
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Scaling up electrochemical CO2 reduction: enhancing the performance of metalloporphyrin complexes in zero-gap electrolyzers†
Wiebke
Wiesner
a,
Jenny Yurley Maldonado
Arias
b,
Julia
Jökel
 b,
Rui
Cao
c and
Ulf-Peter
Apfel
*ab
b,
Rui
Cao
c and
Ulf-Peter
Apfel
*ab
aRuhr-Universität Bochum, Fakultät für Chemie und Biochemie, Anorganische Chemie I, Universitätsstraße 150, 44801 Bochum, Germany. E-mail: ulf.apfel@ruhr-uni-bochum.de
bFraunhofer UMSICHT, Department for Electrosynthesis, Osterfelder Str. 3, 46047 Oberhausen, Germany
cShaanxi Normal University, Key Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Xi’an, 710119, China
First published on 6th November 2024
Abstract
Metalloporphyrins are widely studied in the field of electrochemical CO2 reduction (CO2R), with the main focus on homogenous catalysis. Herein, six metalloporphyrins (M = Fe, Co, Ni, Cu, Zn, Ag) were incorporated in gas diffusion electrodes and used in zero-gap electrolyzers to reach varying FEs for CO of <1% (Fe,Ni), 11% (Cu), 37% (Zn), 75% (Co) and nearly 100% (Ag) at a current density of 50 mA cm−2.
Electrochemical CO2R is a promising approach to convert the greenhouse gas CO2 into useful C1 building blocks for the chemical industry, such as CO or CH4. Therefore, numerous transition metal complexes have been extensively tested for their catalytic efficiency in this reaction, predominantly in laboratory-scale systems. More recently, they also became tested in systems orientated towards practical applications.1,2 A widely investigated complex class on the laboratory-scale are metalloporphyrins, mostly comprising a meso-tetraphenylporphyrin (TPP) based ligand.3 Especially, the iron complex demonstrated an excellent performance in homogeneous catalytic systems towards the generation of CO at low overpotentials.4,5 Additionally a zinc porphyrin also showed the ability to homogeneously convert CO2 to CO.6 Similarly, cobalt porphyrins have shown efficacy in converting CO2 to formate in solution.7 In recent years, multiple studies were conducted on the investigation of the immobilization of porphyrins upon electrocatalysis. Different approaches were carried out within these reports: immobilization to carbon nanotubes (CNTs) via non-covalent interactions or covalent attachment to CNTs,8–12 as well as incorporation into gas diffusion electrodes (GDEs) with a carbon black support. Most of these studies were performed in H-type cells9,10,12–14 and occasionally in flow-cells.11,15 For instance, hydroxy substituted Fe(TPP) derivatives which have been attached to CNTs, covalently and non-covalently, maintained their high activity reaching an FE for CO between 90% and 96% in aqueous solutions.9,10 In H-Type cell experiments, Co(TPP) immobilized on a GCE surface also exhibited a good catalytic performance towards CO formation with product selectivity over 90%.12 Moreover, copper based systems applied in a two compartment cell even showed higher reduction products, like methane or ethylene with faradaic efficiencies (FEs) of 27% and 17%, respectively.11,13,16
Despite these important advances, information on the applicability and on how to utilize heterogenized molecular systems in the application of orientated conditions, such as higher current densities, omittance of mass transport limitations and potentially scalable systems, are limited. Moreover, until now research regarding nickel or silver based porphyrins as electrocatalysts for CO2 reduction is limited.3 Within this work, we report the incorporation of six different metal TPPs (M(TPP), Fig. 1a) complexes (M = Fe, Co, Ni, Cu, Zn, and Ag) into GDEs which have been applied into modular and industrially relevant zero-gap electrolyzers at elevated current densities (Fig. 1b). Due to the solely gaseous cathode environment in zero-gap setups, the often occurring problems with GDEs, such as electrode flooding, are mostly prevented in this setup. Further, the mass transport of CO2 is improved due to the omittance of a liquid catholyte, and the gas is immediately delivered to the catalytic sites.17,18 A clear comparison between zero-gap and H-type cells can be found in the ESI† (Table S1). Moreover, the overall metal loading on the electrode surface can be lowered while maintaining the same activity when metal complexes are used.19
 | ||
| Fig. 1 (a) scheme of the structure of the used complexes M(TPP); (b) photo of the used zero-gap electrolyzer during electrolysis. | ||
The metalloporphyrins M(TPP) used in this study were synthesized according to published procedures.20–24 For the manufacturing of the GDEs, catalytic inks were prepared consisting of CNTs in the presence of a Nafion binder, as well as carbon black (SuperP) in the presence of Sustainion XA-9 as the binder, which were drop cast onto a carbon cloth after sonication. Herein, the carbon black is more interesting for industrial applications due to its low cost compared to that of CNTs, the latter of which are the state of the art carbon support for porphyrin complexes in the literature.3,9,10,20 During the electrolysis experiments within this work, electrodes with a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (M(TPP): carbon support) using a loading of 0.25 mg cm−2 of the M(TPP) were investigated. For more details regarding the electrode preparation, see ESI.† The zero-gap electrolyzer was equipped with a Ni-foam anode and an anion exchange membrane (AEM) made of PiperION (40 μm thickness) which directly connects the anode and cathode sides. The cathode side was fed with humidified gaseous CO2 (100% relative humidification), whereas on the anode side, 1 M KOH was cycled as the anolyte (20 mL min−1). Electrolysis was performed for 1 h at 50 mA cm−2 followed by 1 h at 100 mA cm−2 at 25 °C without exchanging any electrolyzer parts in between the change of current density. To determine the product gas composition, GC injections were performed at every 30 min of catalysis (for averaged values, see Tables S2 and S3, ESI†). The determined faradaic efficiencies (FEs) after 1 h of electrolysis are shown in Fig. 2a and b, respectively.
:2 (M(TPP): carbon support) using a loading of 0.25 mg cm−2 of the M(TPP) were investigated. For more details regarding the electrode preparation, see ESI.† The zero-gap electrolyzer was equipped with a Ni-foam anode and an anion exchange membrane (AEM) made of PiperION (40 μm thickness) which directly connects the anode and cathode sides. The cathode side was fed with humidified gaseous CO2 (100% relative humidification), whereas on the anode side, 1 M KOH was cycled as the anolyte (20 mL min−1). Electrolysis was performed for 1 h at 50 mA cm−2 followed by 1 h at 100 mA cm−2 at 25 °C without exchanging any electrolyzer parts in between the change of current density. To determine the product gas composition, GC injections were performed at every 30 min of catalysis (for averaged values, see Tables S2 and S3, ESI†). The determined faradaic efficiencies (FEs) after 1 h of electrolysis are shown in Fig. 2a and b, respectively.
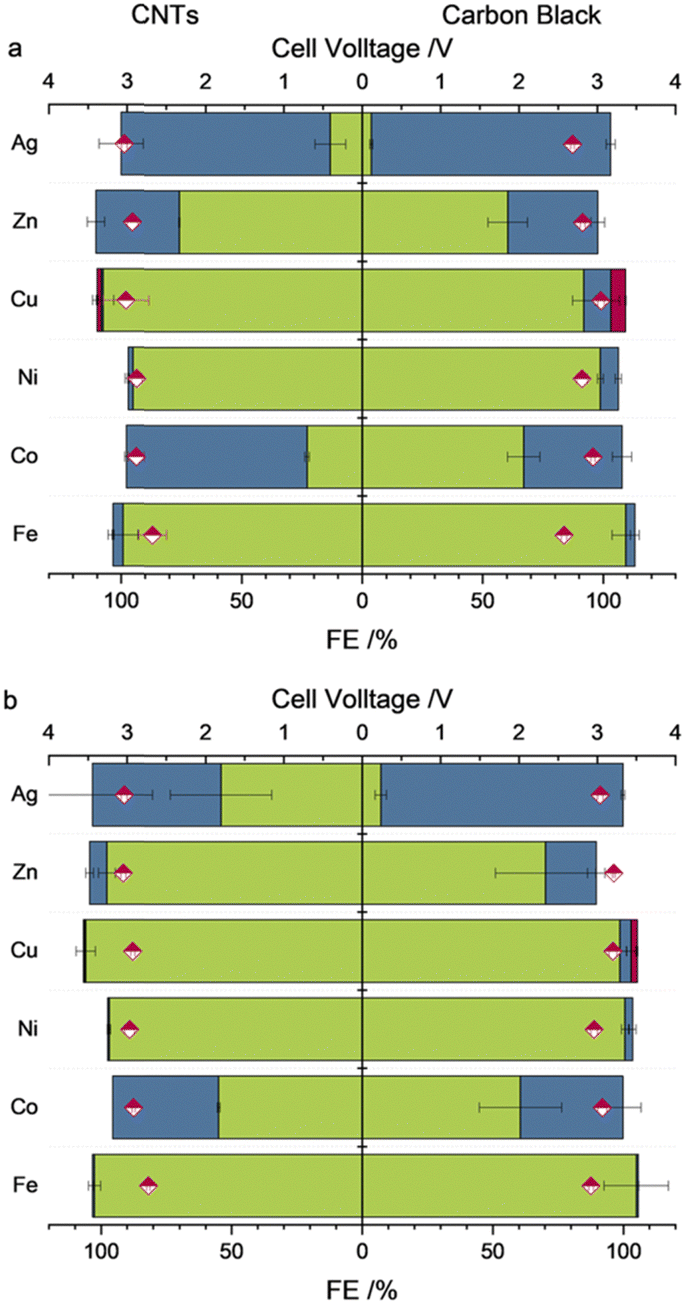 | ||
| Fig. 2 Observed averaged cell voltages (pink diamonds) and FEs for H2 (green), CO (blue) and CH4 (pink) with either MWCNTs (left half of graph) or carbon black (right half of graph) as carbon support after 1 h of electrolysis at (a) 50 mA cm−2 or (b) 100 mA cm−2. | ||
The different M(TPP) complexes show highly variable activities towards the formation of CO ranging from below 1% (Fe,Ni), 11% (Cu) accompanied by 6% FE for CH4, 37% (Zn), 75% (Co) and nearly 100% (Ag) at an applied current density of 50 mA cm−2. These altering reactivities cannot be attributed to differing electrode morphology when using different catalysts, since the SEM measurements performed pre- and post-electrolysis showed similar surface structures of the electrodes, including metal complex particles submerged in the used carbon support (Fig. S1–S8, ESI†). For instance, the homogenously highly active Fe(TPP) complex does not show any activity towards CO generation under the herein applied conditions, reaching FEs below 1%. To get an insight on the present metal species on the electrode surface and potential modifications during electrolysis, X-ray photoelectron spectroscopy (XPS) of the electrodes prior and after catalysis was performed. Prior to catalysis, the XPS spectrum of the Fe 2p3/2 orbital shows a peak at 711.6 eV, which is in accordance with XPS spectra reported for iron porphyrin complexes.9,10,15 Nevertheless, a reliable fitting is not possible due to the low intensity. The intensity of the Fe 2p XPS spectrum after electrolysis (Fig. S10, ESI†) is even lower and therefore does not allow us to make a statement as to the presence or not of an iron species, while in the XPS analysis of the membrane surface (Fig. S21, ESI†), iron was observed on it. These XPS results indicate that the complex decomposes during catalysis and potentially gets demetallized under the applied conditions, which differ strongly from those reported to date. During additional experiments at a decreased current density of 10 mA cm−2, the Fe(TPP) retains its high activity towards CO generation (>90%) within the first 30 min of catalysis. However, a decline in activity was observed over the 2-hour electrolysis period, further suggesting that the catalyst is unstable under the harsher conditions compared to those in homogeneous experiments (Table S3, ESI†). Another catalyst which exhibited nearly no activity towards the generation of CO or any other C1 reduction products is Ni(TPP). This is in accordance with studies performed on nickel porphyrins in electrochemical CO2R since nickel porphyrin complexes are not broadly known for efficient CO2R without further ligand modifications.25 XPS analysis of the pre- and post-electrolysis GDEs indicated that the Ni might have been washed out during catalysis since the intensity of the Ni 2p orbital binding energies is very low in the post-catalysis electrode, while again the signals of the N 1s orbitals of the porphyrin nitrogens are not affected by catalysis (Fig. S11 and S12, ESI†). The vanishing of the metal centres for the Fe(TPP) and Ni(TPP) after electrolysis offer a possible explanation for their low catalytic activities. The complexes appear to be unstable under the applied conditions. Even though the corresponding copper complex Cu(TPP) also mainly favours the hydrogen evolution reaction (HER) over CO2R, it is also able to produce CO with an FE of 11% and CH4 in the noteworthy amount of 6% at an applied current density of 50 mA cm−2, a behaviour that supports previous studies.13,26 The pre-electrolysis Cu 2p3/2 XPS spectra show a peak at 934.3 eV with corresponding satellites at higher binding energies. (Fig. 3) The presence of a Cu(II) species is verified by the characteristic shape of the Cu 2p satellite peak. During electrolysis, the generation of a reduced metal species can be observed, indicated by an emerging peak at a lower binding energy of 933.2 eV, while the Cu(II) species stays as the main copper species present.27 It can be assumed that this species is hindering the CO2R since the formation of C1 reduction products is decreased with a higher current density and longer catalysis duration. Additionally, Zn(TPP) maintains its ability to reduce CO2 to CO under the herein applied harsh electrolysis conditions, reaching FEs up to 37% at 50 mA cm−2. Even though the FE for CO decreases with an increase of current density, the metal centre stays unaffected during catalysis, as indicated by XPS showing an unaltered peak for the Zn 2p3/2 orbital at 1022.3 eV. (Fig. 3) Additionally, an Auger parameter for the zinc centre of 2010.01 eV was obtained, which further confirms its oxidation state.28 Further, Co(TPP) stands out as the only non-noble metal porphyrin complex tested in this study, which shows a higher selectivity for CO formation compared to that of H2, reaching an FE of up to 75% at 50 mA cm−2, and maintaining the high activity towards CO2R, which was also observed in H-type cell experiments.12 Moreover, it is noteworthy that the Co complex is the only one within this series showing an enhanced performance in the presence of MWCNTs as the carbon support. Under the same catalysis conditions with a carbon black support the FE for CO is nearly 35% lower. Just as for the Zn complex, no decomposition of the Co(II) centre was observed in XPS, keeping the binding energies for the Co 2p3/2 orbital at 781 eV (Fig. 3) which is also in accordance with reported XPS values for Co porphyrin complexes.29 Nevertheless, the silver-based catalyst achieved by far the greatest performance towards CO2R reaching an FE of nearly 100% towards CO formation when a current density of 50 mA cm−2 is applied and carbon black is used as the carbon support. Further, after applying a current density of 100 mA cm−2 the FE for CO only decreased to 92%, maintaining its high activity. While not only showing the highest performance towards CO2 reduction, the experiments using the Ag(TPP) with a carbon black support were accompanied by the lowest cell voltage of 3.0 V at a current density of 100 mA cm−2 for all complexes which are active in CO2R. Nevertheless, this transition metal complex seems to transform into metallic silver particles during electrolysis, which could be stabilized by the nitrogen rich ligand environment. In the post-electrolysis XPS, new bands evolved at lower binding energies for the Ag 3d5/2 and 3d3/2 orbital signals, which resemble a reduced species (Fig. 3).19 However, making a definite statement on the silver oxidation state is not possible, since the intensity of the silver Auger peaks in the corresponding XPS is too low to make a reliable fitting (Fig. S20, ESI†). One similarity of all t complexes, except Co(TPP), is that they all perform better in the presence of the more cost-efficient carbon black, which could be attributed to a better distribution of the carbon particles over the electrode surface. In SEM measurements (Fig. S1–S8, ESI†) it was seen that the CNTs rather tended to form agglomerates instead of an even layer across the complete electrode area. Furthermore, in the literature it is known that Co(TPP), which is the only active catalyst with empty d-orbitals within this study, can induce electronic interactions with CNTs via interactions of their π-systems. This could facilitate the electron transfer from the carbon support towards the empty d-orbitals of the catalytically active centre.30–32 An increase of the current density also always results in an increased HER for all the complexes.
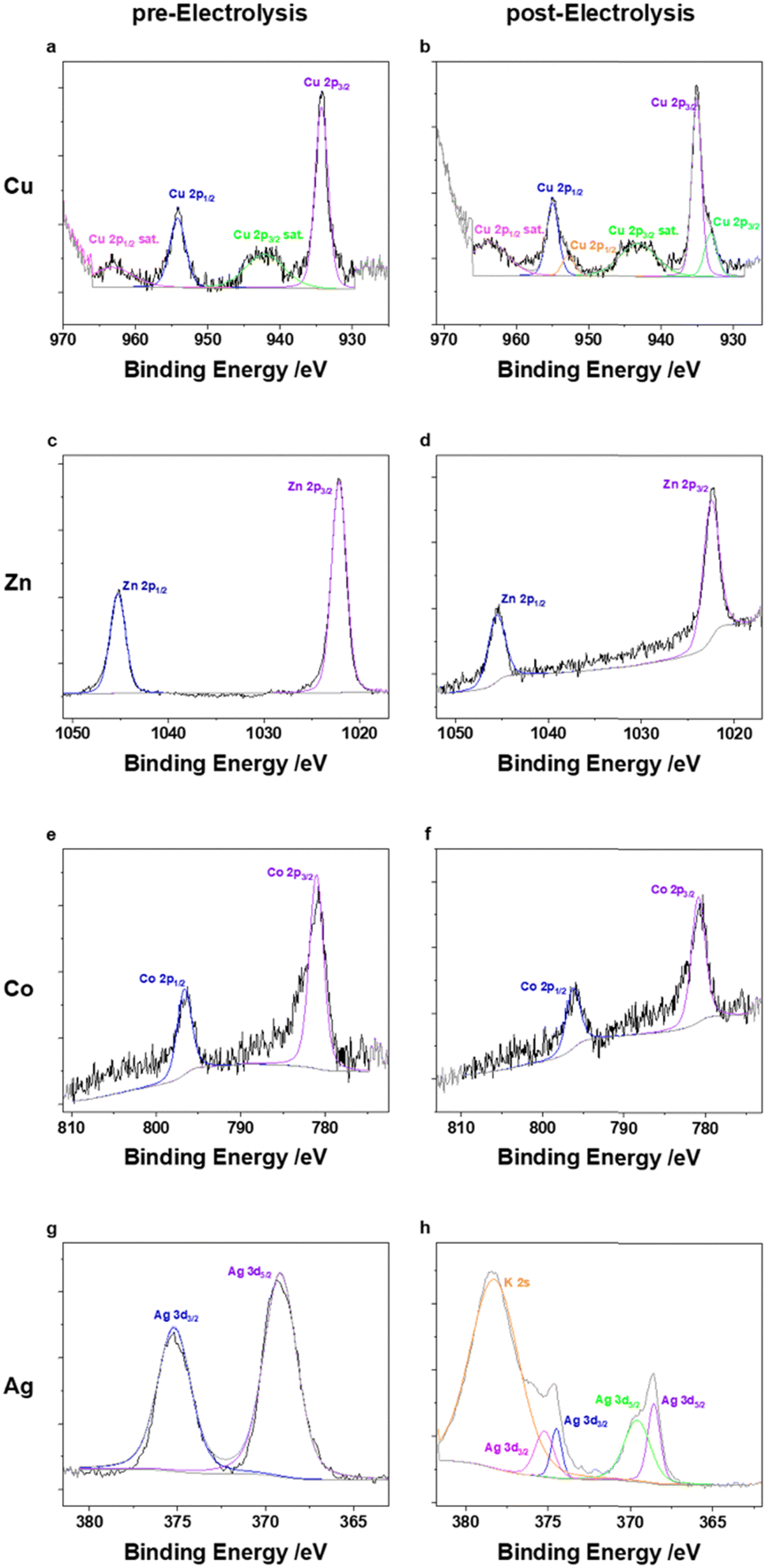 | ||
| Fig. 3 Fitted XPS spectra for the Ag 3d (a) and (e), Cu 2p (b) and (f), Zn 2p (c) and (g) and Co 2p (d) and (h) orbitals. Spectra on the left side belong to the electrode pre-electrolysis, spectra on the right side to the electrode post-electrolysis. | ||
In conclusion, this study emphasizes the that results obtained with homogeneous or immobilized catalysts in a liquid catholyte are not easily transferable to application-ready setups. This is due to the significantly altered local reaction environments, which include pronounced changes in CO2 and H2O concentration as well as pH gradients, coupled with more severe catalytic conditions. Catalysts well-known for their efficiency and selectivity in CO2 reduction in solution or immobilized in single-cell set ups, such as the prominent Fe(TPP), do not demonstrate activity under application-orientated settings. Furthermore, it was shown that the most suitable carbon support needs to be determined each time to boost the catalyst performance as much as possible. Contrary to previously reported studies on M(TPP)s as catalysts for electrochemical CO2 reduction, two complexes not typically noted for this reaction showed the highest performance in a zero-gap assembly: the Co(TPP) achieved an FE of 40% at 100 mA cm−2 and Ag(TPP) achieved an FE of 92% at 100 mA cm−2 while employing moderate cell voltages of 3 V. Under the herein applied conditions, the Fe(TPP) and Ni(TPP) do not exhibit a significant FE for CO generation, which can be attributed to a possible demetallation of the complex during electrolysis, whereas the Zn(TPP) still reaches an Fe for CO of 19% at 100 mA cm−2. Additionally it is noteworthy that the Cu(TPP) produces CH4 with an FE of 6% at an applied current density of 50 mA cm−2.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- S. Gu, A. N. Marianov, T. Lu and J. Zhong, Chem. Eng. J., 2023, 470, 144249 CrossRef CAS.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed.
- J. Jiang, A. J. Matula, J. R. Swierk, N. Romano, Y. Wu, V. S. Batista, R. H. Crabtree, J. S. Lindsey, H. Wang and G. W. Brudvig, ACS Catal., 2018, 8, 10131–10136 CrossRef CAS.
- K. Takahashi, K. Hiratsuka, H. Sasaki and T. Shinobu, Chem. Lett., 1979, 305–308 CrossRef CAS.
- G. Xu, H. Lei, G. Zhou, C. Zhang, L. Xie, W. Zhang and R. Cao, Chem. Commun., 2019, 55, 12647–12650 RSC.
- A. Maurin and M. Robert, J. Am. Chem. Soc., 2016, 138, 2492–2495 CrossRef CAS PubMed.
- A. Maurin and M. Robert, Chem. Commun., 2016, 52, 12084–12087 RSC.
- M. N. Hossain, R. Khakpour, M. Busch, M. Suominen, K. Laasonen and T. Kallio, ACS Appl. Energy Mater., 2023, 6, 267–277 CrossRef CAS.
- X. Hu, M. H. Rønne, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Angew. Chem., Int. Ed., 2017, 56, 6468–6472 Search PubMed.
- Z. Weng, J. Jiang, Y. Wu, Z. Wu, X. Guo, K. L. Materna, W. Liu, V. S. Batista, G. W. Brudvig and H. Wang, J. Am. Chem. Soc., 2016, 138, 8076–8079 CrossRef CAS PubMed.
- Y. Wu, J. Jiang, Z. Weng, M. Wang, D. L. J. Broere, Y. Zhong, G. W. Brudvig, Z. Feng and H. Wang, ACS Cent. Sci., 2017, 3, 847–852 CrossRef CAS.
- M. Abdinejad, C. Dao, X.-A. Zhang and H. B. Kraatz, J. Energy Chem., 2021, 58, 162–169 CrossRef CAS.
- P. Yu, X. Lv, Q. Wang, H. Huang, W. Weng, C. Peng, L. Zhang and G. Zheng, Small, 2023, 19, 2205730 CrossRef CAS PubMed.
- A. Gawel, T. Jaster, D. Siegmund, J. Holzmann, H. Lohmann, E. Klemm and U.-P. Apfel, iScience, 2022, 25, 104011 CrossRef CAS PubMed.
- L. Hoof, N. Thissen, K. Pellumbi, K. Junge Puring, D. Siegmund, A. K. Mechler and U.-P. Apfel, Cell Rep. Phys. Sci., 2022, 3, 100825 CrossRef CAS.
- K. Pellumbi, D. Krisch, C. Rettenmaier, H. Awada, H. Sun, L. Song, S. A. Sanden, L. Hoof, L. Messing, K. J. Puring, D. Siegmund, B. R. Cuenya, W. Schöfberger and U.-P. Apfel, Cell Rep. Phys. Sci., 2023, 4, 101746 CrossRef CAS.
- Y. Okabe, S. K. Lee, M. Kondo and S. Masaoka, J. Biol. Inorg. Chem., 2017, 22, 713–725 CrossRef CAS.
- I. K. Attatsi, W. Zhu and X. Liang, New J. Chem., 2020, 44, 4340–4345 RSC.
- S. A. Vail, D. I. Schuster, D. M. Guldi, M. Isosomppi, N. Tkachenko, H. Lemmetyinen, A. Palkar, L. Echegoyen, X. Chen and J. Z. H. Zhang, J. Phys. Chem. B, 2006, 110, 14155–14166 CrossRef CAS.
- J. Śniechowska, P. Paluch and M. J. Potrzebowski, RSC Adv., 2017, 7, 24795–24805 RSC.
- M. So, V. A. L. Roy, Z. Xu, S. S. Chui, M. Yuen, C. Ho and C. Che, Chem. – Asian J., 2008, 3, 1968–1978 CrossRef CAS.
- M. Abdinejad, L. F. B. Wilm, F. Dielmann and H. B. Kraatz, ACS Sustainable Chem. Eng., 2021, 9, 521–530 CrossRef CAS.
- Y. Wang, M. Liu, G. Gao, Y. Yang, R. Yang, H. Ding, Y. Chen, S. Li and Y. Lan, Angew. Chem., Int. Ed., 2021, 60, 21952–21958 Search PubMed.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- M. Zhu, J. Chen, L. Huang, R. Ye, J. Xu and Y. Han, Angew. Chem., Int. Ed., 2019, 58, 6595–6599 Search PubMed.
- X. Lu, B. Dereli, T. Shinagawa, M. Eddaoudi, L. Cavallo and K. Takanabe, Chem. Catal., 2022, 2, 1143–1162 CrossRef CAS.
- S. Dou, L. Sun, S. Xi, X. Li, T. Su, H. J. Fan and X. Wang, ChemSusChem, 2021, 14, 2126–2132 CrossRef CAS PubMed.
- F. Z. Bouanis, M. Bensifia, I. Florea, S. Mahouche-chergui, B. Carbonnier, D. Grande, C. Léonard, A. Yassar and D. Pribat, Org. Electron., 2021, 96, 106212 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04497e |
| This journal is © The Royal Society of Chemistry 2024 |