
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiscale characterizations of structural evolution in mesoporous CeO2†
Tianyu
Li
 and
Efrain E.
Rodriguez
*
and
Efrain E.
Rodriguez
*
Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742-2115, USA. E-mail: efrain@umd.edu
First published on 24th June 2024
Abstract
In situ ultra-small-angle and wide-angle X-ray scattering enables simultaneous tracking of the structural parameters of mesoporous CeO2 from the atomic scale to the micron-size scale. This multiscale approach provides a path to better understand structure–property relationships in mesoporous polycrystalline materials under dynamic conditions such as high temperature cycling.
Just as describing the structure of a protein necessitates experiments at multiple length scales, elucidating the structure of a functional material at different length scales, from the atomic to the macroscopic, is also required to understand its properties and performance. For example, the atomic structure, particle morphology and packing manner of electrode materials can all influence Li-ion battery performance.1–3 In addition, the structures at different scales might also influence each other. For example, the size of the particles or grains, which are at the micron or nano scale, might determine the atomic structure of the material.4,5
Mesoporous materials illustrate this problem of multiple length scales since their performance is affected by the atomic structure constituting the pore walls, the specific pore topology, and finally the particle morphology. Mesoporous metal oxides are found in a wide variety of applications and fields such as heterogeneous catalysis and rechargeable batteries due to their high surface areas and favorable mass transport characteristics.6,7 Nowadays, it is quite routine to characterize their structures at different length scales via ex situ methods. However, when those mesoporous materials are utilized in dynamic process (e.g., charging/discharging in a battery or a catalytic cycle at elevated temperatures), their structures undoubtedly change, thereby altering the material's performance. In that case, ex situ experiments become less relevant to evaluate performance under real working conditions. To address such a shortcoming, researchers have developed multiple in situ structural characterization techniques.8,9 Though it remains a challenge to perform simultaneous, multilength scale structural analyses in situ, such experiments should yield unprecedented insight into the complex interrelations among various features of functional material at different scales.
Due to their high importance in heterogenous catalysis, the materials we explored in the study are mesoporous ceria (CeO2) in the SBA-15 and KIT-6 structures prepared via nanocasting.10 The structure of mesoporous CeO2 needs to be described at different scales. At the angstrom-scale, Ce and O atoms order via ionic bonds to form a crystalline phase with the fluorite-type structure. At the nanoscale, CeO2 crystallites form nano-size grains that are further “stacked” into the ordered mesoporous structure. Finally, at the microscale, the mesoporous structures grow large enough to form near-micron-sized particles. These different scales can be characterized in a traditional, ex situ fashion. Indeed, we show such results for our mesoporous CeO2 as independent verification such as powder X-ray diffraction, small angle X-ray scattering and transmission electron microscopy (the ex situ TEM are displayed in Fig. S1 and S2 (ESI†) to confirm the multi-scale structures of our CeO2 samples).
In this communication the main experiment that goes beyond the ex situ methodology is the in situ scattering under heating conditions of our CeO2 samples from atomic scale to micron scale. An ultra-small-angle X-ray scattering (USAXS) instrument with a wide angle scattering (WAXS) capability provides an extreme range in length scales,11,12 ideal for simultaneous structural characterization of mesoporous materials. To this end, we utilized a synchrotron X-ray based USAXS instrument equipped with WAXS detectors on beamline 9-ID at the advanced photon source (Argonne National Laboratory). The Q-range of the combined USAXS/WAXS is 8 × 10−5 Å−1–6 Å−1, which gives characteristic scales ranging from 6 × 10−6 m to 1 × 10−10 m, i.e., four orders of magnitude, in real space. This broad range is extensive enough to cover the nanostructures and microstructures of the studied materials.
We show one isotherm of the in situ USAXS and WAXS heating experiment in Fig. 1. We first demonstrate in the SBA-15 templated mesoporous CeO2 how the different regions of the USAXS/WAXS data correlate to the real space structures. At the extremely low Q region of the USAXS data (Q < 0.001 Å−1), the curve reflects the micro-scale to meso-scale (>100 nm) structure information of the mesoporous material particles. By fitting a simple unified model (Fig. 1a) (composed of a Guinier part and a power law tail),13,14 the radius of gyration (Rg) of the particle can be estimated and should be comparable to the particle size.
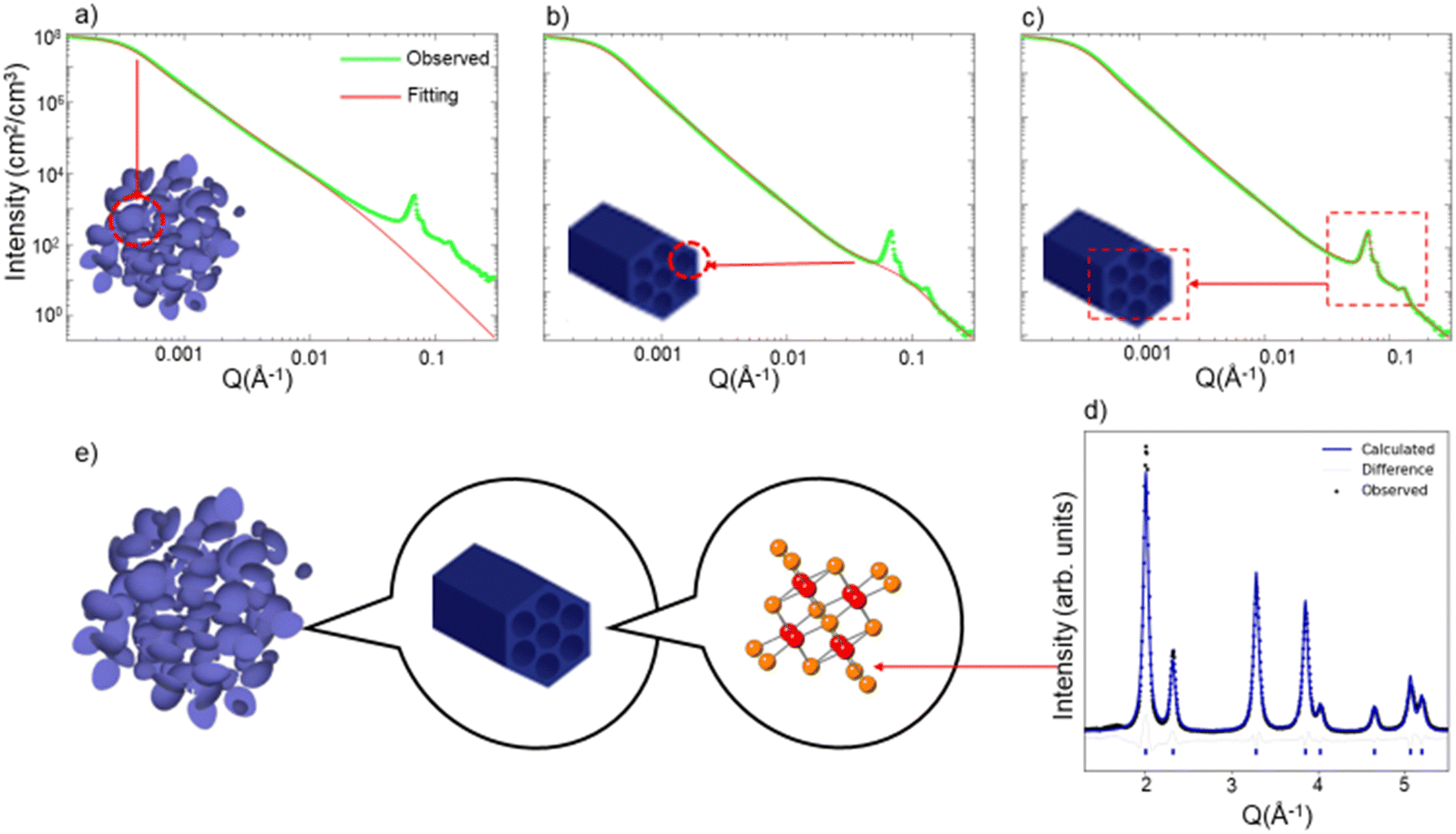 | ||
| Fig. 1 USAXS data fitted considering (a) Only particle size, (b) particle size and mesopore size, (c) particle size, mesopore size and ordered pore structures. (d) WAXS/XRD pattern with Rietveld refinement. (e) Illustration of the structure of mesoporous CeO2 at different scales. Data are collected on SBA-15 templated mesoporous CeO2 at 50 °C. USAXS data is plotted at log scale for both axes. | ||
At the high-Q region (Q > 0.01 Å−1), two features are noted. First, the uptrend of the curve at high Q region does not match fitted curve when only particle structure is considered in the fitting model (Fig. 1a). By adding a second unified model with much smaller Rg (∼30 Å), the curve at low-Q region can be well fit, as is displayed in Fig. 1b. We attribute this uptrend in the curve to the scattering due to each individual mesopore, i.e., the pore's form factor. The pore size of the mesoporous CeO2 is measured from TEM image (Fig. S1, ESI†), which is around 30–40 Å, comparable to the fitted Rg value obtained from the high-Q region. The second feature at the high-Q region is the appearance of several sharp peaks, which is a result of the diffraction from the ordered mesopore structures. These diffraction peaks contain information such as manner of the ordering (symmetries and distance between lattice planes) and degree of the ordering. The diffraction peaks shown in USAXS data correspond to the hexagonal pores of SBA-15 CeO2 and can be indexed to the p6mm plane group, while the mesopores of KIT-6 CeO2 are described by the Ia3d space group.10 The diffraction peaks in USAXS data can be fit with certain peak shapes individually (Fig. 1c), and peak position and half width information can be extracted simultaneously.
Outside of the USAXS data, the WAXS data (Q > 1 Å−1) contains the information about atomic arrangement in the materials. By performing Rietveld refinement analysis15 with the diffraction patterns (Fig. 1d), we can track the change of the crystal structure, crystallinity (grain size) and lattice expansion.
For the in situ structure characterization upon heating, the USAXS/WAXS patterns were continuously taken at different temperature steps. Throughout the experiment, our mesoporous CeO2 powder samples were heated in air. In the first experiment, we heated the sample up to 1000 °C and cooled it down at the same rate down to 100 °C. The collected USAXS/WAXS patterns at different temperatures were fitted with the previously mentioned methods to extract important structural parameters at different length scales.
Fig. 2a and b display the in situ USAXS and WAXS patterns of SBA-15 templated CeO2 powders upon heating to 1000 °C. A two-dimensional (2D) representation or contour plot of the data is provided in Fig. S4 and S5 (ESI†). Visually, the diffraction peaks in the USAXS start to deform at 800 °C, indicating the collapse of the ordered pore structure. The bump (indicated with black arrows in Fig. 2a), which we attribute to the scattering from individual pores, shifts to the lower Q region at 800 °C as well. This shift to lower Q implies that the size of mesopores becomes larger. In the WAXS (Fig. 2b), the diffraction peaks start to become sharper at 800 °C, suggesting an increase of crystalline grain size. The red curves in Fig. 2c shows change of the Rg of pores (from USAXS data fitting) and crystalline grain size (from WAXS Rietveld refinement) upon heating/cooling. Both values start to show an uptrend at around 700 °C and this uptrend becomes more obvious at 800 °C.
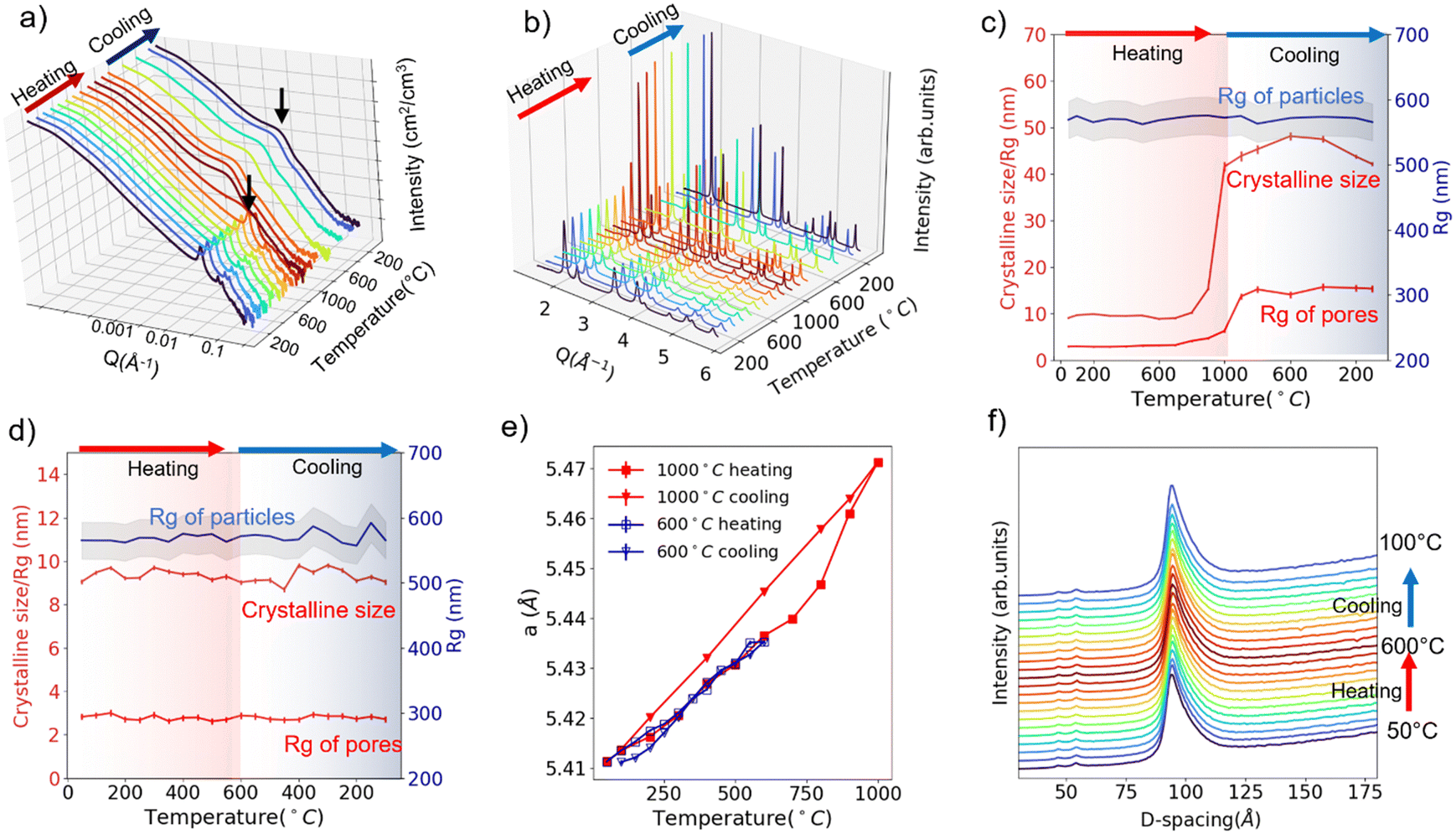 | ||
| Fig. 2 In situ USAXS patterns (a) and WAXS patterns(b) under heating/cooling to/from 1000 °C. Extracted grain size, Rg of pores and Rg of particles under heating/cooling to/from 1000 °C (c) and under heating/cooling to/from 600 °C (d). Lattice expansion under heating/cooling to/from 1000 °C and under heating/cooling to/from 600 °C (e). (100) Diffraction peak from the ordered mesopores under heating/cooling to/from 600 °C (f). Data are collected for SBA-15 templated mesoporous CeO2. USAXS data (a) is plotted at log scale for Q and intensity. | ||
The in situ USAXS/WAXS experiments indicate that the collapse of the ordered pore structure, enlargement of the pore size and the recrystallization of the crystalline grains correlates to each other since they happen at nearly the same pace. We infer that the heating triggered the recrystallization process of the crystalline grains, which causes the fusion of the initial grains into larger grains. Crystalline CeO2 is known to have a relatively high melting point (2400 °C). However, in the mesoporous CeO2, CeO2 exists in a nanocrystalline form (∼10 nm), which will lead to a significantly enhanced surface tension and thus a lower melting point.16 Thus, the fusion of CeO2 nano grains happens at a much lower temperature than the actual melting point. And this fusion process between the “building blocks” of ordered mesopore structure leads to both the collapse of the ordered structure and the appearance of larger pores. The in situ TEM heating experiment up to 1000 °C (Fig. S6, ESI†) shows the evidence of fusion of crystalline grains and appearance of larger pores due to the fusion, which agrees well with the USAXS/WAXS data. Interestingly, the USAXS in the low-Q region does not show any significant changes upon heating to 1000 °C. The fitted Rg value for particles remains nearly unchanged during the whole experiment, as is indicated by the blue curve in Fig. 2c. Such an observation suggests the recrystallization/fusion only happens between crystalline grains in the porous structure but not in the packed mesoporous particles up to 1000 °C. Upon cooling, the Rg of pores and crystalline grain size maintains its value and does not recover back to the original state, which is intuitively reasonable as the recrystallization is not a reversible process.
The 1000 °C in situ heating experiment shows that the SBA-15 templated mesoporous CeO2 seems to be durable upon heating up to 600 °C. Thus, we performed another in situ characterization on new powder samples with a 600 °C heating and cooling cycle. USAXS and WAXS data are displayed in Fig. S7 and S8 (ESI†), and the fitted size parameters are displayed in Fig. 2d. No significant changes were observed in both scattering patterns and fitted parameters, suggesting a good thermal and cycling stability of the structure at all scales up to 600 °C.
The lattice parameters can also be extracted from Rietveld refinement with the WAXS pattern. The lattice parameter changes upon 600 °C heating/cooling cycle and 1000 °C heating/cooling cycle are plotted in Fig. 2e. The thermal expansion of the lattice is observed. However, it is obvious the thermal expansion for 1000 °C cooling is larger than 1000 °C heating or 600 °C heating/cooling (α = 1.172 × 10−5 °C−1vs. 0.848 × 10−5 °C−1). As mentioned previously, during the 1000 °C cooling step, the ordered mesoporous structure is lost and crystalline size enlarged. We thus conclude that the mesoporous structure alleviates the thermal expansion of CeO2, which might be a result of strain from the porous structure or surface tension from the nano-sized grains.17,18
Theoretically, the expansion of the pore structure or particles can also be tracked by monitoring the diffraction peak shifts in the USAXS patterns. Fig. 2f displays the tracking of (100) diffraction peak (USAXS data) of the hexagonal pore structure upon 600 °C heating/cooling. A shift from 93.9 Å to 94.4 Å (USAXS resolution 0.1 Å−1) is observed upon heating to 600 °C from 50 °C, corresponding to a thermal expansion coefficient of 0.97 × 10−5 °C−1, which is comparable to the value (0.848 × 10−5 °C−1) extracted from atomic lattice parameter change. Cooling leads to the shift back to smaller d-spacing, indicating the pore structure thermal expansion is reversible up to 600 °C. However, we are not able to determine whether the pore structure thermal expansion or atomic lattice thermal expansion is more significant (0.97 × 10−5 °C−1vs. 0.848 × 10−5 °C−1) due to the resolution limit of the USAXS experiment at the time.
Similar in situ heating USAXS/WAXS experiments are also performed on KIT-6 templated mesoporous CeO2. The USAXS/WAXS patterns and fitted parameters for 1000 °C heating/cooling are 600 °C heating/cooling are presented on Fig. S11 and S12 (ESI†), respectively. Though both Rg of pores and crystalline grain size start to become larger at 700 °C (Fig. S11c, ESI†), same as SBA-15 templated mesoporous CeO2, the diminishment of diffraction peak from ordered structure is only visible beyond 900 °C (800 °C for SBA-15 templated mesoporous CeO2) (Fig. S11a, ESI†), suggesting the Ia3d ordered pore structure is more durable towards heating than p6mm ordered pore structure. The recrystallization/fusion similarly only happens between crystalline grains in the porous structure but not packed mesoporous particles up to 1000 °C as both high-Q region of USAXS data and the fitted Rg for particles do not show significant change (Fig. S11a and c, ESI†).
The enlargement of Rg for the pore upon heating seems to be more significant than the SBA-15 templated CeO2, as indicated by the shift of the bump and fitted Rg value change (from 3 to 25 nm vs. from 3 to 10 nm, shown on Fig. S11c, ESI† and Fig. 2c). For 600 °C heating/cooling cycles, KIT-6 templated mesoporous CeO2 shows similar good thermal and cycling stability (Fig. S12, ESI†). The lattice parameter changes upon 600 °C heating/cooling cycle and 1000 °C heating/cooling cycle are also extracted and plotted in Fig. S13 (ESI†). The smaller lattice thermal expansion on ordered porous structure is again observed (α = 1.197 × 10−5 °C−1 for 1000 °C cooling vs. 0.847 × 10−5 °C−1 for others). Fig. S14 (ESI†) displays the tracking of d-spacing of (211) diffraction peak (USAXS data) of the cubic pore structure upon 600 °C heating/cooling. A shift from 92.3 Å to 93.0 Å is observed upon heating to 600 °C from 50 °C, corresponding to a thermal expansion coefficient of 1.37 × 10−5 °C−1. Again, we are not able to use this value to make any conclusive comparisons due to the resolution limit.
In summary, we demonstrate how in situ structural characterization of mesoporous CeO2 powder samples at different scales can be simultaneously obtained via synchrotron X-ray based USAXS/WAXS. Our study introduces an effective approach to monitor the evolution of particle size, pore structures, pore size, grain size and atomic structure of the mesoporous materials under dynamic conditions. Furthermore, we highlight that the structures of materials at different scales can influence each other, a factor that has not received sufficient attention in previous studies on mesoporous materials. Future research should focus more on understanding the interplay between structures at different scales and exploring its impact on material performance. Future experiments could expand beyond heating/cooling cycles to include gas flowing for catalytic reactions and charging/discharging for batteries. The multiscale in situ structural characterization will provide deep insights of the correlation between structural features at different scales and their synergistic impact on materials performance. Exploring the impact of rare metal cluster/single atom loading on the structural behaviors of mesoporous oxides will be also of great interest, as the loading can significantly influence the surface energy and strain of mesoporous materials, which, in turn, can affect their structural evolution under dynamic conditions.
This work is supported by the Department of Defense (grant HDTRA1-19-1-0001) and Department of Commerce (grant 70NANB20H139). Use of the Advanced Photon Source at Argonne National Laboratory was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. We thank Dr Jan Ilavsky, and Dr Ivan Kuzmenko at beamline 9ID, for their help with USAXS data collection and sample preparation.
Data availability
The data supporting this article have been included as part of the ESI.†In situ scattering data sets for this article are available at Open Science Framework at 10.17605/OSF.IO/2K4XA.Conflicts of interest
The authors declare no competing financial interest.References
- A. Van Der Ven, J. Bhattacharya and A. A. Belak, Acc. Chem. Res., 2013, 46, 1216–1225 CrossRef CAS PubMed.
- D.-W. Chung, P. R. Shearing, N. P. Brandon, S. J. Harris and R. E. García, J. Electrochem. Soc., 2014, 161, A422–A430 CrossRef CAS.
- M. K. Devaraju and I. Honma, Adv. Energy Mater., 2012, 2, 284–297 CrossRef CAS.
- M. Yashima, T. Hoshina, D. Ishimura, S. Kobayashi, W. Nakamura, T. Tsurumi and S. Wada, J. Appl. Phys., 2005, 98, 014313 CrossRef.
- J. Chen, L. Fan, Y. Ren, Z. Pan, J. Deng, R. Yu and X. Xing, Phys. Rev. Lett., 2013, 110, 115901 CrossRef PubMed.
- U. Ciesla and F. Schüth, Microporous Mesoporous Mater., 1999, 27, 131–149 CrossRef CAS.
- Y. Ren, Z. Ma and P. G. Bruce, Chem. Soc. Rev., 2012, 41, 4909–4927 RSC.
- L. Liu, Trends Chem., 2021, 3, 898–901 CrossRef CAS.
- Q. Meyer, Y. Zeng and C. Zhao, Adv. Mater., 2019, 31, 1901900 CrossRef CAS PubMed.
- S. C. Laha and R. Ryoo, Chem. Commun., 2003, 2138–2139 RSC.
- J. Ilavsky, F. Zhang, R. N. Andrews, I. Kuzmenko, P. R. Jemian, L. E. Levine and A. J. Allen, J. Appl. Crystallogr., 2018, 51, 867–882 CrossRef CAS PubMed.
- J. Ilavsky, F. Zhang, A. J. Allen, L. E. Levine, P. R. Jemian and G. G. Long, Metall. Mater. Trans. A, 2013, 44, 68–76 CrossRef CAS.
- G. Beaucage, J. Appl. Crystallogr., 1995, 28, 717–728 CrossRef CAS.
- J. Ilavsky and P. R. Jemian, J. Appl. Crystallogr., 2009, 42, 347–353 CrossRef CAS.
- R. W. Cheary and A. Coelho, J. Appl. Crystallogr., 1992, 2, 109–121 CrossRef.
- D. A. Porter, K. E. Easterling and K. E. Easterling, Phase Transformations in Metals and Alloys (Revised Reprint), CRC Press, 2009 Search PubMed.
- Q. Li, H. Zhu, L. Hu, J. Chen and X. Xing, Acc. Chem. Res., 2019, 52, 2694–2702 CrossRef CAS PubMed.
- S. Elomari, M. D. Skibo, A. Sundarrajan and H. Richards, Compos. Sci. Technol., 1998, 58, 369–376 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc02128b |
| This journal is © The Royal Society of Chemistry 2024 |