for further functionalization, were obtained in moderate yields 43%, 45% and 45%, respectively. More electron rich arenes were viable, but in reduced yields. For example, tolyl and the piperonyl, indolyl, and isoquinoyl heteroaryl anilines all participated, but in attenuated yields. The migratory aryl group required at least one strong electron withdrawing group, but additional substituents could be installed in the flanking position (17p–17s). We were able to successfully migrate the trifluoromethanesulfonyl derivative (17t), in place of the activating nitro group.
We next inspected the generality of alkyne substrates by employing bis(4-nitrophenyl)amine 14b as model substrate (Scheme 3). Different alkyl substituted alkynyl ester derivatives reacted smoothly, gving the corresponding products 17u–17x in 34–81% yields. We could link the common biologically relevant molecules L-menthol, (−)-borneol, α-tocopherol, ergocalciferol, and stigmasterol through the ester moiety, incorporating these moieties into the enaminoate products 17y–17C. Alkynes bearing different electron-withdrawing groups, such as thioester and ketone, were also feasible in this reaction, affording products 17D and 17E in reduced yields of 39% and 22%.
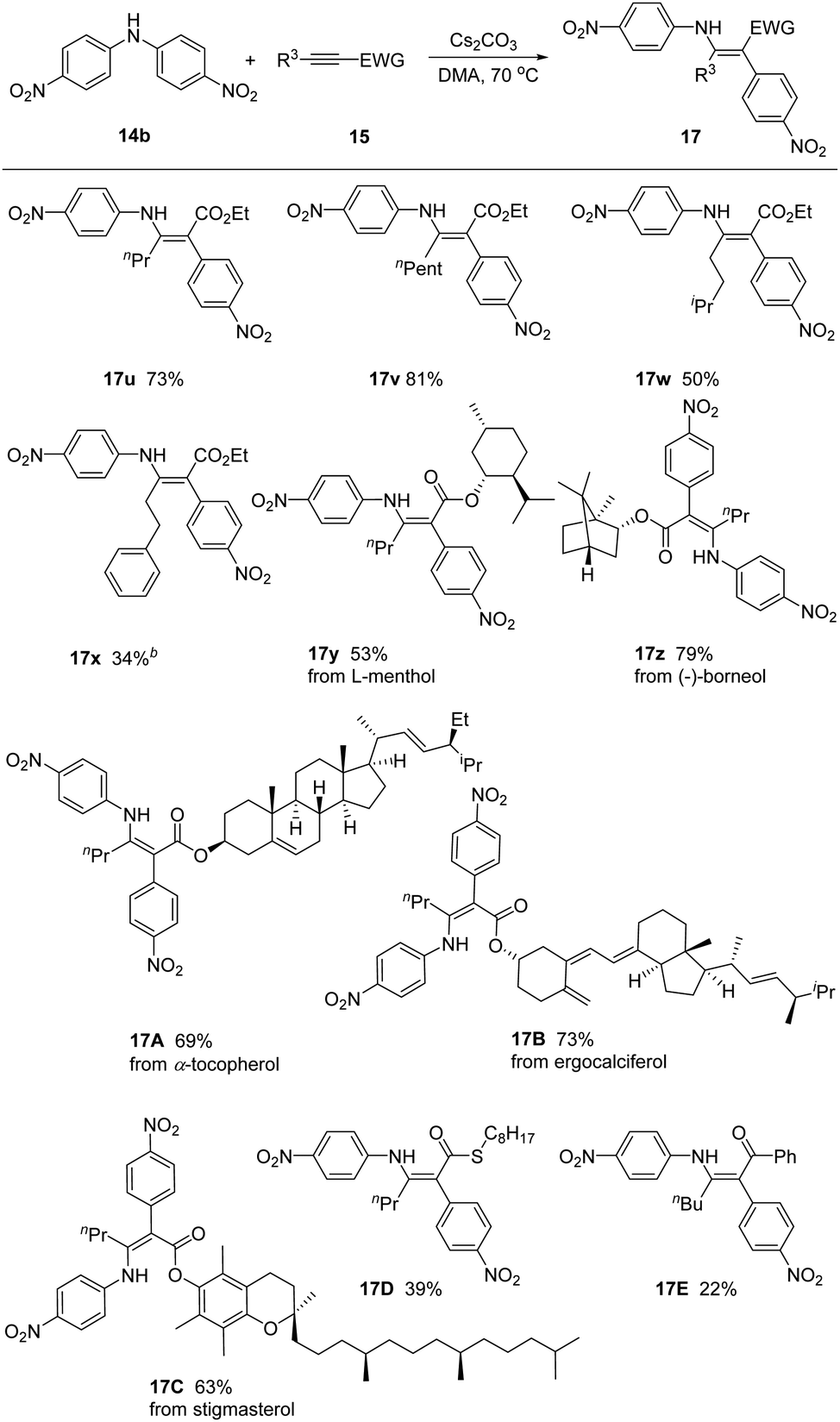 | ||
| Scheme 3 Scope of alkynes. a14b (1.2 equiv.), 15 (1.0 equiv.) and Cs2CO3 (1.5 equiv.) in DMA at 70 °C. | ||
To demonstrate the practicality of this method for harnessing aniline arylation, we conducted a series of transformations on the enaminoate 17a (Scheme 4). We could access indoline and pyrazolone heterocycles 18 and 19 through treatment with iodine, and p-methoxyphenylhydrazine hydrochloride, respectively.
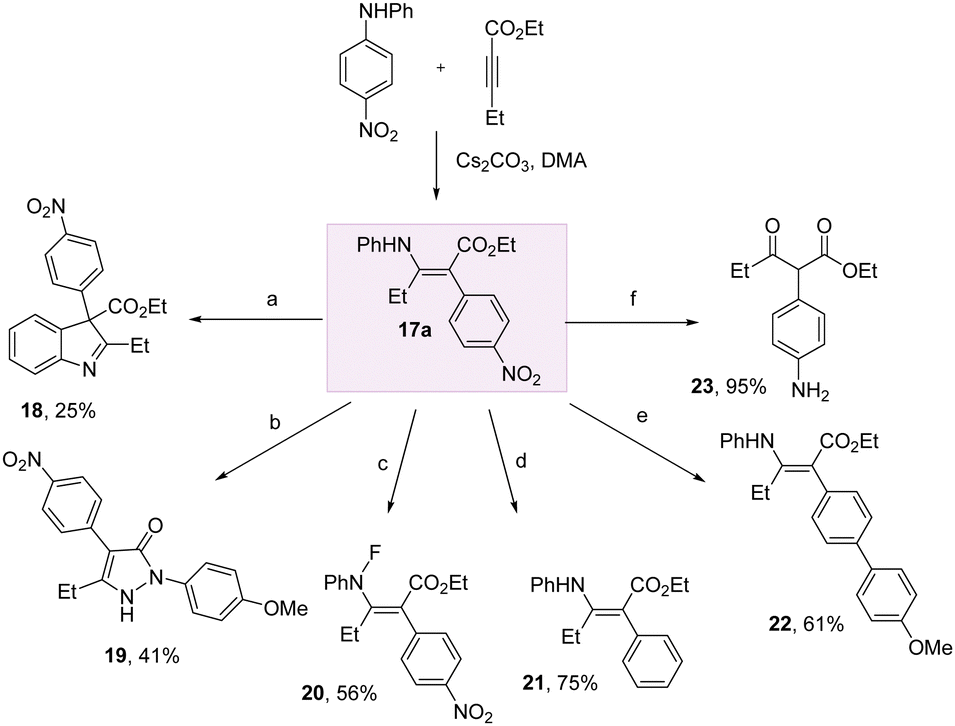 | ||
| Scheme 4 Enaminoate transformations. aI2, K2CO3, DMF, 100 °C, 1 h; b4-methoxyphenylhydrazine hydrochloride, EtOH, reflux, 16 h; cselectfluor, DCM/H2O, r.t., 4 days; dPd(acac)2, Brettphos, K3PO4, iPrOH, 1,4-dioxane, 130 °C, overnight; ePd(acac)2, Brettphos, K3PO4, 18-crown-6, (4-methoxyphenyl)boronic acid, 1,4-dioxane, 130 °C, 16 h; fFe, NH4Cl, EtOH/H2O, 60 °C, 12 h. | ||
Fluorination of the N–H bond of 17a with selectfluor in DCM/H2O successfully delivered 20 in 56% yield. The nitro group could be removed entirely to give the phenyl derivative 21 using Nakao's reductive palladium method. Likewise, a nitro-Suzuki was successful to give the biaryl 22 in 61% yield. The electron-poor nitroarene could be easily transformed into the electron rich aniline using Fe/NH4Cl in EtOH/H2O at 60 °C, with concomitant hydrolysis of the enamine to give the keto-ester 23.
In conclusion, we have developed an intermolecular aminoarylation of alkynes using anilines. The reaction allows cheap aniline building blocks to be used as arylating agents for a range of enamine syntheses, with the products directed to diverse heterocyclic products. Further applications of this process are underway in our laboratory.
We thank the Leverhulme Trust for funding (LT RPG-2021-281). Dr Avantika Hasija (University of Manchester) is thanked for X-Ray crystallographic analysis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Reviews:
(a) A. R. Allen, E. A. Noten and C. R. J. Stephenson, Chem. Rev., 2022, 122, 2695–2751 CrossRef CAS PubMed
; (b) D. M. Whalley and M. F. Greaney, Synthesis, 2022, 1908–1918 CAS
- Selected examples of tandem STRs:
(a) Y. Guoren and Z. Zheng, Synth. Commun., 1997, 27, 1455–1463 CrossRef
- X.-Q. Chu, D. Ge, Y.-Y. Cui, Z.-L. Shen and C.-J. Li, Chem. Rev., 2021, 121, 12548–12680 CrossRef CAS PubMed
-
(a) E. A. Noten, C. H. Ng, R. M. Wolesensky and C. R. J. Stephenson, Nat. Chem., 2024, 16, 599–606 CrossRef CAS PubMed
- T. Sephton, A. Charitou, C. Trujillo, J. M. Large, S. Butterworth and M. F. Greaney, Angew. Chem., Int. Ed., 2023, 62, e202310583 CrossRef CAS PubMed
-
(a) S. B. Blakey and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 6046–6047 CrossRef CAS PubMed
-
(a) G. Liu, Y. Shen, Z. Zhou and X. Lu, Angew. Chem., Int. Ed., 2013, 52, 6033–6037 CrossRef CAS PubMed
- P. T. G. Rabet, S. Boyd and M. F. Greaney, Angew. Chem., Int. Ed., 2017, 56, 4183–4186 CrossRef CAS PubMed
-
(a) V. Lupi, M. Penso, F. Foschi, F. Gassa, V. Mihali and A. Tagliabue, Chem. Commun., 2009, 5012–5014 RSC
- Deposition number 2342281† contains the supplementary crystallographic data for compound 17a.
- P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2000, 122, 10405–10417 CrossRef CAS
- H. Bai, H. Zhang, Y. Guo, H. Chen, D. Wei, S. Li, Y. Zhu and W. Zhang, Org. Chem. Front., 2019, 6, 125–133 RSC
Footnote |
| † Electronic supplementary information (ESI) available: All preparative procedures, characterisation data, and NMR spectra. CCDC 2342281. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc01935k |
| This journal is © The Royal Society of Chemistry 2024 |