
Chemical oxidation of a double-twisted nanographene†
Zhiyu
Zhang
 a,
Yoshifumi
Hashikawa
*b and
Chaolumen
*a
a,
Yoshifumi
Hashikawa
*b and
Chaolumen
*a
aCollege of Chemistry and Chemical Engineering, Inner Mongolia University, Hohhot, 010021, China. E-mail: chaolumen@imu.edu.cn
bInstitute for Chemical Research, Kyoto University, Uji, Kyoto 611-0011, Japan. E-mail: hashi@scl.kyoto-u.ac.jp
First published on 30th October 2024
Abstract
Chemical oxidation of a double-twisted nanographene led to quantitative generation of corresponding dicationic species, which exhibited an intense absorption band tailing to 1500 nm wherein two orthogonally arranged π-skeletons contributed to transitions and a high dissymmetry factor of −0.0209 was recorded at 1443 nm in the NIR-II region.
Twisted nanographenes (NGs) have just emerged as a novel class of non-planar NGs, while the role of the twist operation is still under exploration.1,2 In most cases, installation of multiple [n]helical units is a way to prepare twisted NGs. Therefore, this type of NGs is often regarded as conventional helical NGs with paying less attention to the role of twist operation.3–10 Nevertheless, twisted NGs possessing a large π-surface were recently revealed.11,12 However, it is, in general, difficult to prepare the sole conformation during the synthesis of twisted NGs so that the products are likely obtained as a mixture of multiple conformations including twisted, wavy, and even combined geometries.13 For instance, in 2020, Wang and co-workers reported the synthesis of C150 NGs which were obtained as a conformational mixture containing unidirectionally-twisted (ut), alternatively-twisted (at), and randomly-twisted geometries.14 At nearly the same time, a research group led by Maçôas and Campaña reported C114 NG and its analogue containing a heptagonal ring, the former of which contains ut and at geometries with a ratio of 1.2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, while the latter was conformationally pure.15
:1, while the latter was conformationally pure.15
Recently, we reported conformation-selective synthesis of a double-twisted NG featuring two orthogonally arranged π-skeletons centered at a contorted pyrene core. This twisted NG exhibited a robust conformational stability with retaining D2 symmetry.16 In that work, the NG was found to undergo two-electron ejection in a redox process with producing an electrochemically stable dicationic species (12+) (Fig. 1). Herein, we report the generation of the dicationic species by chemical oxidation and clarify the influence of twisted geometry on its chiroptoelectronic properties.
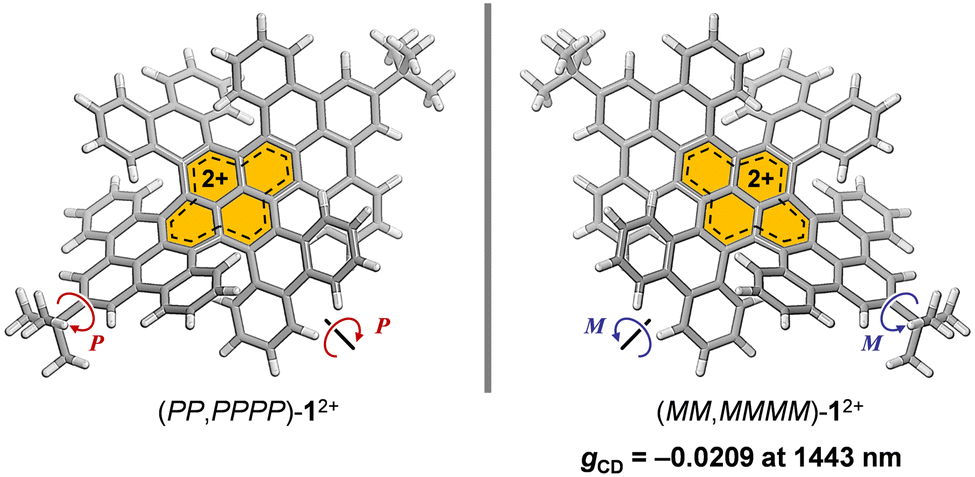 | ||
| Fig. 1 A dicationic double-twisted NG (12+) studied herein. | ||
Chemical oxidation of 1 was performed with 2.2 equiv. of NOSbF6 in C2H4Cl2 under a N2 atmosphere (Fig. 2a). After addition of the oxidant at 0 °C, the solution was allowed to warm to room temperature. During 2 h, the compound was mostly precipitated. The resulting solid was collected by centrifugation using C2H4Cl2/n-hexane (1:20) to afford 12+[SbF6−]2 in 83% isolated yield as a brown solid. It should be noted that 12+[SbF6−]2 was stable in CD2Cl2 at room temperature under N2 for at least 19 days (Fig. S4, ESI†) as confirmed by 1H NMR showing nine sets of sharp signals (Fig. 2b). Upon comparing the chemical shifts of 12+[SbF6−]2 with those for neutral 1, significant upfield shifts were found for protons labeled with H(1), H(6), and H(9), while opposite shifts were confirmed for those labeled with H(3), H(4), and H(5), indicating a change in local aromaticity at each hexagon. These protons were fully assigned according to through-space correlation peaks observed by NOESY (Fig. 2c). Owing to the dicationic charge, 13C signals were overall shifted down-field, while two characteristic signals showing considerable shifts appeared at δ 148.53 and 144.35 ppm (Fig. 2d). The simulated 13C spectrum of 12+ (GIAO-B3LYP/6-311 + G(2d,p)//B3LYP-D3/6-31G(d); Fig. S5, ESI†) reproduces the spectral features with the two signals being assignable to the pyrene carbons. This implies that the cationic charge is strongly distributed on the pyrene moiety in 12+.
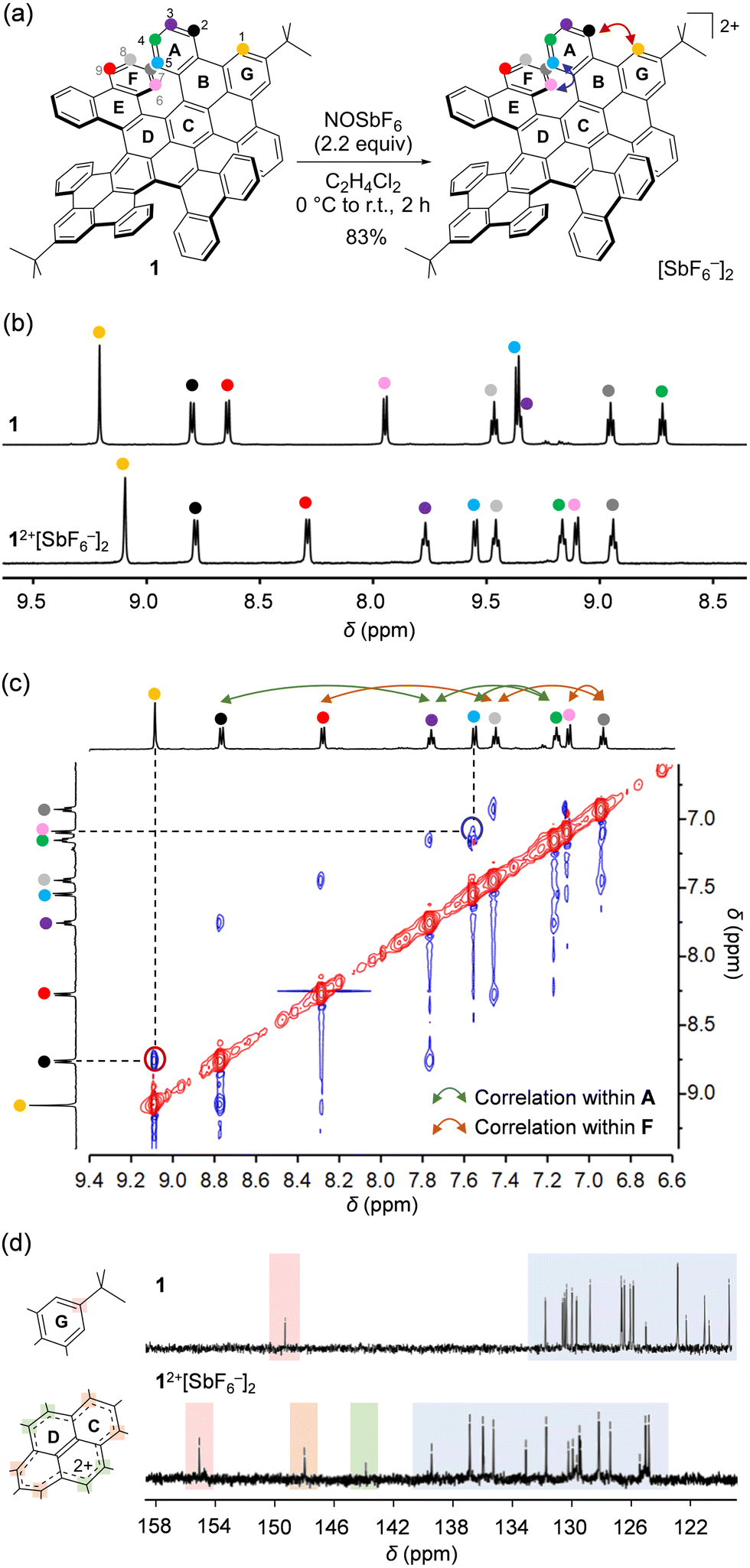 | ||
| Fig. 2 (a) Chemical oxidation of 1 (the arrows represent NOE correlations). (b) 1H NMR (600 MHz, CD2Cl2) spectra of 1 and 12+[SbF6−]2. (c) NOESY (600 MHz, CD2Cl2) spectrum of 12+[SbF6−]2. (d) 13C NMR spectra of 1 (151 MHz, CD2Cl2) and 12+[SbF6−]2 (151 MHz, CD2Cl2/CD3CN = 5:1). | ||
To get better insights into the aromatic character of 12+, theoretical calculations were performed for both 1 and 12+. Fig. 3a shows an optimized structure of 12+, showing that H(3) is located 5.634 Å above an edge of ring F and H(4) and H(5) face ring E with distances of 4.350 and 2.361 Å, respectively. Therefore, the chemical shifts of these protons should be affected considerably. The 3D ICSS (iso-chemical shielding surface) plots clearly demonstrate the significant change in the shielding environment at ring C (Fig. 3b). The NICS(0) (nucleus independent chemical shift) value at C changes from −8.2 (1) to +8.6 (12+) upon 2e− oxidation. This change was also verified by ACID (anisotropy of current-induced density) plots where diatropic ring currents at C in 1 are switched to paratropic nature in 12+ (Fig. 3c). The significant change in aromaticity was also confirmed for rings E and G where NICS(0) values varied from −1.4 (1) to +3.0 (12+) and from −9.7 (1) to −7.6 (12+), respectively (Fig. 3b). From these information, the observed up-field shifts for H(1), H(9), and H(6) are ascribed to a weakened deshielding effect caused by decreased aromaticity in G for the former as well as shielding effect from weak antiaromaticity attained by E for the rest. In addition, the down-field shifts for H(3), H(4), and H(5) are due to a weakened shielding effect caused by lowered aromaticity of F for the former as well as a deshielding effect from weak antiaromaticity attained by E for the rest. The electrostatic potential (ESP) map of 12+ exhibits localization of the cationic charge on the pyrene moiety (Fig. 3d), being matched well with the resonance structure (Fig. 3e) expected from 13C spectral feature (Fig. 2d).
Next, absorption spectra were recorded in CH2Cl2 (Fig. 4a). Neutral 1 exhibits a strong absorption band at 592 nm. By chemical oxidation to 12+[SbF6−]2, an intense broadband absorption was observed at 1175 nm (ε = 41583 M−1 cm−1), tailing to 1500 nm in the NIR-II (1000–1700 nm) region. Since 12+[SbF6−]2 is a chiral nanographene with two-fold twist chirality and four-fold helical chirality, we first performed optical resolution of rac-1 to obtain (MM,MMMM)–1 which shows dissymmetry factors (gCD) of 6.5 × 10−3 at 342 nm and 6.9 × 10−4 at 592 nm.16 After chemical oxidation to (MM,MMMM)–12+[SbF6−]2, the circular dichroism (CD) spectrum was recorded in CH2Cl2 (Fig. 4b), being demonstrative of a wide range of chiroptical activities. The highest Cotton effect of (MM,MMMM)–12+[SbF6−]2 was found at 536 nm where gCD is obtained to be +1.9 × 10−3. The slightly higher gCD value of −3.5 × 10−3 was observed at 680 nm with a negative Cotton effect. Notably, the highest gCD value with an order of 10−2 (−2.09%) was detected at 1443 nm in the near-infrared II (NIR-II) region. Such a large anisotropy factor,17–22 especially in the NIR region,23–25 is quite rare among known chiral nanocarbon materials.
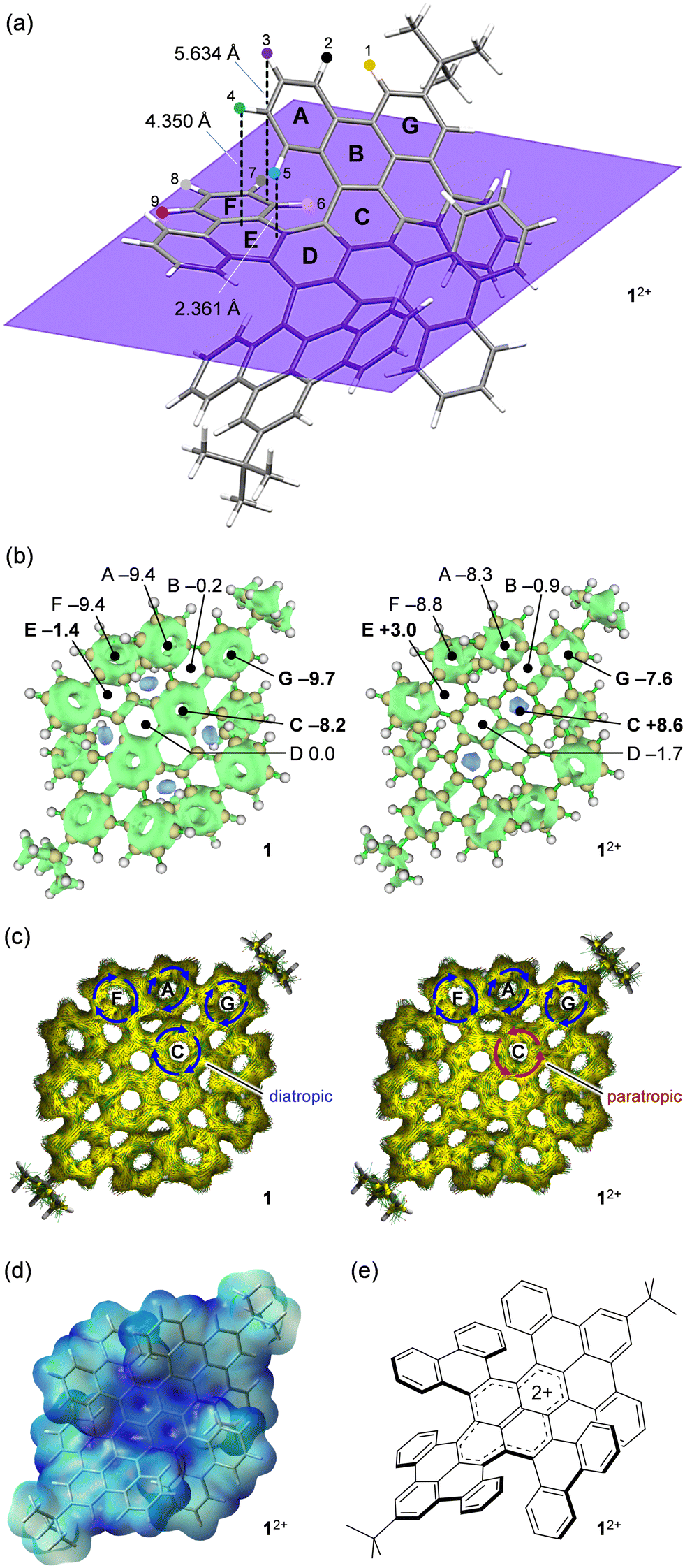 | ||
| Fig. 3 (a) Optimized structure of 12+ (B3LYP-D3/6-31G(d)). (b) 3D ICSS plots of 1 and 12+ with NICS(0) (units in ppm) (HF/6-31 + G(d,p)). (c) ACID plots (B3LYP/6-31G(d)). (d) ESP map of 12+ (B3LYP-D3/6-31G(d)). (e) The most probable resonance structure of 12+. | ||
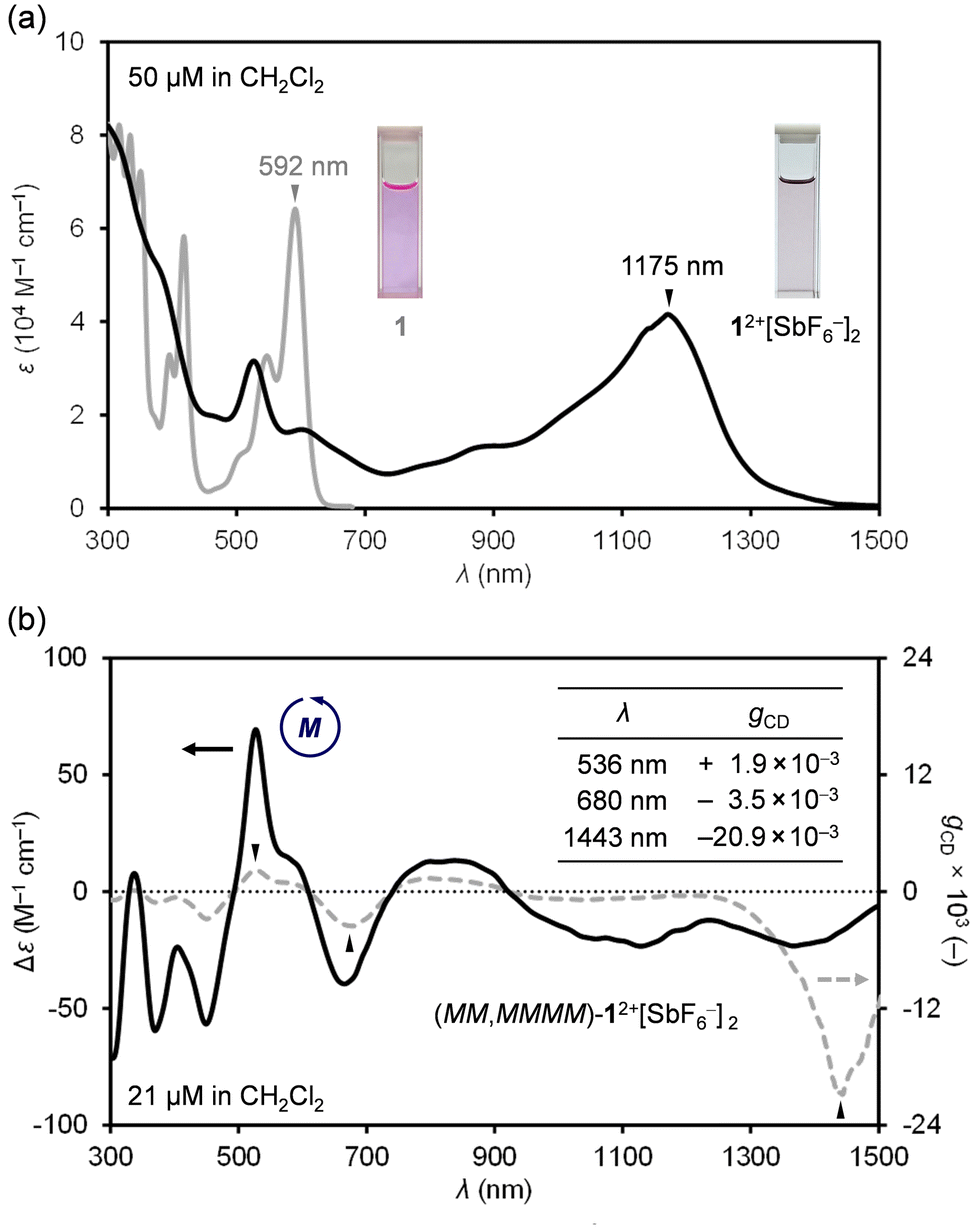 | ||
| Fig. 4 (a) UV-vis-NIR absorption spectra of 1 and 12+[SbF6−]2. (b) CD and gCD spectra (CH2Cl2) of (MM,MMMM)–12+[SbF6−]2. | ||
To verify the origin of high gCD, we computed the optoelectronic nature of (MM,MMMM)–12+[SbF6−]2 (Fig. 5). The intense NIR-II bands were then assigned to S0 → S2 (λcalc = 1172 nm) and S0 → S3 (1135 nm) with significant contributions from excitations corresponding to HOMO–1 → LUMO (92%) and HOMO → LUMO (74%), respectively (Fig. 5a). These transitions are electronically allowed with moderately large oscillator strengths (f) of 0.1728 and 0.3912, respectively, and mostly occur within the terropyrene moiety which is one of the twisted π-skeletons in 12+. The lowest energy transition (S0 → S1) was confirmed at 1380 nm, featuring an excitation, with a 72% contribution, from HOMO-2, which shows significant delocalization of orbital coefficients on the twisted tetrabenzotetracene moiety, to the LUMO with a strong character of the contorted pyrene. The oscillator strength of this transition was suggested to be smaller (f = 0.0156) by a tenth compared with those of S0 → S2 (0.1728) and S0 →S3 (0.3912). This is primarily owing to the smaller magnitude of electric transition dipole moment, |μ| (S0 → S1, 182 × 10−20 esu cm; S0 → S2, 557 × 10−20 esu cm; S0 → S3, 825 × 10−20 esu cm).
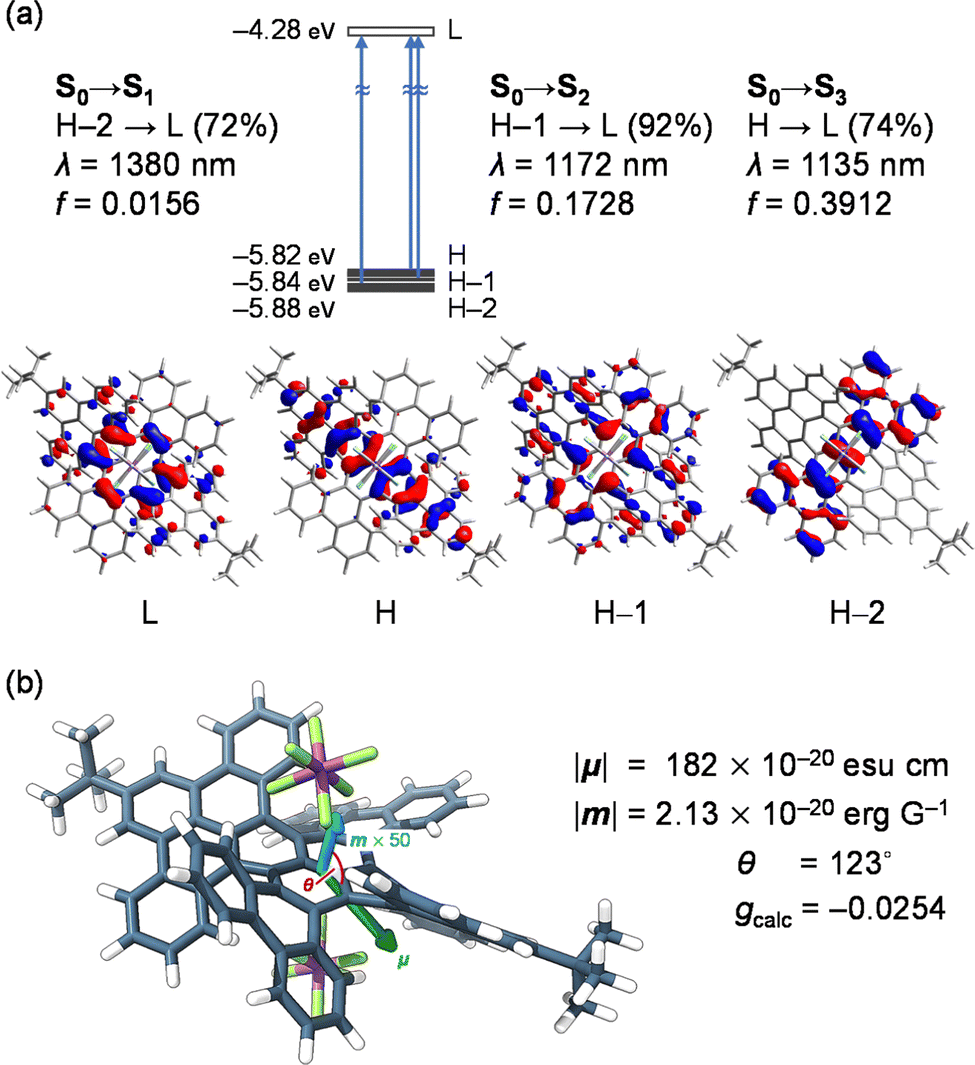 | ||
| Fig. 5 (a) Optical transitions of (MM,MMMM)–12+[SbF6−]2 with molecular orbitals. (b) Pictorial representation of transition dipole moments μ and m. The calculations were performed at the TD CAM-B3LYP-D3/SDD & 6-31G(d)//B3LYP-D3/SDD & 6-31G(d) level of theory (SDD was applied for Sb) and transition energies were calibrated with 0.72.26 | ||
For the lowest energy transition of (MM,MMMM)–12+[SbF6−]2, the size of the magnetic transition dipole moment was estimated to be |m| = 2.13 × 10−20 erg G−1, while the angle between the two transition dipole moments was given as 123°. The arrangement of the two transition dipole moments was therefore illustrated as shown in Fig. 5b. Accordingly, the gcalc was obtained as −0.0254 which is in accordance with the experimental observation (gCD = −0.0209). For common organic molecules, |μ| tends to be larger by ca. 300 times than |m| wherein the physical origin is a ratio between the two transition dipole moments as large as ea0/μB ≈ 274 (in CGS units)27,28 where e is the elementary charge, a0 is the Bohr radius, and μB is the Bohr magneton. Under this circumstance, gcalc is simply described as 4(|m|/|μ|) cos θ which obviously suggests that gcalc is more likely to be perturbed by an electric term for the electromagnetic wave of interest than the magnetic term. The |μ|/|m| ratio of 85.3 for 12+[SbF6−]2 is, however, one order smaller than those for common chiral organic molecules, consequently leading to a large gCD value even in the NIR-II region.
In summary, we synthesized a stable dicationic double-twisted nanographene, 12+[SbF6−]2, wherein the cationic charge is delocalized on the central pyrene core. Notably, owing to the small |μ|/|m| ratio, a high dissymmetry factor of −0.0209 was recorded at 1443 nm in the NIR-II region. The robust stability of the dicationic NG is achieved probably due to steric protection of the central pyrene core by the peripheral π-wings arranged in a propeller manner. This would be of benefit for its usage as an NIR-II chiral chromophore.
Financial support was provided by the National Natural Science Foundation of China (No. 2022161035), the Education Department of Inner Mongolia Autonomous Region (NJYT22089), the Funding Scheme for High-Level Overseas Chinese Students’ Return, International Collaborative Research Program of Institute for Chemical Research (ICR), Kyoto University (2022-39 and 2023-46), the JSPS KAKENHI Grant Number JP22H04538, the Advanced Technology Institute Research Grants 2023, ISHIZUE 2024 of Kyoto University, and the JACI Prize for Encouraging Young Researchers.
Data availability
The data supporting this article are included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- Y.-F. Wu, L. Zhang, Q. Zhang, S.-Y. Xie and L.-S. Zhang, Org. Chem. Front., 2022, 9, 4726–4743 RSC.
- H. V. Anderson, N. D. Gois and W. A. Chalifoux, Org. Chem. Front., 2023, 10, 4167–4197 RSC.
- R. K. Dubey, M. Melle-Franco and A. Mateo-Alonso, J. Am. Chem. Soc., 2022, 144, 2765–2774 CrossRef CAS PubMed.
- A. Bernhardt, D. Čavlović, M. Mayländer, O. Blacque, C. M. Cruz, S. Richert and M. Juríček, Angew. Chem., Int. Ed., 2024, 63, e202318254 CrossRef CAS PubMed.
- S. E. Penty, G. R. F. Orton, D. J. Black, R. Pal, M. A. Zwijnenburg and T. A. Barendt, J. Am. Chem. Soc., 2024, 146, 5470–5479 CrossRef CAS PubMed.
- R. Eichelmann, P. Jeudy, L. Schneider, J. Zerhoch, P. R. Mayer, J. Ballmann, F. Deschler and L. H. Gade, Org. Lett., 2024, 26, 1172–1177 CrossRef CAS.
- R. Bam, W. Yang, G. Longhi, S. Abbate, A. Lucotti, M. Tommasini, R. Franzini, C. Villani, V. J. Catalano, M. M. Olmstead and W. A. Chalifoux, Angew. Chem., Int. Ed., 2024, 63, e202404849 CrossRef CAS PubMed.
- Y. Liu, Z. Li, M.-W. Wang, J. Chan, G. Liu, Z. Wang and W. Jiang, J. Am. Chem. Soc., 2024, 146, 5295–5304 CrossRef CAS PubMed.
- A. Swain, K. Radacki, H. Braunschweig and P. Ravat, Chem. Sci., 2024, 15, 11737–11747 RSC.
- X. Liu, Z. Jin, F. Qiu, Y. Guo, Y. Chen, Z. Sun and L. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202407547 CrossRef CAS PubMed.
- C. Wallerius, O. Erdene-Ochir, E. V. Doeselar, R. Alle, A. T. Nguyen, M. F. Schumacher, A. Lützen, K. Meerholz and S. H. Pun, Precis. Chem, 2024, 2, 488–494 CrossRef CAS.
- B. Borrisov, G. M. Beneventi, Y. Fu, Z. Qiu, H. Komber, Q. Deng, P. M. Greißel, A. Cadranel, D. M. Guldi, J. Ma and X. Feng, J. Am. Chem. Soc., 2024, 146, 27335–27344 CrossRef CAS PubMed.
- Y. Li, Z. Jia, S. Xiao, H. Liu and Y. Li, Nat. Commun., 2016, 7, 11637 CrossRef PubMed.
- S. Ma, J. Gu, C. Lin, Z. Luo, Y. Zhu and J. Wang, J. Am. Chem. Soc., 2020, 142, 16887–16893 CrossRef CAS PubMed.
- S. Castro-Fernández, C. M. Cruz, I. F. A. Mariz, I. R. Márquez, V. G. Jiménez, L. Palomino-Ruiz, J. M. Cuerva, E. Maçôas and A. G. Campaña, Angew. Chem., Int. Ed., 2020, 59, 7139–7145 CrossRef.
- Y. Dong, Z. Zhang, Y. Hashikawa, H. Meng, F. Bai, K. Itami and C. Chaolumen, Angew. Chem., Int. Ed., 2024, 63, e202406927 CrossRef CAS PubMed.
- S. Sato, A. Yoshii, S. Takahashi, S. Furumi, M. Takeuchi and H. Isobe, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13097–13101 CrossRef CAS.
- W. Zheng, T. Ikai, K. Oki and E. Yashima, Nat. Sci, 2022, 2, e20210047 CrossRef CAS.
- Y. Hashikawa, S. Okamoto, S. Sadai and Y. Murata, J. Am. Chem. Soc., 2022, 144, 18829–18833 CrossRef CAS PubMed.
- M. Toya, T. Omine, F. Ishiwari, A. Saeki, H. Ito and K. Itami, J. Am. Chem. Soc., 2023, 145, 11553–11565 CrossRef CAS PubMed.
- Y. Hashikawa, S. Okamoto and Y. Murata, Nat. Commun., 2024, 15, 514 CrossRef CAS PubMed.
- G.-F. Huo, W.-T. Xu, Y. Han, J. Zhu, X. Hou, W. Fan, Y. Ni, S. Wu, H.-B. Yang and J. Wu, Angew. Chem., Int. Ed., 2024, 63, e202403149 CrossRef CAS.
- Y. Nakakuki, T. Hirose, H. Sotome, M. Gao, D. Shimizu, R. Li, J. Hasegawa, H. Miyasaka and K. Matsuda, Nat. Commun., 2022, 13, 1475 CrossRef CAS.
- Y. Hashikawa, S. Sadai, S. Okamoto and Y. Murata, Angew. Chem., Int. Ed., 2023, 62, e202215380 CrossRef CAS PubMed.
- S. T. Bao, S. Louie, H. Jiang, Q. Jiang, S. Sun, M. L. Steigerwald, C. Nuckolls and Z. Jin, J. Am. Chem. Soc., 2024, 146, 51–56 CrossRef CAS PubMed.
- Y. Hashikawa, S. Okamoto and Y. Murata, Commun. Chem., 2020, 3, 90 CrossRef CAS PubMed.
- T. Mori, Chem. Rev., 2021, 121, 2373–2412 CrossRef CAS PubMed.
- H. Kubo, T. Hirose, T. Nakashima, T. Kawai, J. Hasegawa and K. Matsuda, J. Phys. Chem. Lett., 2021, 12, 686–695 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc05264a |
| This journal is © The Royal Society of Chemistry 2024 |