
Diamidocarbene-derived palladium and nickel–sulfur clusters†
Minji
Lee‡
ab,
Hyunju
Noh‡
ab and
Youngsuk
Kim
 *abc
*abc
aDepartment of Chemistry, Pusan National University, Busan, Republic of Korea. E-mail: youngsuk.kim@pusan.ac.kr
bInstitute for Future Earth, Pusan National University, Busan, Republic of Korea
cChemistry Institute for Functional Materials, Pusan National University, Busan, Republic of Korea
First published on 28th October 2024
Abstract
Novel palladium and nickel–sulfur clusters were synthesized using a diamidocarbene-derived carbon disulfide ligand. Structural characterization revealed a tetranuclear metal–sulfur cluster geometry with each metal center exhibiting square-planar coordination. The ligand was redox-active, accommodating oxidation states ranging from 0 to −2.
Metal clusters are compounds consisting of a small number of closely packed metal atoms stabilized by ligands.1 These clusters exhibit unique properties owing to the metal–metal and metal–ligand interactions, distinguishing them from individual atoms and bulk metals.2 Metal–sulfur clusters, incorporating sulfides and/or thiolates as ligands, are particularly notable for their distinctive reactivity and magnetic properties.3 For example, iron–sulfur clusters are widely used as prosthetic groups in biological systems, facilitating electron transfers and catalyzing multi-electron transformations of small molecules (Fig. 1A).4 These clusters often incorporate other transition metals, such as nickel, vanadium, or molybdenum, enhancing their versatility and functionality in biological systems.5 In addition, cobalt–sulfur clusters have recently gained significant attention owing to their unique electronic properties, rendering them promising candidates for novel electronic materials.6 Other metal–sulfur clusters incorporating different transition and main-group metals have also been extensively studied.7
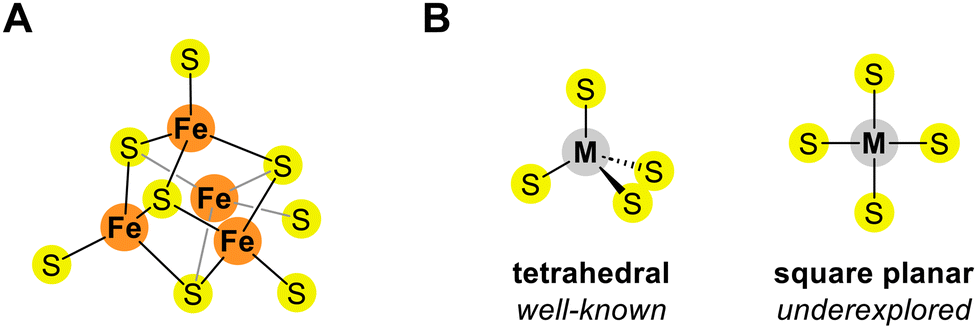 | ||
| Fig. 1 (A) Typical structure of an iron–sulfur cluster consisting of four iron atoms. (B) Tetrahedral and square-planar coordination geometries. | ||
Exploring new geometries and connectivities in metal–sulfur clusters is essential for developing clusters with novel functions and broadening their applications. Thus, the objective of this study was to expand this scope by using tetravalent metal centers with square-planar coordination geometry, such as Pd(II) and Ni(II), which are less commonly used in metal–sulfur cluster chemistry (Fig. 1B).8 Specifically, we synthesized and characterized novel metal–sulfur clusters with square-planar Pd(II) and Ni(II), using a new ligand derived from diamidocarbene (DAC) and carbon disulfide (CS2).
N-heterocyclic carbenes (NHCs) form stable zwitterionic adducts with CS2.9 These imidazolium dithiocarboxylates are versatile ligands for coordinating various transition metals.10 We aimed to extend this chemistry to more electrophilic singlet carbene, such as DAC reported by Bielawski et al.11
The DAC–CS2 adduct (L) was synthesized from the reaction between DAC and CS2 in diethyl ether (Fig. 2A), yielding a red solid with 88% yield. Crystal structure determination revealed, as expected, a molecular structure consistent with zwitterionic DAC–CS2, featuring a CDAC–CCS2 bond length of 1.499(11) Å and an N–C–C–S torsion angle of 90.7(8)°, indicating the single bond character of CDAC–CCS2 (Fig. 2B). These structural parameters were very similar to those of NHC–CS2 adducts, which typically show a CNHC–CCS2 bond length of 1.49 Å and an N–C–C–S torsion angle of 90°.9f
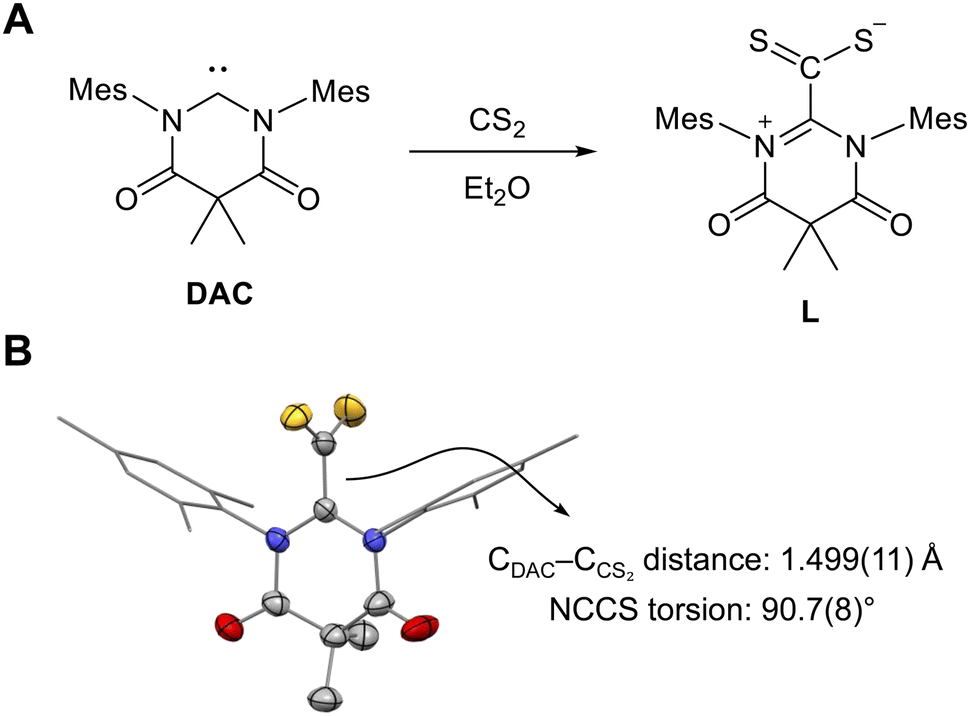 | ||
| Fig. 2 (A) Synthesis of DAC–CS2 adduct L. (B) Solid-state structure of L obtained using single-crystal X-ray crystallography. The thermal ellipsoids are shown at the 50% probability level with carbon (gray), sulfur (yellow), nitrogen (blue), and oxygen (red). Mes substituents are shown as sticks. Hydrogen atoms are omitted for clarity. | ||
After synthesizing the ligand L, we investigated its coordination reactivity with Pd(0) and Ni(0) sources. The reaction of L with tris(dibenzylideneacetone)dipalladium (Pd2dba3) in acetonitrile produced black crystals in 53% yield (Fig. 3A). Single-crystal X-ray crystallography revealed that the molecular structure of the product was an unexpected tetrapalladium cluster (L4Pd4) with a previously unknown Pd4S8 core structure exhibiting pseudo-S4 symmetry (Fig. 3B and Table S1, ESI†). Interestingly, each palladium center displayed distorted square-planar PdS4 coordination, consistent with a Pd oxidation state of +2 (sum of S–Pd–S angles for each palladium center was approximately 354°). Unlike the free L, the DAC–CS2 ligands in the L4Pd4 cluster displayed planar geometry with N–C–C–S torsion angles of 9.3(4)–10.7(4)°. The CDAC–CCS2 bond lengths for the ligands (1.359(4)–1.361(4) Å) were significantly smaller than that of the free L, indicating double-bond character. These structural parameters suggest that each ligand was reduced by two electrons to L2−, completing the charge balance of the cluster. Notably, the analogous reaction of NHC–CS2 adducts with Pd(0) resulted in a one-electron reduction, forming a radical anionic ligand.12 This difference clearly illustrates the stronger electron-accepting ability of DAC compared to typical NHCs.
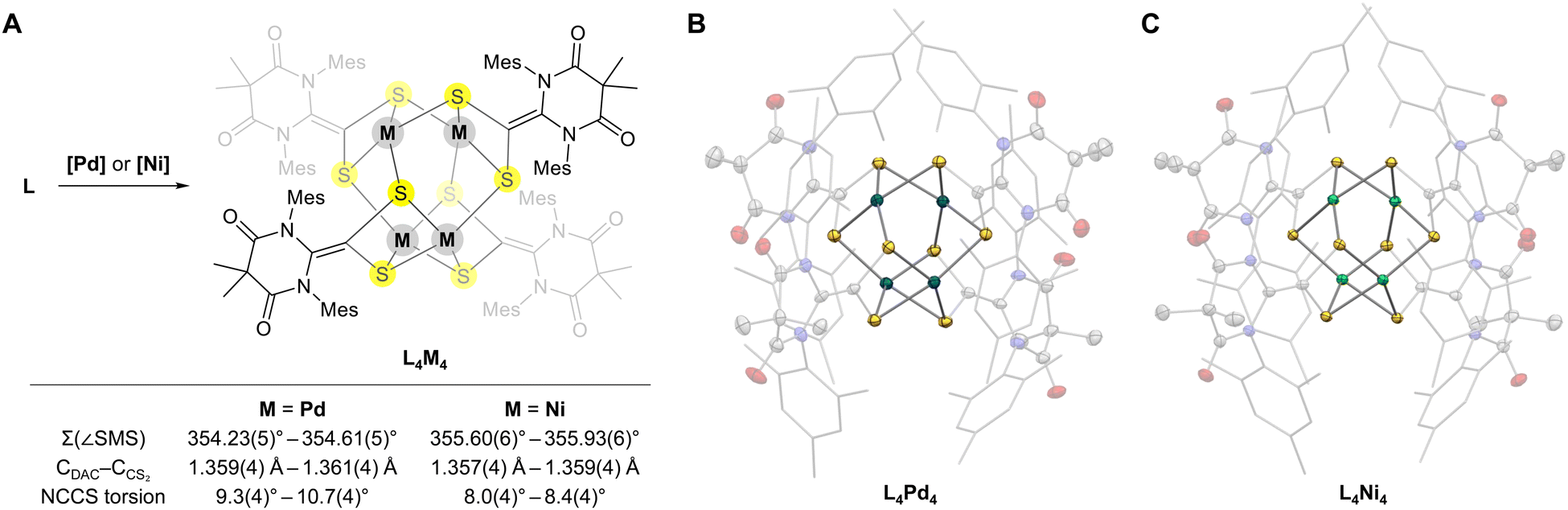 | ||
| Fig. 3 (A) Syntheses of the tetrapalladium (L4Pd4) and tetranickel clusters (L4Ni4). [Pd] = Pd2dba3, [Ni] = Ni(cod)2 (B) Solid-state structure of L4Pd4 and (C) L4Ni4 obtained using single-crystal X-ray crystallography. The thermal ellipsoids are shown at the 50% probability level with palladium (turquoise) and nickel (green). Mes substituents are shown as sticks. Hydrogen atoms, solvent molecules, and disorders are omitted for clarity. | ||
When L was reacted with Ni(cod)2 as a Ni(0) source, brown crystals were formed and isolated in 64% yield. Single-crystal X-ray crystallography revealed the formation of a tetranickel cluster (L4Ni4) with a structure analogous to the L4Pd4 cluster (Fig. 3C and Table S2, ESI†). Each nickel center exhibited a distorted square-planar geometry, similar to that in the L4Pd4 cluster (sum of S–Ni–S angles was approximately 356°). The N–C–C–S torsion angles ranged from 8.0(4)° to 8.4(4)°, and the CDAC–CCS2 bond lengths were 1.357(4)–1.359(4) Å, consistent with the double-bond character of CDAC–CCS2 and dianionic nature (L2−) of the ligands.
Proton nuclear magnetic resonance (1H NMR) spectra of L4Pd4 and L4Ni4 displayed a single set of signals from DAC (Fig. S2 and S3, ESI†), consistent with S4 symmetry, while the desymmetrization of DAC–CS2 peaks indicated the formation of a rigid structure that prevented the rotation of CDAC–CCS2 bonds, consistent with their double-bond character. The addition of free L to the solution of L4Pd4 or L4Ni4 did not result in peak broadening in the 1H NMR spectra, confirming that the clusters retained their structures in solution (Fig. S4 and S5, ESI†).
Interestingly, during the synthesis of L4Ni4, a trinickel cluster byproduct (L4Ni3) was also observed (Fig. 4A). Although this byproduct could not be isolated, we crystallized the cluster and examined its structure (Fig. 4B and Table S3, ESI†). Similar to the L4Ni4 cluster, each nickel center in L4Ni3 exhibited distorted square-planar NiS4 coordination, consistent with the Ni oxidation state of +2. One of the four ligands exhibited different structural parameters. L1–L3 had a geometry similar to those of the ligands in L4Pd4 and L4Ni4 (CDAC–CCS2 bond lengths of 1.338(13)–1.358(13) Å and N–C–C–S torsion angles of 7.8(8)–9.4(7)°), suggestive of a formal dianionic ligand L2−. In contrast, L4 had a CDAC–CCS2 bond length of 1.523(12) Å and an N–C–C–S torsion angle of 76.5(9)°, resembling the free ligand L. These structural parameters indicate the redox-non-innocence of ligand L, confirming its ability to change oxidation states from 0 to −2 depending on the coordination environment (Fig. 4C).
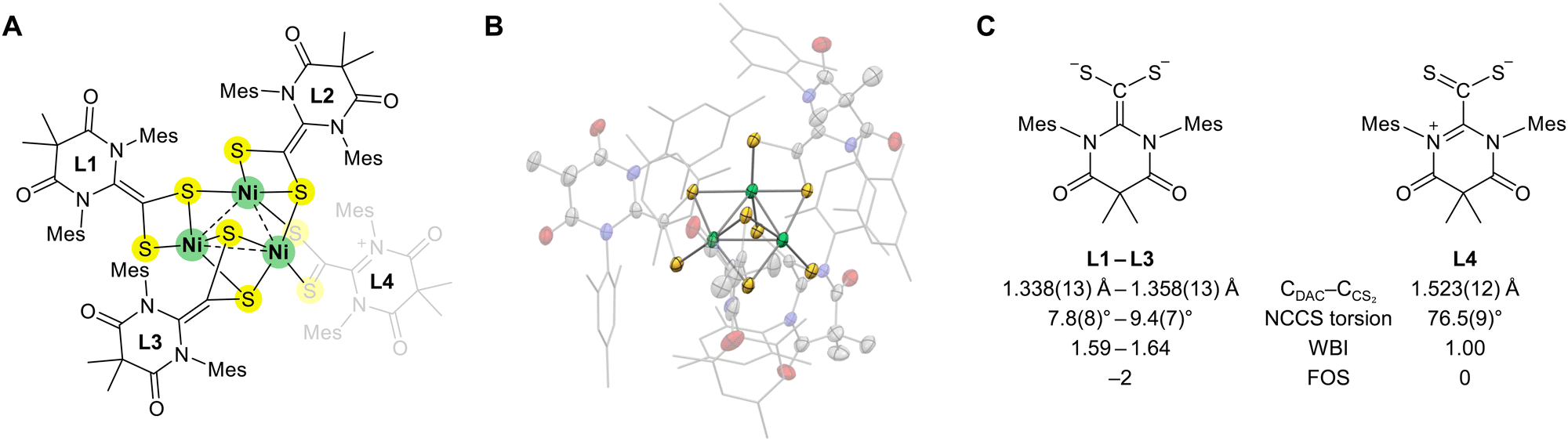 | ||
| Fig. 4 (A) Structure of the trinickel cluster (L4Ni3). (B) Solid-state structure of L4Ni3 obtained using single-crystal X-ray crystallography. The thermal ellipsoids are shown at the 50% probability level with colors as in Fig. 3. Mes substituents are shown as sticks. Hydrogen atoms, solvent molecules, and disorders are omitted for clarity. (C) Ligands in L4Ni3 with different structural parameters. CDAC–CCS2 bond lengths, N–C–C–S torsion angles, Wiberg bond index (WBI) of CDAC–CCS2 bonds, and formal oxidation states (FOSs) are shown. | ||
To further validate our observations and gain insights into the electronic structure of the clusters, density functional theory (DFT) calculations were performed. The optimized geometries (TPSSh/Def2-TZVP) closely matched the experimental structures obtained from X-ray crystallography (Tables S4–S6, ESI†). The ligands in the L4Pd4 and L4Ni4 clusters showed optimized CDAC–CCS2 bond lengths ranging from 1.359 to 1.361 Å and N–C–C–S torsion angles of 2.4° to 3.6°. These values corroborated the crystallographic data indicating the double-bond character of CDAC–CCS2 and planarization of the L2− in the cluster environment. In contrast, the L4 ligand of the L4Ni3 cluster showed the calculated CDAC–CCS2 bond length of 1.480 Å, and N–C–C–S torsion angle of 82.4°, consistent with its zwitterionic electronic structure. Wiberg bond indices (WBIs) were calculated using the natural atomic orbital (NAO) basis set. The CDAC–CCS2 bonds exhibited WBIs in the range of 1.59–1.64, aligning with the observed double-bond character. In contrast, the CDAC–CCS2 bond in the L4 ligand of L4Ni3 cluster had a WBI of 1.0, consistent with the absence of the double-bond character (Fig. 4C).
DFT calculations also reproduced the stretched tetrahedron arrangement of metal atoms in the L4Pd4 and L4Ni4 clusters (Fig. 5). For example, for the L4Pd4 cluster, the Pd1–Pd2 (X-ray: 2.6522(6) Å; DFT: 2.665 Å) and Pd3–Pd4 (X-ray: 2.6526(5) Å; DFT: 2.665 Å) distances were notably smaller than other Pd–Pd distances (X-ray: 3.2845(4)–3.3064(4) Å; DFT: 3.310–3.311 Å). These distances were comparable to the Pd–Pd bond lengths observed in previously reported square-planar Pd(II) dimers (approximately 2.7 Å)13 and aligned closely with the Pd–Pd distances in bulk palladium metal (approximately 2.75 Å).14 Similarly, in the L4Ni4 cluster, the calculated Ni1–Ni2 and Ni3–Ni4 distances of 2.402 Å closely matched the experimental values of 2.4214(6) Å (Ni1–Ni2) and 2.4156(5) Å (Ni3–Ni4). Despite these small intermetallic distances, no significant electronic communication was observed between the metal atoms. The calculated WBIs for Pd1–Pd2 (0.18) in the L4Pd4 cluster and Ni1–Ni2 (0.16) in the L4Ni4 cluster suggest formal metal–metal bond orders close to zero, consistent with the characterization of the metal centers as square planar.
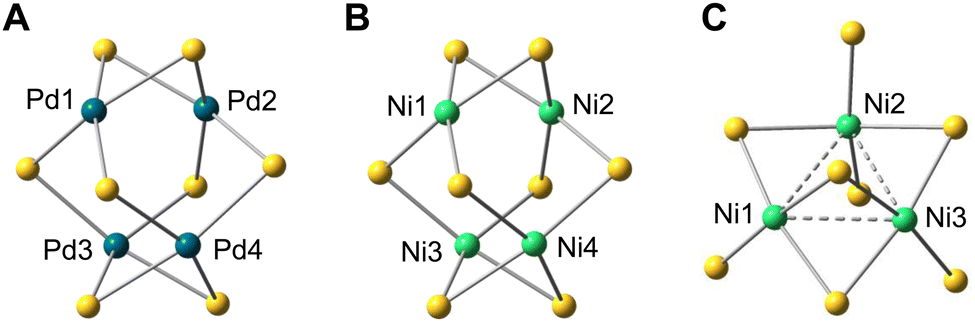 | ||
| Fig. 5 DFT-optimized structure of (A) L4Pd4, (B) L4Ni4, and (C) L4Ni3. Only metal and sulfur atoms are shown. | ||
In summary, we synthesized and characterized novel palladium and nickel–sulfur clusters, L4Pd4 and L4Ni4, using a DAC-derived carbon disulfide ligand. These clusters exhibited a previously unknown tetranuclear metal–sulfur cluster geometry, with each metal center displaying square-planar coordination. X-ray crystallography and DFT calculations revealed the redox-active nature of the ligand, capable of adopting formal oxidation states (FOSs) ranging from 0 to −2. While the FOS of the free ligand is 0, the ligands in the L4Pd4 and L4Ni4 clusters exhibited FOSs of −2. Interestingly, in the L4Ni3 cluster, one ligand retained an FOS of 0, while the other three ligands had FOSs of −2. The distinct structural features and redox properties of these palladium and nickel clusters introduce a novel dimension to metal–sulfur cluster chemistry. The potential applications of these clusters in catalysis and materials science are currently under investigation.
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (Ministry of Science and ICT) (RS-2023-00210804). This research was also supported by Global – Learning & Academic research institution for Master's, PhD students, and Postdocs (LAMP) Program of the National Research Foundation of Korea (NRF) grant funded by the Ministry of Education (RS-2023-00301938). Additionally, this work was supported by the Korea Basic Science Institute (National research Facilities and Equipment Center) grant funded by the Korea government (Ministry of Science and ICT) (RS-2024-00403266). Finally, this work was also supported by Pusan National University Research Grant, 2022. We gratefully acknowledge Dr Dongwook Kim for his assistance with single crystal X-ray structure determination.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for L, L4Pd4, L4Ni4, and L4Ni3 has been deposited at the CCDC (2381957–2381960†) and can be obtained from https://www.ccdc.cam.ac.uk/.Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. Pinkard, A. M. Champsaur and X. Roy, Acc. Chem. Res., 2018, 51, 919–929 CrossRef CAS PubMed; (b) S. A. Claridge, A. W. Castleman, Jr., S. N. Khanna, C. B. Murray, A. Sen and P. S. Weiss, ACS Nano, 2009, 3, 244–255 CrossRef CAS PubMed; (c) Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC.
- (a) X. Roy, C. H. Lee, A. C. Crowther, C. L. Schenck, T. Besara, R. A. Lalancette, T. Siegrist, P. W. Stephens, L. E. Brus, P. Kim, M. L. Steigerwald and C. Nuckolls, Science, 2013, 341, 157–160 CrossRef CAS PubMed; (b) A. M. Champsaur, T. J. Hochuli, D. W. Paley, C. Nuckolls and M. L. Steigerwald, Nano Lett., 2018, 18, 4564–4569 CrossRef CAS PubMed.
- D. Fenske, J. Ohmer, J. Hachgenei and K. Merzweiler, Angew. Chem., Int. Ed., 2003, 27, 1277–1296 CrossRef.
- (a) H. Beinert, R. H. Holm and E. Munck, Science, 1997, 277, 653–659 CrossRef CAS PubMed; (b) A. C. Brown and D. L. M. Suess, in Comprehensive Coordination Chemistry III, ed. E. C. Constable, G. Parkin and L. Que Jr, Elsevier, Oxford, 2021, DOI:10.1016/B978-0-08-102688-5.00053-2, pp. 134–156.
- (a) S. C. Lee, W. Lo and R. H. Holm, Chem. Rev., 2014, 114, 3579–3600 CrossRef CAS PubMed; (b) R. D. Britt, G. Rao and L. Tao, Nat. Rev. Chem., 2020, 4, 542–549 CrossRef CAS PubMed; (c) D. W. N. Wilson, M. S. Fataftah, Z. Mathe, B. Q. Mercado, S. DeBeer and P. L. Holland, J. Am. Chem. Soc., 2024, 146, 4013–4025 CrossRef CAS PubMed.
- (a) A. P. Aydt, B. Qie, A. Pinkard, L. Yang, Q. Cheng, S. J. L. Billinge, Y. Yang and X. Roy, ACS Appl. Mater. Interfaces, 2019, 11, 11292–11297 CrossRef CAS PubMed; (b) V. Chauhan, A. C. Reber and S. N. Khanna, J. Am. Chem. Soc., 2017, 139, 1871–1877 CrossRef CAS PubMed; (c) S. M. Stuczynski, Y. U. Kwon and M. L. Steigerwald, J. Organomet. Chem., 1993, 449, 167–172 CrossRef CAS.
- (a) N. Rinn, I. Rojas-Leon, B. Peerless, S. Gowrisankar, F. Ziese, N. W. Rosemann, W. C. Pilgrim, S. Sanna, P. R. Schreiner and S. Dehnen, Chem. Sci., 2024, 15, 9438–9509 RSC; (b) A. Choy, D. Craig, I. Dance and M. Scudder, J. Chem. Soc., Chem. Commun., 1982, 1246–1247 RSC; (c) J. R. Long, L. S. McCarty and R. H. Holm, J. Am. Chem. Soc., 1996, 118, 4603–4616 CrossRef CAS; (d) Y. V. Mironov, A. V. Virovets, N. G. Naumov, V. N. Ikorskii and V. E. Fedorov, Chem. – Eur. J., 2000, 6, 1361–1365 CrossRef CAS; (e) P. Vaqueiro and M. L. Romero, Chem. Commun., 2007, 3282–3284 RSC; (f) M. A. Shestopalov, Y. V. Mironov, K. A. Brylev, S. G. Kozlova, V. E. Fedorov, H. Spies, H. J. Pietzsch, H. Stephan, G. Geipel and G. Bernhard, J. Am. Chem. Soc., 2007, 129, 3714–3721 CrossRef CAS PubMed.
- (a) V. P. Ananikov, N. V. Orlov, S. S. Zalesskiy, I. P. Beletskaya, V. N. Khrustalev, K. Morokuma and D. G. Musaev, J. Am. Chem. Soc., 2012, 134, 6637–6649 CrossRef CAS PubMed; (b) Z. G. Lin, L. Y. Fan, A. Kondinski, N. Vankova, T. Heine, B. K. Chen, A. Haider, B. Wang, U. Kortz and C. W. Hu, Inorg. Chem., 2016, 55, 7811–7813 CrossRef CAS PubMed; (c) Y. Yamashina, Y. Kataoka and Y. Ura, Inorg. Chem., 2014, 53, 3558–3567 CrossRef CAS PubMed; (d) P. Saha, S. Amanullah and A. Dey, J. Am. Chem. Soc., 2020, 142, 17312–17317 CrossRef CAS PubMed; (e) C. Pohlker, I. Schellenberg, R. Pottgen and S. Dehnen, Chem. Commun., 2010, 46, 2605–2607 RSC; (f) C. Zhang, T. Matsumoto, M. Samoc, S. Petrie, S. Meng, T. C. Corkery, R. Stranger, J. Zhang, M. G. Humphrey and K. Tatsumi, Angew. Chem., Int. Ed., 2010, 49, 4209–4212 CrossRef CAS PubMed; (g) C. Tan, M. Jin, X. Ma, Q. Zhu, Y. Huang, Y. Wang, S. Hu, T. Sheng and X. Wu, Dalton Trans., 2012, 41, 8472–8476 RSC.
- (a) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed; (b) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed; (c) P. Bellotti, M. Koy, M. N. Hopkinson, F. Glorius, P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem., 2021, 5 Search PubMed; (d) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed; (e) S. Park and Y. Kim, Bull. Korean Chem. Soc., 2023, 44, 1004–1007 CrossRef CAS; (f) L. Delaude, A. Demonceau and J. Wouters, Eur. J. Inorg. Chem., 2009, 1882–1891 CrossRef CAS; (g) G. Kuchenbeiser, M. Soleilhavoup, B. Donnadieu and G. Bertrand, Chem. – Asian J., 2009, 4, 1745–1750 CrossRef CAS PubMed.
- (a) N. Touj, F. Mazars, G. Zaragoza and L. Delaude, Beilstein J. Org. Chem., 2023, 19, 1947–1956 CrossRef CAS PubMed; (b) S. Naeem, L. Delaude, A. J. White and J. D. Wilton-Ely, Inorg. Chem., 2010, 49, 1784–1793 CrossRef CAS PubMed; (c) L. Delaude, X. Sauvage, A. Demonceau and J. Wouters, Organometallics, 2009, 28, 4056–4064 CrossRef CAS; (d) S. Naeem, A. L. Thompson, L. Delaude and J. D. Wilton-Ely, Chem. - Eur. J., 2010, 16, 10971–10974 CrossRef CAS PubMed; (e) T. F. Beltran, G. Zaragoza and L. Delaude, Dalton Trans., 2016, 45, 18346–18355 RSC; (f) M. Zain Aldin, G. Zaragoza, W. Deschamps, J. D. Tomani, J. Souopgui and L. Delaude, Inorg. Chem., 2021, 60, 16769–16781 CrossRef CAS PubMed; (g) J. Ortmeyer, U. Flörke, G. Henkel, R. Wilhelm and A. Neuba, Eur. J. Inorg. Chem., 2017, 3191–3197 CrossRef CAS; (h) J. A. Cabeza, P. García-Álvarez and M. Guadalupe Hernández-Cruz, Eur. J. Inorg. Chem., 2012, 2928–2932 CrossRef CAS; (i) T. F. Beltran, G. Zaragoza and L. Delaude, Dalton Trans., 2017, 46, 13002–13009 RSC.
- (a) T. W. Hudnall and C. W. Bielawski, J. Am. Chem. Soc., 2009, 131, 16039–16041 CrossRef CAS PubMed; (b) T. W. Hudnall, J. P. Moerdyk and C. W. Bielawski, Chem. Commun., 2010, 46, 4288–4290 RSC; (c) J. P. Moerdyk, D. Schilter and C. W. Bielawski, Acc. Chem. Res., 2016, 49, 1458–1468 CrossRef CAS PubMed; (d) V. Cesar, S. Labat, K. Miqueu, J. M. Sotiropoulos, R. Brousses, N. Lugan and G. Lavigne, Chem. - Eur. J., 2013, 19, 17113–17124 CrossRef CAS PubMed; (e) G. A. Blake, J. P. Moerdyk and C. W. Bielawski, Organometallics, 2012, 31, 3373–3378 CrossRef CAS; (f) L. L. Liu, J. Zhou, L. L. Cao, R. Andrews, R. L. Falconer, C. A. Russell and D. W. Stephan, J. Am. Chem. Soc., 2018, 140, 147–150 CrossRef CAS PubMed; (g) J. P. Moerdyk and C. W. Bielawski, Nat. Chem., 2012, 4, 275–280 CrossRef CAS PubMed; (h) R. Pei, W. Chang, L. He, T. Wang, Y. Zhao, Y. Liang and X. Wang, Nat. Commun., 2024, 15, 7943 CrossRef CAS PubMed; (i) R. Pei, L. He, Y. Zhao and X. Wang, J. Am. Chem. Soc., 2023, 145, 21733–21737 CrossRef CAS PubMed.
- S. Park, J. Y. Hwang, J. Shin and Y. Kim, J. Am. Chem. Soc., 2024, 146, 28508–28515 CAS.
- (a) A. Kobayashi, T. Kojima, R. Ikeda and H. Kitagawa, Inorg. Chem., 2006, 45, 322–327 CrossRef CAS PubMed; (b) J. F. Berry, E. Bill, E. Bothe, F. A. Cotton, N. S. Dalal, S. A. Ibragimov, N. Kaur, C. Y. Liu, C. A. Murillo, S. Nellutla, J. M. North and D. Villagran, J. Am. Chem. Soc., 2007, 129, 1393–1401 CrossRef CAS PubMed.
- J. W. Arblaster, Selected values of the crystallographic properties of the elements, ASM International, Materials Park, Ohio, 2018 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, X-ray crystal data, UV-vis spectra, DFT calculation results. CCDC 2381957–2381960. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc04582c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |