
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbene transfer reactivity from a nickelacyclobutane†
María L. G.
Sansores-Paredes
 a,
Martin
Lutz
b and
Marc-Etienne
Moret
*a
a,
Martin
Lutz
b and
Marc-Etienne
Moret
*a
aOrganic Chemistry and Catalysis, Institute for Sustainable and Circular Chemistry, Faculty of Science, Utrecht University, Universiteitsweg 99, 3584 CG, Utrecht, The Netherlands. E-mail: m.moret@uu.nl
bStructural Biochemistry, Bijvoet Centre for Biomolecular Research, Utrecht University, 3584 CG, Utrecht, The Netherlands
First published on 30th September 2024
Abstract
A formal carbene-transfer reaction from an isolated nickelacyclobutane to an isocyanide to form a ketenimine is reported. DFT calculations support a stepwise 1,1-insertion/fragmentation pathway without a carbene intermediate. This unusual reactivity suggests a potential new role as “carbene reservoir” for nickelacyclobutanes, which are typically seen as intermediates in catalytic cyclopropanation.
Metallacyclobutanes are often invoked as intermediates in catalytic cyclopropanation and olefin metathesis.1–8 Generally formed by [2+2] cycloaddition of a metal-carbene and an olefin, they are versatile intermediates that can undergo reductive elimination yielding cyclopropanes, [2+2] cycloreversion yielding a metal carbene and an olefin, and insertion of a neutral fragment yielding a metallacyclopentane (Fig. 1).1–8 As part of environmentally-motivated research efforts on base metal catalysis,9,10 Ni-catalyzed cyclopropanation has seen promising developments, where nickelacyclobutanes are proposed as key intermediates.5–8,11–23 To further our understanding of the reactivity of these species, we recently described the preparation of a pentacoordinated nickelacyclobutane embedded in a diphosphine pincer ligand.7 We found that exogenous ligands could selectively induce cyclopropanation (with the π-acceptor CO) or olefin-metathesis-like opening (with the σ-donor MeCN), in contrast with previously reported square planar nickelacyclobutanes.8,16,22,24
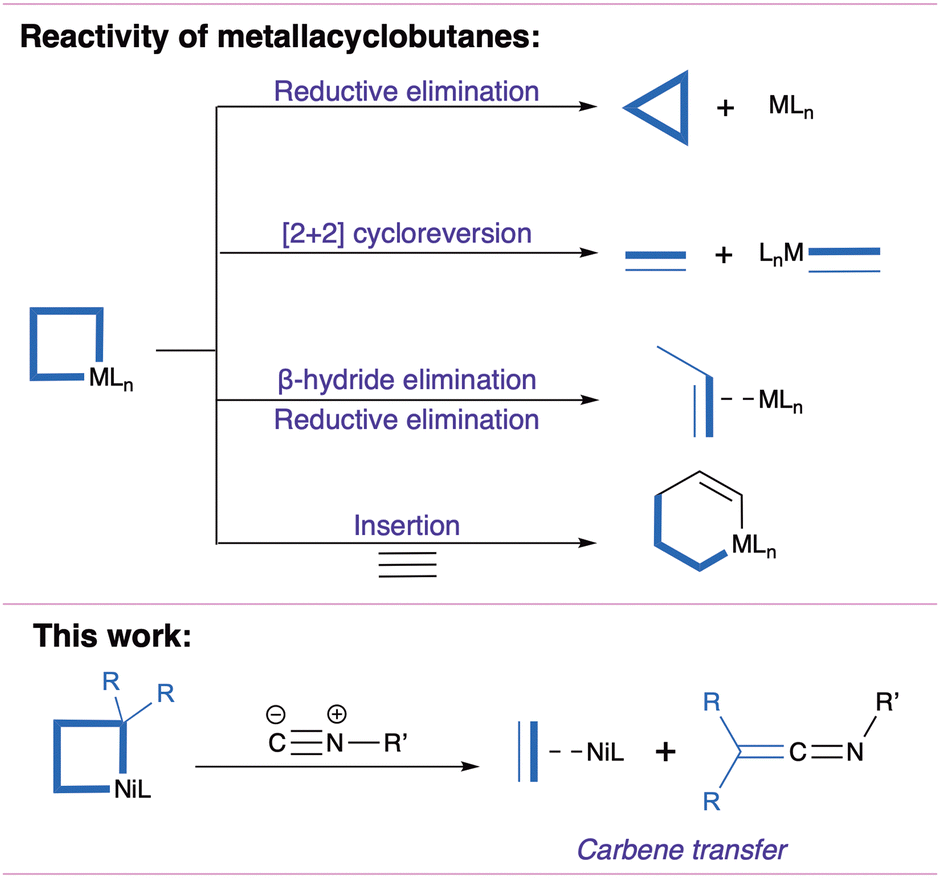 | ||
| Fig. 1 Reactivity of metallacyclobutanes and present work. | ||
Here we report on an unexpected reactive pathway induced by coordination of t-butyl isocyanide (CNtBu, R–NC): a formal carbene transfer generating a ketenimine (Fig. 1) and an olefin complex. While this process could be thought of as the result of [2+2] cycloreversion followed by coupling of the resulting nickel carbene and the isocyanide,25–30 DFT calculations support a distinct mechanism involving a nickelacyclopentane intermediate formed by 1,1-insertion of CNtBu in a Ni–C bond. It suggests that these intermediates could act as “carbene reservoirs” and undergo carbene transfer reactions without prior [2+2] cycloreversion.
Reaction of the pentacoordinate nickelacyclobutane 1 with two equiv of CNtBu in C6D6 initially resulted in rapid coordination of CNtBu in apical position to yield 1-CNtBu (Scheme 1). This is evidenced by a downfield shift and sharpening of the 1H NMR signal corresponding to the CH2 group from δ 4.40 ppm in 1 to 4.78 ppm in 1-CNtBu, and by a sharpening of the 31P{1H} NMR signal to a singlet at 27.6 ppm (ESI,† Section S3). Both observations are consistent with the displacement of the π-interacting tolyl group by an isocyanide molecule to form a symmetrical structure and parallel those made upon coordination of CO.7
 | ||
| Scheme 1 Carbene transfer with 2 eq. t-butylisocyanide. | ||
A 1H NMR spectrum recorded after 2 h (ESI,† Section S2.2) showed the appearance of the olefin complex (PhbppeH,H)Ni(CNtBu) 2 and ketenimine t-BuN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CC(p-Tol)2, as a result of the transfer of the C(p-Tol)2 carbene fragment to an isocyanide molecule. The reaction was complete after 23 h. Additionally, a small amount of 1,1-di(p-tolyl)ethylene was observed (∼10%) suggesting [2+2] cycloreversion as a minor pathway. Complex 2 was identified by a 31P{1H} NMR singlet at δ(C6D6) 25.7 ppm and a characteristic 1H NMR signal at δ 3.65 ppm (t, JH,P = 2.2 Hz, 2H), that both match those of a sample of 2 independently synthesized from PhbppeH,H, Ni(cod)2 and CNtBu (ESI,† Section 1.3 and 3). An ATR-FTIR spectrum of the reaction mixture (ESI,† Section S2.2) confirms the presence of complex 2 and displays the characteristic NCC stretching peak of t-BuNCC(p-Tol)2 as a strong signal at 2005 cm−1.31,32 The identity of the organic product is bolstered by the presence of ketenimine peak at 183.5 ppm in the APT 13C{1H} spectrum of the reaction mixture (ESI,† Section S2.2).27,31,33 Using a large excess of CNtBu (55 equivalents) did not result in any substantial changes in the reactivity (ESI,† Section S2.3). No further reaction was observed when the isolated product 2 was exposed to bis(4-tolyl)diazomethane, indicating that the CNtBu ligand in 2 binds too strongly for catalytic turnover to be accessible with this system.
CC(p-Tol)2, as a result of the transfer of the C(p-Tol)2 carbene fragment to an isocyanide molecule. The reaction was complete after 23 h. Additionally, a small amount of 1,1-di(p-tolyl)ethylene was observed (∼10%) suggesting [2+2] cycloreversion as a minor pathway. Complex 2 was identified by a 31P{1H} NMR singlet at δ(C6D6) 25.7 ppm and a characteristic 1H NMR signal at δ 3.65 ppm (t, JH,P = 2.2 Hz, 2H), that both match those of a sample of 2 independently synthesized from PhbppeH,H, Ni(cod)2 and CNtBu (ESI,† Section 1.3 and 3). An ATR-FTIR spectrum of the reaction mixture (ESI,† Section S2.2) confirms the presence of complex 2 and displays the characteristic NCC stretching peak of t-BuNCC(p-Tol)2 as a strong signal at 2005 cm−1.31,32 The identity of the organic product is bolstered by the presence of ketenimine peak at 183.5 ppm in the APT 13C{1H} spectrum of the reaction mixture (ESI,† Section S2.2).27,31,33 Using a large excess of CNtBu (55 equivalents) did not result in any substantial changes in the reactivity (ESI,† Section S2.3). No further reaction was observed when the isolated product 2 was exposed to bis(4-tolyl)diazomethane, indicating that the CNtBu ligand in 2 binds too strongly for catalytic turnover to be accessible with this system.
More insights into the reaction mechanism were provided by an experiment with a lower amount (1.5 equiv.) of CNtBu (Scheme 2). A slight excess was found necessary to ensure full initial conversion to 1-CNtBu. Monitoring the reaction over time by 31P{1H} NMR again showed gradual conversion of 1-CNtBu (δ(C6D6) 27.6 ppm) to compound 2 (δ(C6D6) 25.7 ppm) at early stages. However, a new P-containing species (3) appeared as a slightly broad singlet at δ(C6D6) 18.5 ppm after 1 h and was present in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1 ratio with 2 after 18 h when all 1-CNtBu was consumed (ESI,† Section S2.1). At this time, the concentration of ketenimine was approximately equal to the sum of those of complexes 2 and 3 according 1H NMR integration (Fig. S3, ESI†). As before, a small amount of 1,1-di(p-tolyl)ethylene (∼10%) was observed. Complex 3 is proposed to be a (PhbppeH,H)Ni(L) type Ni(0) complex (e.g. L = C6D6) on the basis of its NMR characteristics. Namely, a broad 1H NMR singlet at δ(C6D6) 3.79 ppm is consistent with a Ni-bound olefinic CH2 group. 1H-31P HMBC confirmed that the signal at δ(C6D6) 3.79 ppm is related to the 31P{1H} NMR peak at 18.5 ppm. The identity of complex 3 was further confirmed by quenching the reaction mixture with 1.5 equivalents of PPh3, which resulted in full conversion of complex 3 to (PhbppeH,H)Ni(PPh3) (4) while complex 2 remained unaffected (Fig. S6 and S7, ESI†). Complex 4 was identified by comparison with a sample independently synthesized from the PhbppeH,H ligand, Ni(cod)2, and PPh3 (ESI,† Section 1.3 and 3), and it is molecular structure was confirmed by X-ray crystal structure determination in addition to its spectroscopic identification (ESI,† Section 4). These results indicate that the second equivalent of CNtBu in Scheme 1 is not required for the carbene transfer step itself, but simplifies the final reaction mixture by capturing the formed Ni(0) fragment.
:1.1 ratio with 2 after 18 h when all 1-CNtBu was consumed (ESI,† Section S2.1). At this time, the concentration of ketenimine was approximately equal to the sum of those of complexes 2 and 3 according 1H NMR integration (Fig. S3, ESI†). As before, a small amount of 1,1-di(p-tolyl)ethylene (∼10%) was observed. Complex 3 is proposed to be a (PhbppeH,H)Ni(L) type Ni(0) complex (e.g. L = C6D6) on the basis of its NMR characteristics. Namely, a broad 1H NMR singlet at δ(C6D6) 3.79 ppm is consistent with a Ni-bound olefinic CH2 group. 1H-31P HMBC confirmed that the signal at δ(C6D6) 3.79 ppm is related to the 31P{1H} NMR peak at 18.5 ppm. The identity of complex 3 was further confirmed by quenching the reaction mixture with 1.5 equivalents of PPh3, which resulted in full conversion of complex 3 to (PhbppeH,H)Ni(PPh3) (4) while complex 2 remained unaffected (Fig. S6 and S7, ESI†). Complex 4 was identified by comparison with a sample independently synthesized from the PhbppeH,H ligand, Ni(cod)2, and PPh3 (ESI,† Section 1.3 and 3), and it is molecular structure was confirmed by X-ray crystal structure determination in addition to its spectroscopic identification (ESI,† Section 4). These results indicate that the second equivalent of CNtBu in Scheme 1 is not required for the carbene transfer step itself, but simplifies the final reaction mixture by capturing the formed Ni(0) fragment.
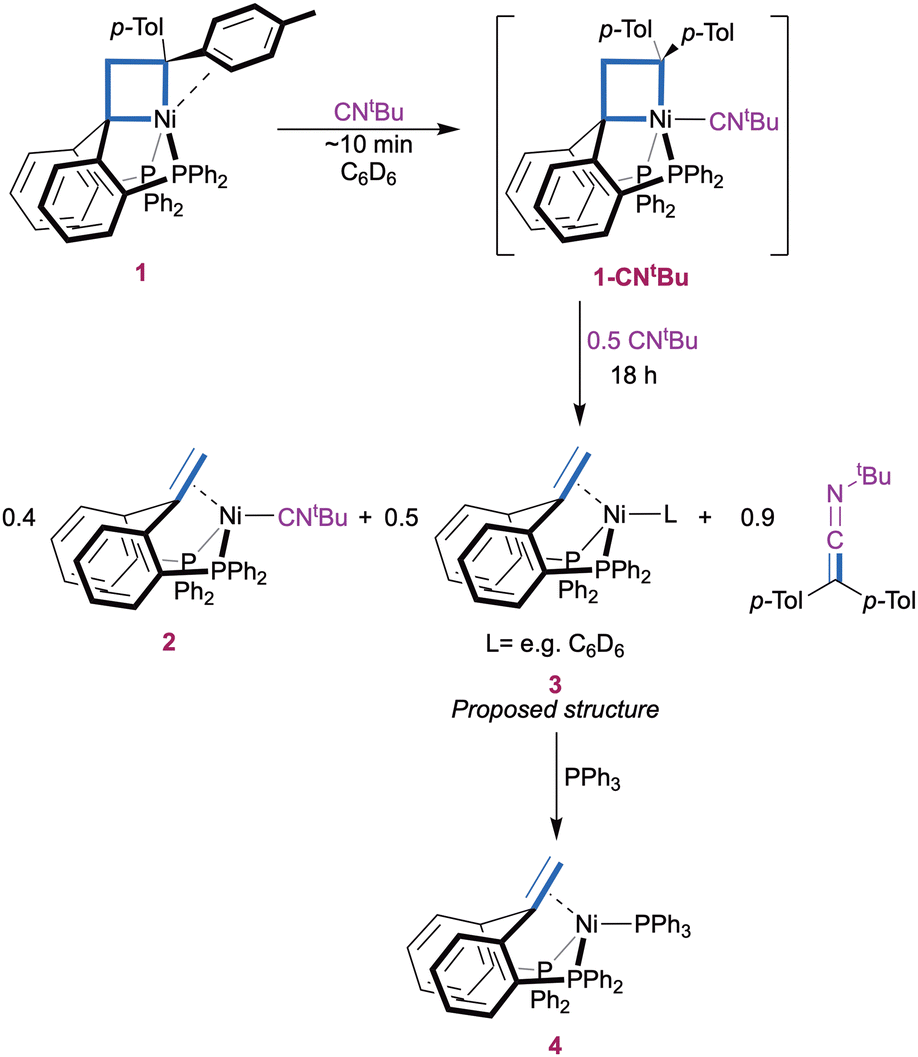 | ||
| Scheme 2 Carbene transfer with 1.5 eq. t-butylisocyanide. | ||
Ketenimines are versatile compounds in organic synthesis,29,31,34–36 which can be synthesized, amongst other, by (catalytic) coupling of a metal carbene and an isocyanide molecule.25–30 This could suggest a mechanism in which reversible [2+2] cycloreversion of the pentacoordinated nickelacyclobutane generates a carbene fragment that is intercepted by the isocyanide reagent. A similar [2+2] cycloreversion has been proposed by Miyashita to explain the reaction of the tetracoordinated nickelacyclobutane (PPh3)2Ni(2,2-dimethylpropa-1,3-diyl) with CO or cyclohexene to generate ketene (OCCH2) or the cyclopropanation product bicyclo[4.1.0]heptane, respectively.21,22,37 In a somewhat related report, Neely and coworkers described an iron azametallacyclobutene with a significant iron carbene resonance, which reacts with isocyanide and CO to form ketenimines and ketenes.38 On the other hand, isocyanides have also been known to undergo migratory insertion with metallacyclobutanes to yield metallacyclopentanes for several metals.8,39–44 In the next section, we assess the feasibility of these different processes using DFT calculations45 performed using a slightly truncated model with Ph groups instead of p-Tol.
We found the process with the lowest overall barrier to start with 1,1-insertion of the isocyanide ligand in a M–C bond of the metallacyclobutane to expand the ring (Fig. 2a). Starting from complex 1-CNtBu, insertion to yield nickelacyclopentane 5 (with the nitrogen lone pair opposite to nickel) is energetically accessible (ΔG‡ = 25.6 kcal mol−1), followed by reductive fragmentation (ΔG‡ = 25.7 kcal mol−1) forming complex 6. Change of coordination of the ketenimine from η2(C,C) to η1(N) yields complex 6 a more stable isomer (−1.9 kcal mol−1, ESI† Section S5.2). If an excess of isocyanide is available, ligand exchange to form complex 2 yields an overall free energy release of −19.8 kcal mol−1. Alternative routes starting with 1,1-insertion were found to be less favorable (ESI,† Section S5.3). The overall barrier of 25.7 kcal mol−1 is at the upper bound for a slow process at room temperature and significantly lower than all other considered pathways.46
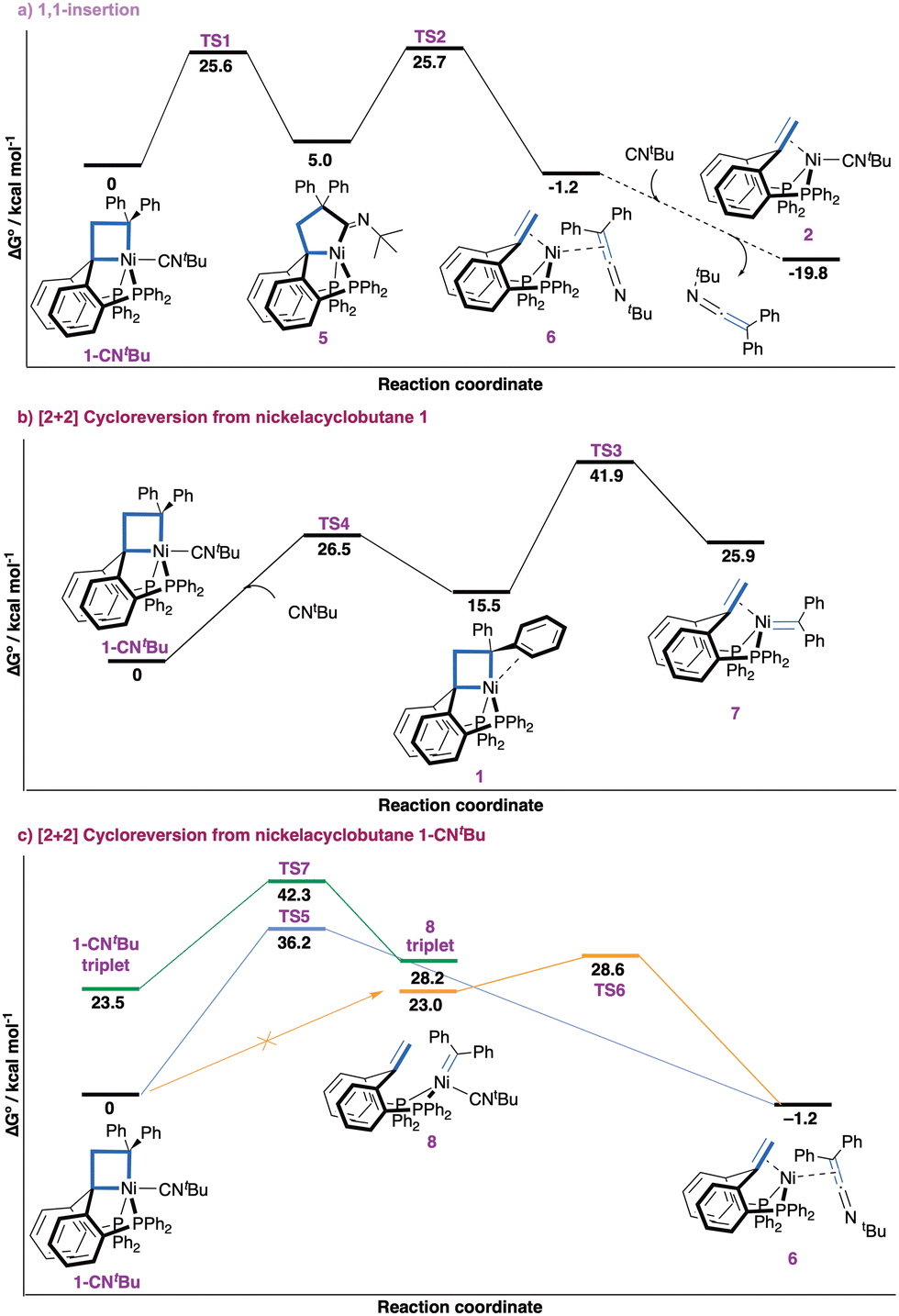 | ||
| Fig. 2 ΔG° energy profiles for the reactivity of 1-CNtBuvia 1,1-insertion (a) or [2+2] cycloreversion (b and c) mechanisms. Dashed lines connect intermediates between which no transition state was optimized. | ||
Pathways involving a [2+2] cycloreversion process yielding a nickel carbene intermediate were calculated to be energetically inaccessible (Fig. 2b). First, the formation of the putative carbene/olefin species 7 from the observed adduct 1-CNtBu after ligand dissociation is kinetically inaccessible. Initial decoordination of CNtBu to form nickelacyclobutane 1 is endergonic by 15.5 kcal mol−1 and hampered by a barrier of 26.5 kcal mol−1. [2+2] Cycloreversion to 7 comes with an additional endergonicity of 10.4 kcal mol−1 and a prohibitively high overall barrier (ΔG‡ = 41.9 kcal mol−1). An alternative process starting with decoordination of one phosphine arm of nickelacyclobutane 1 was discarded due to the high energy of this ligand dissociation (27.6 kcal mol−1, ESI† Section S5.3). Second, we investigated whether the carbene fragment could be directly transferred to the CNtBu ligand in 1-CNtBu (Fig. 2c). A transition state for concerted carbene transfer was located yielding complex 6 (−1.2 kcal mol−1), but the associated barrier is prohibitively high (ΔG‡ = 36.2 kcal mol−1). Third, a nickel carbene complex 8 bearing an isocyanide ligand was found to be relatively high in energy (23.0 kcal mol−1). Attempts to locate a transition state for the [2+2] cycloreversion yielding 8 from 1-CNtBu were unsuccessful. A potential energy surface (PES) scan suggests there is actually no transition state connecting complex 8 to 1-CNtBu (Fig. S35, ESI†). Rather, the ketenimine complex 6 appears to be an intermediate in the hypothetical transformation of 1-CNtBu into 8. This suggests complex 8 is not an intermediate of the process. Additionally, we disfavor complex 8 as a plausible intermediate due to the high free energy (ΔG‡ = 28.6 kcal mol−1) of the transition state for the formation of ketenimine complex 6 from 8. Fourth, the possibility of two-state reactivity involving the triplet state was also considered,47 but the [2+2] cycloreversion process in triplet state was associated with a prohibitively high barrier (ΔG‡ = 42.3 kcal mol−1). Finally, the direct carbene transfer and [2+2] cycloreversion process starting from a tetracoordinated nickelacyclobutane (1-CNtBu-noP) resulting from decoordination of one phosphine arm was computed (ESI,† Section 5.4). Both processes were found unfeasible with overall barriers of ΔG‡ = 52.1 kcal mol−1 and ΔG‡ = 48.3 kcal mol−1, respectively. Hence, no energetically accessible pathway for direct carbene transfer with or without a nickel carbene intermediate was identified, further supporting the sequential 1,1-insertion/reductive fragmentation as operative mechanism for the observed formal carbene transfer reaction.
The contrasting reactivity of 1-CNtBu (carbene transfer) and 1-CO (cyclopropane formation)7 is surprising in view of the isoelectronic character of CO and isocyanides. To obtain additional insights, the different decomposition pathways were investigated computationally for both compounds (ESI,† Sections S5.5 and S5.6). For 1-CNtBu the transition state for cyclopropane formation by reductive elimination was found to be prohibitively high in energy (ΔG‡ = 33.9 kcal mol−1) in good agreement with experiment. The calculated barrier of 22.5 kcal mol−1 for [2+2] cycloreversion yielding (PCcarbeneP)Ni(CNtBu) is ca. 3 kcal mol−1 lower than that for isocyanide insertion. The disparity is at odds with experimental observations but within the typical error range of DFT calculations. Additionally, the experimental observation of a small amount of 1,1-di(p-tolyl)ethene alongside the carbene transfer process is consistent with a small difference between the barriers for [2+2] cycloreversion and insertion. For 1-CO, cyclopropane formation is the favoured reaction pathway with an overall barrier of 23.8 kcal mol−1 in good agreement with experiment. The [2+2] cycloreversion process is higher in energy by 4 kcal mol−1 and insertion pathway is higher by 6.1 kcal mol−1. These differences highlight the high sensitivity of the pentacoordinated nickelacyclobutane 1 towards the electronic nature of the exogeneous ligand in apical position, the stronger π-accepting character of CO markedly favouring reductive elimination of a cyclopropane unit.
In summary, we disclose an unusual carbene transfer reaction from a pentacoordinated nickelacyclobutane to a molecule of CNtBu yielding a ketenimine. DFT calculations support a mechanistic pathway that does not involve a nickel carbene intermediate but instead a nickelacyclopentane resulting from 1,1-insertion of CNtBu into a Ni–C bond. These results further highlight the importance of the coordination environment of nickelacyclobutane intermediates for selective reactions. The possibility to access carbene-like reactivity without an actual carbene intermediate (e.g. generated by [2+2] cycloreversion) suggests a possible use of metallacyclobutanes as “carbene reservoirs” to tame unstable metal carbene fragments.
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement No 715060). The X-ray diffractometer has been financed by the Netherlands Organization for Scientific Research (NWO). This work made use of the Dutch national e-infrastructure with the support of the SURF Cooperative using grant no. EINF-3520. The authors thank Storm van der Voort, for his assistance in the synthesis of complex 4; and Dr Andrea Darú, for his assistance with DFT calculations.
Data availability
The supplementary data of this article have been included in the ESI† contains synthesis and characterization of new compounds, additional experiments and computational details. CCDC number 2365514† contains supplementary crystallographic data that can be obtained at the Cambridge Crystallographic Data Centre.Conflicts of interest
There are no conflicts to declare.Notes and references
- R. H. Grubbs and A. Miyashita, Metallacycles in Organotransition Metal Chemistry. Fundamental Research in Homogeneous Catalysis, Springer US, Boston MA, 1978, 9, 207–220 Search PubMed.
- P. W. Jennings and L. L. Johnson, Chem. Rev., 1994, 94, 2241–2290 CrossRef.
- O. M. Ogba, N. C. Warner, D. J. O’Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC.
- T. P. Montgomery, A. M. Johns and R. H. Grubbs, Catalysts, 2017, 7, 87 CrossRef.
- K. D. Kitiachvili, D. J. Mindiola and G. L. Hillhouse, J. Am. Chem. Soc., 2004, 126, 10554–10555 CrossRef PubMed.
- S. A. Künzi, R. Gershoni-Poranne and P. Chen, Organometallics, 2019, 38, 1928–1938 CrossRef.
- M. L. G. Sansores-Paredes, S. Voort, M. Lutz and M.-E. Moret, Angew. Chem., Int. Ed., 2021, 60, 26518–26522 CrossRef PubMed.
- M. L. G. Sansores-Paredes, P. M. Pérez-García and M.-E. Moret, Eur. J. Inorg. Chem., 2023, e202300192 CrossRef CAS.
- R. J. M. Klein Gebbink and M.-E. Moret, Non-Noble Metal Catalysis, Wiley, 2019 Search PubMed.
- M. C. Haibach, A. R. Ickes, A. M. Wilders and S. Shekhar, Org. Proc. Res. Dev. U. S. A., 2020, 24, 2428–2444 CrossRef CAS.
- J. M. Sarria Toro, T. Den Hartog and P. Chen, Chem. Commun., 2014, 50, 10608–10610 RSC.
- R. Waterman and G. L. Hillhouse, J. Am. Chem. Soc., 2003, 125, 13350–13351 CrossRef CAS PubMed.
- D. J. Yarrow, J. A. Ibers, Y. Tatsuno and S. Otsuka, J. Am. Chem. Soc., 1973, 95, 8590–8597 CrossRef CAS.
- J. Xu, N. B. Samsuri and H. A. Duong, Chem. Commun., 2016, 52, 3372–3375 RSC.
- M. Liu, N. Le and C. Uyeda, Angew. Chem., Int. Ed., 2023, 62, e202308913 CrossRef CAS.
- K. D. Kitiachvili, D. J. Mindiola and G. L. Hillhouse, J. Am. Chem. Soc., 2004, 126, 10554–10555 CrossRef CAS.
- S. A. Künzi, J. M. Sarria Toro, T. Den Hartog and P. Chen, Angew. Chem., Int. Ed., 2015, 54, 10670–10674 CrossRef.
- R. H. Grubbs, A. Miyashita, M. Liu and P. Burk, J. Am. Chem. Soc., 1978, 100, 2418–2425 CrossRef CAS.
- R. H. Grubbs, A. Miyashita, M.-I. M. Liu and P. L. Burk, J. Am. Chem. Soc., 1977, 99, 3863–3864 CrossRef CAS.
- R. H. Grubbs and A. Miyashita, J. Am. Chem. Soc., 1978, 100, 7418–7420 CrossRef CAS.
- A. Miyashita and R. H. Grubbs, Tetrahedron Lett., 1981, 22, 1255–1256 CrossRef CAS.
- A. Miyashita, M. Ohyoshi, H. Shitara and H. J. Nohira, Organomet. Chem., 1988, 338, 103–111 CrossRef CAS.
- E. S. Kline, R. H. Hauge, Z. H. Kafafi and J. L. Margrave, Organometallics, 1988, 7, 1512–1516 CrossRef CAS.
- D. J. Harrison, A. L. Daniels, I. Korobkov and R. T. Baker, Organometallics, 2015, 34, 5683–5686 CrossRef CAS.
- R. Aumann, Angew. Chem., Int. Ed. Engl., 1988, 27, 1456–1467 CrossRef.
- A. Grass, N. S. Dewey, R. L. Lord and S. Groysman, Organometallics, 2019, 38, 962–972 CrossRef CAS.
- C. A. Laskowski and G. L. Hillhouse, Chem. Sci., 2011, 2, 321–325 RSC.
- Z. Tang, S. Mandal, N. D. Paul, M. Lutz, P. Li, J. I. Van Der Vlugt and B. De Bruin, Org. Chem. Front., 2015, 2, 1561–1577 RSC.
- A. K. Maity, M. Zeller and C. Uyeda, Organometallics, 2018, 37, 2437–2441 CrossRef CAS PubMed.
- X. Bi, Z. Liu, S. Cao, J. Wu, G. Zanoni and P. Sivaguru, ACS Catal., 2020, 10, 12881–12887 CrossRef.
- M. Bayat, D. Gheidari and M. Mehrdad, Arab. J. Chem., 2022, 15, 104098 CrossRef.
- B. M. Hakey, D. C. Leary, J. C. Martinez, J. M. Darmon, N. G. Akhmedov, J. L. Petersen and C. Milsmann, Organometallics, 2022, 16, 2268–2280 CrossRef.
- J. Firl, W. Runge, W. Hartmann and H.-P. Utikal, Chem. Lett., 1975, 51–54 CrossRef.
- M. Alajarin, M. Marin-Luna and A. Vidal, J. Org. Chem., 2012, 29, 5637–5653 Search PubMed.
- T. R. Roose, D. S. Verdoorn, P. Mampuys, E. Ruijter, B. U. W. Maes and R. V. A. Orru, Chem. Soc. Rev., 2022, 51, 5842–5877 RSC.
- P. Lu and Y. Wang, Chem. Soc. Rev., 2012, 41, 5687–5705 RSC.
- A. Miyashita, H. Shitara and H. Nohira, J. Chem. Soc., Chem. Commun., 1985, 13, 850–851 RSC.
- C. A. Richards, N. P. Rath and J. M. Neely, Inorg. Chem., 2022, 61, 13266–13270 CrossRef CAS PubMed.
- J. Campora, P. Palma and E. Carmona, Coord. Chem. Rev., 1999, 193–195, 207–281 CrossRef CAS.
- C. Valero, M. Grehl, D. Wingbermuehle, L. Kloppenburg, D. Carpenetti, G. Erker and J. L. Petersen, Organometallics, 1994, 13, 415–417 CrossRef CAS.
- G. Greidanus-Strom, C. A. G. Carter and J. M. Stryker, Organometallics, 2002, 21, 1011–1013 CrossRef CAS.
- D. D. Wick, T. O. Northcutt, R. J. Lachicotte and W. D. Jones, Organometallics, 1998, 17, 4484–4492 CrossRef CAS.
- W. Henderson, R. D. W. Kemmitt, L. J. S. Prouse and D. R. Russell, J. Chem. Soc., Dalt. Trans., 1990, 3, 781–789 RSC.
- F. J. Berg and J. L. Petersen, Organometallics, 1993, 12, 3890–3895 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallin, 2016 Search PubMed. The full citation appears as ref. 9 in the ESI†.
- H. Ryu, J. Park, H. K. Kim, J. Y. Park, S. T. Kim and M. H. Baik, Organometallics, 2018, 37, 3228–3239 CrossRef CAS.
- D. Schröder, S. Shaik and H. Schwarz, Acc. Chem. Res., 2000, 33, 139–145 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2365514. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc04273e |
| This journal is © The Royal Society of Chemistry 2024 |