
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVersatile dehydrogenation of carbonyls enabled by an iodine(III) reagent†
Bence B.
Botlik
 ,
Patrick
Finkelstein
,
Ann-Sophie K.
Paschke
,
Julia C.
Reisenbauer
and
Bill
Morandi
*
,
Patrick
Finkelstein
,
Ann-Sophie K.
Paschke
,
Julia C.
Reisenbauer
and
Bill
Morandi
*
Laboratorium für Organische Chemie, ETH Zürich, Vladimir Prelog Weg 3, HCI, 8093 Zürich, Switzerland. E-mail: morandib@ethz.ch
First published on 2nd August 2024
Abstract
We report the utilisation of an iodine(III) reagent to access α,β-unsaturated carbonyls from the corresponding silyl enol ethers of ketones and aldehydes, and from enol phosphates of lactones and lactams. The transformation is rapid, scalable, and can be carried out in one pot, directly dehydrogenating saturated carbonyls.
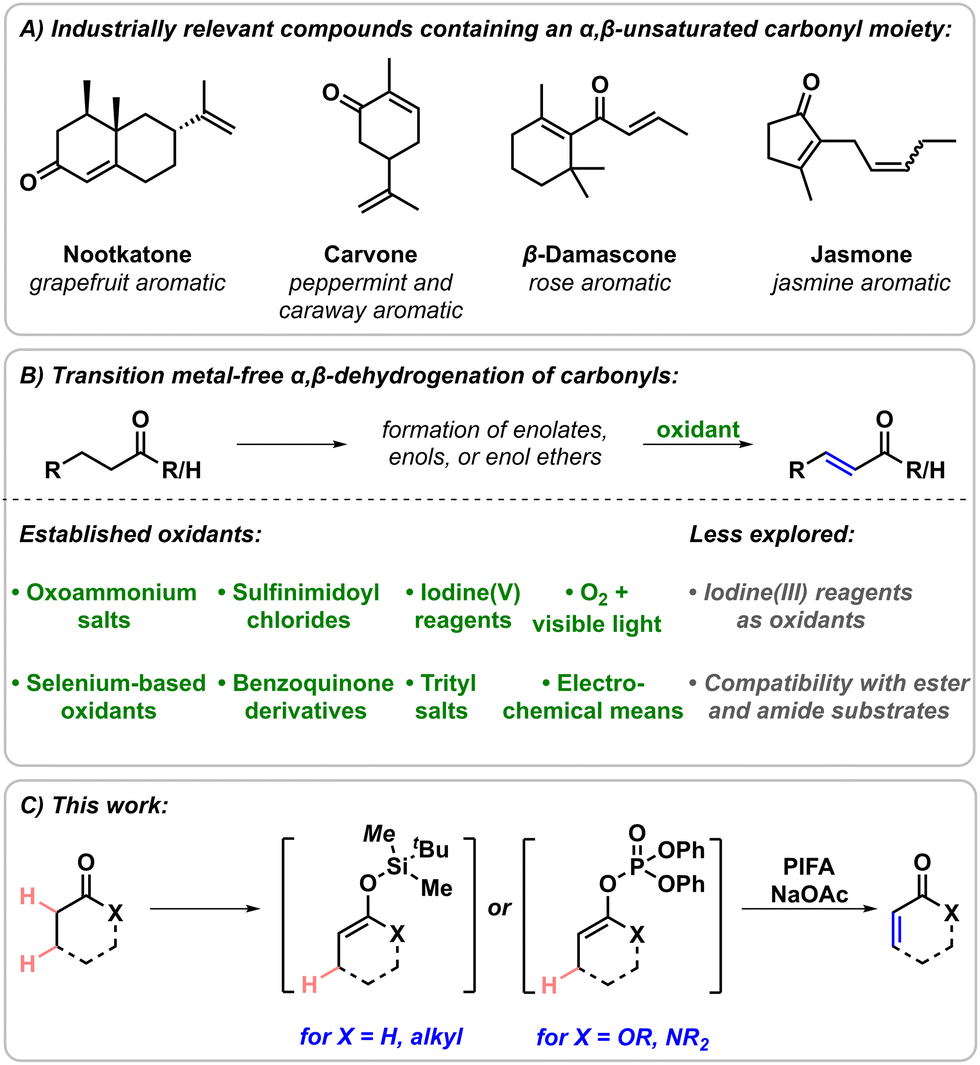
Converting saturated carbonyl compounds into the corresponding α,β-unsaturated derivatives is a key transformation in synthetic organic chemistry.1 The products of these dehydrogenation processes are valuable intermediates and versatile building blocks in the synthesis of natural products as well as industrially relevant targets, and ubiquitous functional moieties in various materials.2,3 The relevance of α,β-unsaturated carbonyls is particularly reflected by their abundance among aroma compounds and odorants, thus they are of special interest for the fragrance industry (Scheme 1A).4
 | ||
| Scheme 1 (A) Relevance of α,β-unsaturated carbonyl compounds. (B) Previous examples of transition metal-free carbonyl dehydrogenation. (C) This work. | ||
In addition, their important role as useful synthetic intermediates is showcased by the large number of established interconversions for these functional handles that further enable their derivatisation. For instance, conjugate additions, cross couplings, regioselective α-functionalisations, and alkene difunctionalisation reactions open different avenues to access a broad range of products from α,β-unsaturated carbonyls.2,5
Most known dehydrogenation methods formally proceed via two consecutive chemical transformations. Typically, in the first step, an enol or an enolate is formed, which is either directly transformed into the desired unsaturated product, or is trapped in situ generating the corresponding silyl enol ether. This strategy allows for the activation of the α-position and controls the regioselectivity of the subsequent dehydrogenation step mediated by an oxidant, resulting in the formation of the corresponding desaturated products. To date, most of the developed carbonyl α,β-dehydrogenation reactions employ transition metals.1,6–15 In contrast, transition metal-free approaches are comparatively less established. Besides a recent electrochemical approach,16 other common strategies rely on the utilisation of oxoammonium salts,17 benzoquinone derivatives,18 selenium compounds,19 trityl salts20 or sulfinimidoyl derivatives21 as oxidants (Scheme 1B). Additionally, methods implementing iodine(V) reagents have also been reported, both using either silyl enol ethers or the saturated carbonyls as starting scaffolds.22,23 However, these methods are often limited in their scopes to the dehydrogenation of ketones and aldehydes, and commonly require long reaction times and elevated temperatures. Methods relying on iodine(III) reagents as oxidants are scarce, and to date have been restricted to specific systems, such as the flavanone to flavone transformations,24 and the double dehydrogenation of cyclic β-ketocarbonyls.25 Moreover, silyl enol ethers usually exhibit different reactivities with iodine(III) reagents and in most cases result in the formation of α-functionalised ketones, commonly generating α-ketoacetate derivatives,26 α-hydroxyketones,27 α-methoxyketones,28 and α-sulfonyloxyketones.29 Our group recently disclosed that iodine(III) reagents facilitate the nitrogen atom insertion into the silyl enol ether of indanone and related scaffolds in the presence of an external nitrogen atom source.30–33 Based on these results, similar substrates possessing silyl enol ether functional handles were investigated towards their reactivity with iodine(III) reagents. However, a mechanistically distinct reactivity was observed in various cases, leading to the formation of the corresponding dehydrogenation products. This is hypothesised to be the result of utilising the understudied combination of a particularly strong hypervalent iodine oxidant and an equivalent amount of base, with easily oxidisable silyl enol ether reaction partners. Herein, the optimisation and development of a general, rapid, and operationally simple iodine(III)-mediated dehydrogenation method is presented, which grants access to α,β-unsaturated carbonyls from silyl enol ethers of ketones and aldehydes, and from enol phosphates of lactones and lactams (Scheme 1C).
Initial optimisation of the transformation was performed using tert-butyldimethylsilyl-(TBS)-protected cyclohexenol 1a as the model substrate (Table 1). Acetone was found to be the most effective solvent for the reaction, with acetonitrile and tetrahydrofuran (THF) also providing moderate yields of the dehydrogenation product 2a, while other examined solvents showed significantly lower conversion to the desired product. Bis(trifluoroacetoxy)iodobenzene (PIFA) was the most suitable oxidant, as the use of other, less oxidising iodine(III) reagents either afforded the desired product in low yields or no product formation was detected. In addition, various inorganic bases were found to be compatible with the initial screening conditions, resulting in nearly identical yields, however, amine bases reduced the desired reactivity. Using oxidant loadings lower than 2 equivalents was detrimental to the yield, however, further increasing the amount of oxidant in the reaction also did not improve the overall yield. Variation of the silyl groups incorporated in the starting material indicated the superiority of the TBS group, as tri-isopropylsilyl (TIPS) enol ethers were transformed with significantly lower yields, and in general low reactivity was observed in the case of other silyl groups, such as tri-methylsilyl and tri-ethylsilyl groups. Finally, the effects of molarity and temperature were examined, and the highest yields of product 2a were obtained when the reaction was performed at 0 °C using a more dilute solution (0.125 M with respect to substrate 1a in acetone). With the optimal conditions in hand, we examined the scope of the transformation (Scheme 2). Cyclic aliphatic ketones 2a and 2b were both obtained in approximately 60% 1H NMR yield, which corresponds well to the isolated yield of 2a. Similarly, other silyl enol ethers exhibiting larger ring sizes were also found to be compatible with the reaction and the macrocyclic product 2c was isolated in moderate 45% yield. Chromanone derivatives 2d and 2e were obtained in good yields (77% and 69%, respectively), as well as phenyl-conjugated alkenes, including 2g and Nabumeton-derived product 2f. In contrast, chalcone 2h was isolated in lower yield, likely due to the decomposition of the silyl enol ether under the reaction conditions, resulting the saturated carbonyl as a major side product. In the case of indanone 1i, however, the corresponding dehydrogenation product 2i could not be observed. Aldehyde-derived starting scaffolds exhibited comparable reactivity to that of ketone-derived ones. The compatibility of aldehydes with our method was examined through a number of examples that are important to the fragrance industry.4 Lilial-derived 2j and 2j′ were obtained in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of regioisomers and were isolated in a combined 56% yield, whereas cinnamaldehyde 2k was isolated in 72% yield. Similarly, citronellal 1l transformed into dehydrogenated product 2l in the presence of another alkene handle in moderate yield, resulting in the formation of both the E and Z alkenes in a 2:1 ratio. Finally, the formation of the desaturated aliphatic aldehyde 2m was also observed. Apart from ketone and aldehyde substrates, lactones and lactams were also found to be suitable substrates for the reaction. As the silyl enol ethers of lactones and lactams are unstable and prone to hydrolysis, the more stable enol phosphate derivatives were employed. Coumarin (2n) was obtained in high yield (78%), while its substituted derivative 2o was isolated in reduced yield. The desaturation of a non-conjugated aliphatic lactone could also be achieved, resulting in the formation of product 2p in 43% 1H NMR yield. When employing an open chain enol phosphate, the desired product 2q was formed and co-isolated with the α-trifluoroacetylated side product 2q′. Additional examples showcasing the reaction's translation to enol phosphates derived from lactams were tested and the corresponding dehydrogenation products 2r and 2s were isolated in 68% and 33%, respectively. The non-conjugated enol phosphate 1t, however, did not yield the corresponding dehydrogenation product 2t.
:1 ratio of regioisomers and were isolated in a combined 56% yield, whereas cinnamaldehyde 2k was isolated in 72% yield. Similarly, citronellal 1l transformed into dehydrogenated product 2l in the presence of another alkene handle in moderate yield, resulting in the formation of both the E and Z alkenes in a 2:1 ratio. Finally, the formation of the desaturated aliphatic aldehyde 2m was also observed. Apart from ketone and aldehyde substrates, lactones and lactams were also found to be suitable substrates for the reaction. As the silyl enol ethers of lactones and lactams are unstable and prone to hydrolysis, the more stable enol phosphate derivatives were employed. Coumarin (2n) was obtained in high yield (78%), while its substituted derivative 2o was isolated in reduced yield. The desaturation of a non-conjugated aliphatic lactone could also be achieved, resulting in the formation of product 2p in 43% 1H NMR yield. When employing an open chain enol phosphate, the desired product 2q was formed and co-isolated with the α-trifluoroacetylated side product 2q′. Additional examples showcasing the reaction's translation to enol phosphates derived from lactams were tested and the corresponding dehydrogenation products 2r and 2s were isolated in 68% and 33%, respectively. The non-conjugated enol phosphate 1t, however, did not yield the corresponding dehydrogenation product 2t.
|
|
||
|---|---|---|
| Entry | Deviation from abovea | Yieldb of 2a (%) |
| a Reaction conditions: silyl enol ether (0.05 mmol), PIFA (0.10 mmol), NaOAc (0.10 mmol), acetone-d6 (0.125 M), 0 °C, 10 min, then room temperature (r.t.), 30 min. b Yields determined by 1H NMR analysis of the crude reaction mixtures, using mesitylene as the internal standard. | ||
| 1 | None | 60 |
| 2 | MeCN instead of acetone | 38 |
| 3 | THF instead of acetone | 43 |
| 4 | PIDA instead of PIFA | 0 |
| 5 | HTIB instead of PIFA | 5 |
| 6 | KOH instead of NaOAc | 59 |
| 7 | KTFA instead of NaOAc | 58 |
| 8 | Et3N instead of NaOAc | 48 |
| 9 | 1 equiv. PIFA | 42 |
| 10 | 3 equiv. PIFA | 60 |
| 11 | TIPS instead of TBS | 28 |

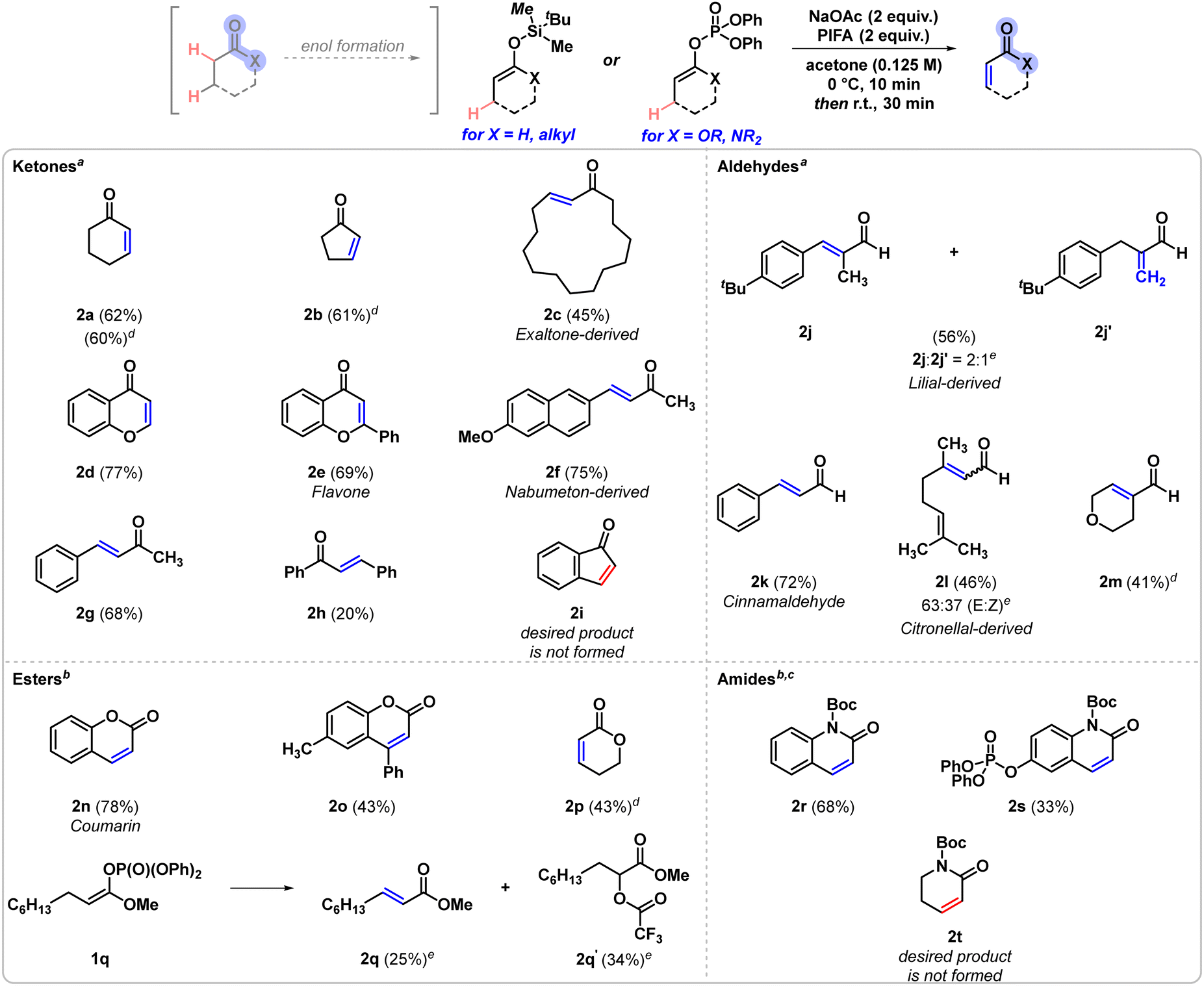 | ||
Scheme 2 Substrate scope. The yields are isolated yields of the reaction, unless indicated otherwise. aSilyl enol ethers were used as starting materials. b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Enol phosphates were used as starting materials. cThe reaction was carried out in acetonitrile instead of acetone. dReaction carried out on 0.05-mmol scale, yields determined by 1H NMR analysis of the crude reaction mixtures, using mesitylene internal standard. eIsomeric ratios were determined by 1H NMR analysis. For details, see ESI.† Enol phosphates were used as starting materials. cThe reaction was carried out in acetonitrile instead of acetone. dReaction carried out on 0.05-mmol scale, yields determined by 1H NMR analysis of the crude reaction mixtures, using mesitylene internal standard. eIsomeric ratios were determined by 1H NMR analysis. For details, see ESI.† | ||
We hypothesised that the reaction could be carried out in a one-pot manner directly using the saturated carbonyl compounds as starting materials, which would greatly improve the synthetic utility of the developed transformation. Therefore, we set out to examine the one-pot dehydrogenation through a representative example of each four carbonyl classes (Scheme 3A). To our delight, ketone 1d′′ yielded the desired product 2d in 51% yield upon using THF solvent for the silyl enol ether formation step and a THF:acetone = 1:9 solvent mixture in the subsequent dehydrogenation step. Similarly, the direct dehydrogenation of carbocyclic ketone 1c′′ was also demonstrated. Using the same conditions, aldehyde 1k′′ afforded the corresponding dehydrogenation product 2k in 48% isolated yield. Furthermore, the possibility for the one-pot dehydrogenation of lactones was demonstrated by using 1n′′, which afforded product 2n in 60% isolated yield. Key to success was the implementation of THF as the reaction solvent, as it is compatible with both the enol phosphate formation and the subsequent oxidation step, alongside using increased amounts of PIFA and implementing KTFA as the base. By using an analogous strategy, the dehydrogenation of lactam 1r′′ also proceeded in a one-pot fashion to afford dehydrogenated lactam 2r. These results possess considerable synthetic importance, as transition metal-free dehydrogenation methods of lactones and lactams are scarce.1,34 Even though higher overall yields are observed when using the two-step approach, the combined yields over the two steps are comparable to the yields of the one-pot transformation. The scalability of the reaction was also examined, and the formation of cinnamaldehyde (2k) was conducted on a gram-scale (14.0 mmol of starting material; Scheme 3B), providing the desired product in 73% isolated yield, which is identical to the result obtained for the 1-mmol scale.
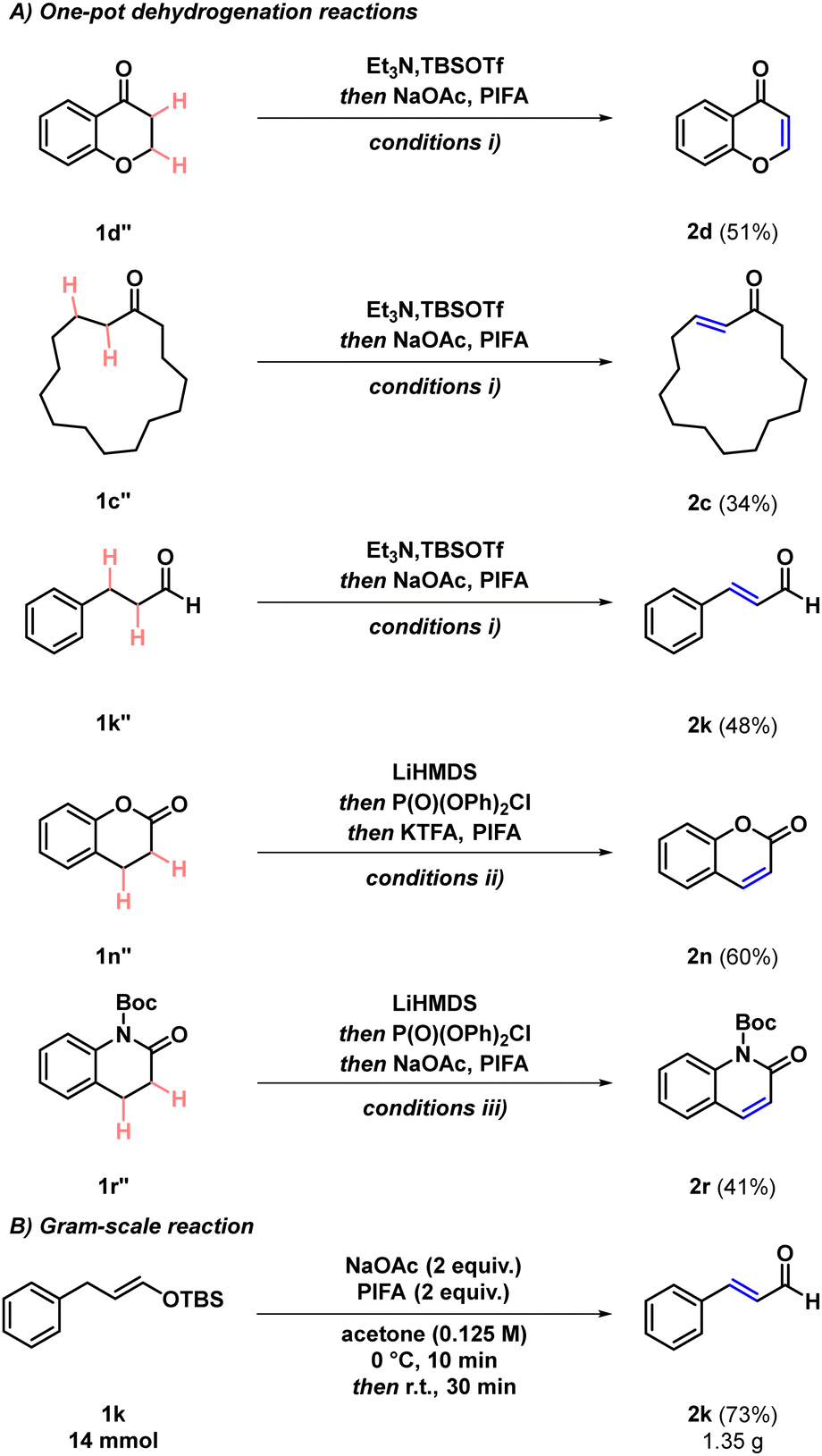 | ||
| Scheme 3 Further experiments. (A) One-pot dehydrogenation reactions. Conditions: (i) Et3N (1.5 equiv.), TBSOTf (1.2 equiv.), THF (1 M), r.t., 1 h; then NaOAc (2 equiv.), PIFA (2 equiv.), THF:acetone = 1:9 (0.1 M), 0 °C, 10 min; then r.t., 30 min. (ii) LiHMDS (1.0 equiv.), dry THF (0.125 M), −78 °C, 30 min; then P(O)(OPh)2Cl (1.0 equiv.), −78 °C to r.t., 1 h; then KTFA (4 equiv.), PIFA (4 equiv.), r.t., 30 min. (iii) LiHMDS (1.1 equiv.), dry THF (0.125 M), −78 °C, 30 min; then P(O)(OPh)2Cl (1.1 equiv.), −78 °C to r.t., 1 h; then NaOAc (2 equiv.), PIFA (2 equiv.), THF:MeCN= 1:1 (0.06 M), 0 °C, 10 min; then r.t., 30 min. (B) Scale-up experiment. | ||
In conclusion, we have developed an operationally simple carbonyl dehydrogenation method, complementing already existing methodologies for the formation of α,β-unsaturated ketones, aldehydes, lactones, and lactams, by using a readily available iodine(III) oxidant under basic conditions. Furthermore, we have demonstrated the excellent scalability of the reaction to gram-scale as well as the feasibility of a one-pot approach to rapidly access α,β-desaturated products directly from the corresponding saturated carbonyls.
We thank Prof. Ori Green (Technion) for valuable discussions. This work was supported by ETH Zürich and the Swiss National Science Foundation (SNSF 184658). B. B. B. and J. C. R. acknowledge a fellowship from the Scholarship Fund of the Swiss Chemical Industry (SSCI). A. S. P. acknowledges a fellowship from the Fonds der Chemischen Industrie (FCI). We thank the NMR and MS (MoBiAS) service departments at ETH Zürich for technical assistance and the Morandi group for critical proofreading of the manuscript.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Gnaim, J. C. Vantourout, F. Serpier, P.-G. Echeverria and P. S. Baran, ACS Catal., 2021, 11, 883–892 CrossRef
.
- H. Chen, L. Liu, T. Huang, J. Chen and T. Chen, Adv. Synth. Catal., 2020, 362, 3332–3346 CrossRef
- G. P. Rosa, A. M. L. Seca, M. do, C. Barreto and D. C. G. A. Pinto, ACS Sustainable Chem. Eng., 2017, 5, 7467–7480 CrossRef
-
D. Pybus and C. Sell, The Chemistry of Fragrances, Royal Society of Chemistry, 2007 Search PubMed
-
S. Patai, The Chemistry of enones, Wiley, 1989 Search PubMed
- Y. Chen, J. P. Romaire and T. R. Newhouse, J. Am. Chem. Soc., 2015, 137, 5875–5878 CrossRef PubMed
- D. Huang, Y. Zhao and T. R. Newhouse, Org. Lett., 2018, 20, 684–687 CrossRef CAS PubMed
- M. Chen and G. Dong, J. Am. Chem. Soc., 2017, 139, 7757–7760 CrossRef CAS
- M.-M. Wang, X.-S. Ning, J.-P. Qu and Y.-B. Kang, ACS Catal., 2017, 7, 4000–4003 CrossRef CAS
- Y. Shang, X. Jie, K. Jonnada, S. N. Zafar and W. Su, Nat. Commun., 2017, 8, 2273 CrossRef PubMed
- M. Chen, A. J. Rago and G. Dong, Angew. Chem., Int. Ed., 2018, 57, 16205–16209 CrossRef CAS PubMed
- Z. Wang, Z. He, L. Zhang and Y. Huang, J. Am. Chem. Soc., 2018, 140, 735–740 CrossRef CAS
- D. Huang, S. M. Szewczyk, P. Zhang and T. R. Newhouse, J. Am. Chem. Soc., 2019, 141, 5669–5674 CrossRef CAS PubMed
- Y. Zhao, Y. Chen and T. R. Newhouse, Angew. Chem., Int. Ed., 2017, 56, 13122–13125 CrossRef PubMed
- D. Huang, S. M. Szewczyk, P. Zhang and T. R. Newhouse, J. Am. Chem. Soc., 2019, 141, 5669–5674 CrossRef PubMed
- S. Gnaim, Y. Takahira, H. R. Wilke, Z. Yao, J. Li, D. Delbrayelle, P.-G. Echeverria, J. C. Vantourout and P. S. Baran, Nat. Chem., 2021, 13, 367–372 CrossRef PubMed
- M. Hayashi, M. Shibuya and Y. Iwabuchi, Org. Lett., 2012, 14, 154–157 CrossRef PubMed
- E. A. Braude, L. M. Jackman and R. P. Linstead, J. Chem. Soc., 1954, 3548–3563 RSC
- K. B. Sharpless, R. F. Lauer and A. Y. Teranishi, J. Am. Chem. Soc., 1973, 95, 6137–6139 CrossRef CAS
- M. E. Jung, Y.-G. Pan, M. W. Rathke, D. F. Sullivan and R. P. Woodbury, J. Org. Chem., 1977, 42, 3961–3963 CrossRef CAS
- T. Mukaiyama, J. Matsuo and H. Kitagawa, Chem. Lett., 2000, 1250–1251 CrossRef CAS
- K. C. Nicolaou, D. L. F. Gray, T. Montagnon and S. T. Harrison, Angew. Chem., Int. Ed., 2002, 41, 996–1000 CrossRef
- K. C. Nicolaou, Y.-L. Zhong and P. S. Baran, J. Am. Chem. Soc., 2000, 122, 7596–7597 CrossRef
- O. Prakash, S. Pahuja and R. M. Moriarty, Synth. Commun., 1990, 20, 1417–1422 CrossRef
- S.-S. Liu, L. Wang, Y.-N. Duan, A. Yu and C. Zhang, Sci. China: Chem., 2019, 62, 597–601 CrossRef
- I. Brunolevskaya, K. Kusainova and A. Kashin, Zh. Org. Khim., 1988, 24, 358 Search PubMed
- R. M. Moriarty, M. P. Duncan and O. Prakash, J. Chem. Soc., Perkin Trans. 1, 1987, 1781–1784 RSC
- R. M. Moriarty, O. Prakash, M. P. Duncan, R. K. Vaid and H. A. Musallam, J. Org. Chem., 1987, 52, 150–153 CrossRef CAS
- R. M. Moriarty, R. Penmasta, A. K. Awasthi, W. R. Epa and I. Prakash, J. Org. Chem., 1989, 54, 1101–1104 CrossRef CAS
- P. Finkelstein, J. C. Reisenbauer, B. B. Botlik, O. Green, A. Florin and B. Morandi, Chem. Sci., 2023, 14, 2954–2959 RSC
- J. C. Reisenbauer, O. Green, A. Franchino, P. Finkelstein and B. Morandi, Science, 2022, 377, 1104–1109 CrossRef CAS
- J. C. Reisenbauer, A.-S. K. Paschke, J. Krizic, B. B. Botlik, P. Finkelstein and B. Morandi, Org. Lett., 2023, 25, 8419–8423 CrossRef CAS
- B. B. Botlik, M. Weber, F. Ruepp, K. Kawanaka, P. Finkelstein and B. Morandi, Angew. Chem., Int. Ed., 2024, e202408230 Search PubMed
- P. Spieß, M. Berger, D. Kaiser and N. Maulide, J. Am. Chem. Soc., 2021, 143, 10524–10529 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc02609h |
| This journal is © The Royal Society of Chemistry 2024 |