
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO insertion enabled γ-C(sp3)–H heteroarylative carbonylation of tertiary alcohols via heteroaryl migration†
Xin
Qi
ab,
Yuanrui
Wang
ab and
Xiao-Feng
Wu
 *abc
*abc
aDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: xwu2020@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 101408, China
cLeibniz-Institut für Katalyse e.V., Albert-Einstein-Straße 29a, 18059 Rostock, Germany
First published on 4th November 2024
Abstract
The direct functionalization of remote C(sp3)–H is valuable but challenging, and is even more difficult to achieve than the γ-C(sp3)–H functionalization of alcohols. Among the strategies, hydrogen atom transfer (HAT) is one of the solutions for such transformations. Herein, we designed a migration reaction involving carbon monoxide, forming an alkoxy radical by photocatalysis, and used carbon monoxide to extend the carbon chain to provide a site for the migration of heteroaryl groups, which makes 1,4-HAT more advantageous, and we relied on this strategy to successfully achieve the synthesis of 1,4-dicarbonyl compounds by γ-C(sp3) functionalization of alcohols.
Direct functionalization of C(sp3)–H bonds is an ideal synthetic method because of its straightforward and atomically efficient nature in the construction of carbon–carbon or carbon–heteroatomic bonds, which has led to its use in modern chemistry for the synthesis of a wide variety of molecules in biologically active natural products, pharmaceuticals, and functional materials using inexpensive and accessible stocks.1–5 The α- and β-C(sp3)–H-functionalization can be readily accomplished through transition metal catalysis, which has also been extensively investigated for a range of aliphatic substrates.6–8 Nevertheless, functionalization reactions targeting more distant positions, such as γ- and δ-C, present a greater challenge. Such functionalization reactions are more difficult to achieve due to the difficulty of forming kinetically unstable intermediates.
In recent years, as chemists have gained a deeper understanding of free radical chemistry, the use of free radicals in organic synthesis has become more prevalent. Attempts have also been made to utilize the free radical pathway to achieve a number of transformations that were previously challenging to attain. Among the radicals, it is well known that oxygen and nitrogen radicals can be used for various synthetic transformations.9–16 The use of oxygen or nitrogen radicals for 1,5-hydrogen atom transfer (1,5-HAT) can figure out the direct functionalization of the remote C(sp3)–H bond (Fig. 1, eqn (a)). The oxygen or nitrogen radical can initially be generated through photocatalysis, which initiates a 1,5-hydrogen atom transfer. This process involves the transfer of a hydrogen atom from the carbon to an oxygen or nitrogen radical, leading to the formation of a highly reactive carbon radical. Subsequently, the radical is captured by another radical partner, thereby installing a functional group at δ-C. The implementation of this strategy results in the functionalization of the inactivated C(sp3)–H bond. The transition state formed by the transfer of the hydrogen atom of γ-C is less stable than the six-membered ring transition state formed by the H atom transfer of the δ-C; hence the 1,4-HAT is much less studied than 1,5-HAT.
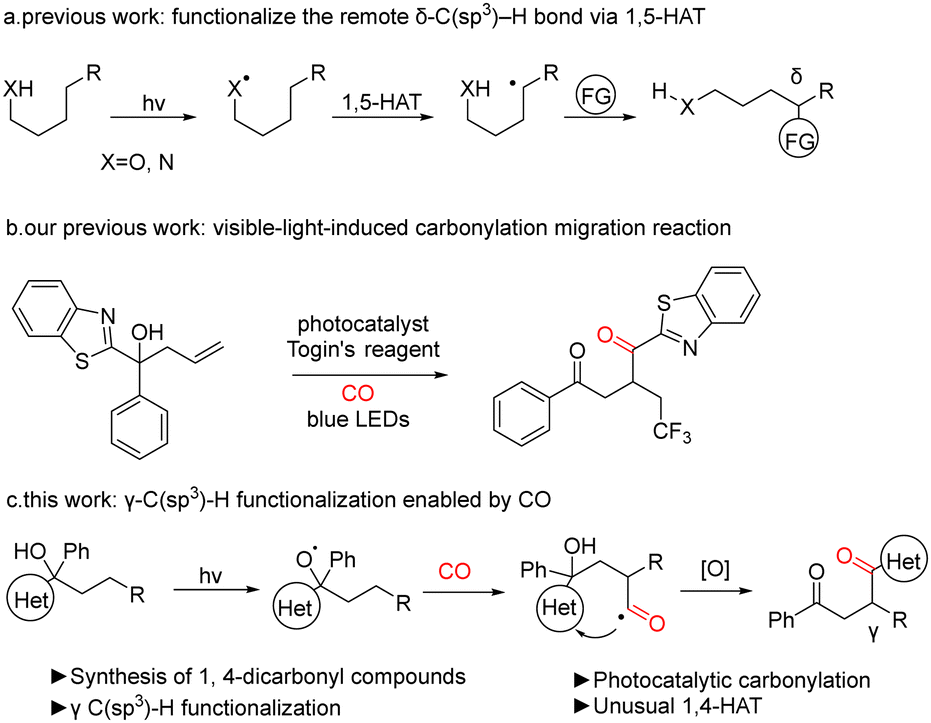 | ||
| Fig. 1 Direct functionalization of remote C(sp3)–H and experimental design. | ||
Since the experimental preparation of carbon monoxide in the 18th century, this gas has been used as a cheap and readily available source of C1 for a variety of carbonylation reactions. The coupling reactions using CO as a carbonyl source are one of the most efficient and direct methods for the synthesis of aldehydes, ketones, acids, and many other derivatives of carboxylic acids.17–20 Among the numerous industrial scale applied carbonylation processes, the carbonylation of methanol with CO is currently the main production method worldwide for acetic acid production.21–23
Previously we have developed radical migration reactions induced by visible light with carbon monoxide insertion (Fig. 1, eqn (b)).24 We propose that the insertion of carbon monoxide serves as a hinge for the migration of the heteroaryl group and that migration promotes more efficient capture of CO, which is a key factor in the carbonylation reaction. On this basis, we envisage the use of CO to extend the carbon chain to provide a site for heteroaryl 1,4-migration, which promotes CO capture, leading to the synthesis of 1,4-dicarbonyl compounds by γ-C(sp3)–H functionalization of alcohols (Fig. 1, eqn (c)).
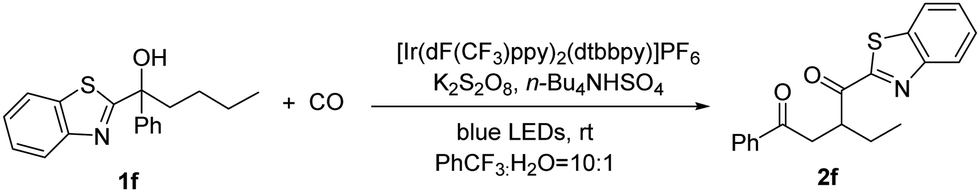
We synthesized a tertiary alcohol containing butyl and benzothiazole groups (1f) as the model substrate for optimizing this migration reaction. Blue LEDs were chosen as the light source and high oxidation potential [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 as the photocatalyst. A persulfate-type oxidant is essential and cannot be substituted by using PIDA, and we finally chose K2S2O8 as the oxidant (Table 1, entries 4 and 5). Here, the compatibility between the photocatalyst and oxidant could be an explanation for this result. The introduction of alternative phase transfer catalysts did not supersede the efficacy of n-Bu4NHSO4 (Table 1, entries 6 and 7), which is postulated to be the optimal phase transfer catalyst for this reaction. After comparing several solvents, PhCF3 was identified as the most suitable option here (Table 1, entry 8). This might benefit from the weak interaction between the fluorine atom and proton of the OH group in the substrate. The addition of a small quantity of water is essential for the dissolution of the persulfate (Table 1, entry 9). Furthermore, no product was formed in the absence of light (Table 1, entry 10).
|
|
||
|---|---|---|
| Entry | Variation of the reaction conditions | Yieldc (%) |
| a Reaction conditions: 1 (0.1 mmol), photocatalyst (3 mol%), K2S2O8 (0.25 mmol), Bu4NHSO4 (0.05 mmol), PhCF3 (1.5 mL), H2O (150 μl) at rt for 36 h under CO (50 bar). b PhCF3 (2.0 mL), H2O (200 μl). c Yield was determined by GC; the isolated yield is given in parentheses. | ||
| 1 | None | 50(48) |
| 2 | Without photocatalyst | 0 |
| 3 | Without CO | 0 |
| 4 | Na2S2O8 instead of K2S2O8 | 46 |
| 5b | PIDA, PIFA instead of K2S2O8 | 0 |
| 6 | 18-Crown-6-ether instead of n-Bu4NHSO4 | 10 |
| 7 | n-Bu4NCl instead of n-Bu4NHSO4 | 45 |
| 8 | Acetone, toluene, CH3CN instead of PhCF3 | 0 |
| 9 | Without H2O | 0 |
| 10 | Without light | 0 |
After determining the optimized reaction conditions, we subsequently studied the substrate scope of this transformation (Scheme 1). To reduce the amount of non-carbonylic migration products and increase the yield of the target product, we replaced the butyl group (1f) with a propyl group (1a), as this avoids 1,5-HAT. Changing the substituent on the aryl group, it can be seen that the reaction can still take place. The introduction of a strong electron-withdrawing group like trifluoromethyl did not affect the reaction (2b), and substrates containing methyl or phenyl groups on the benzene ring also underwent a smooth carbonyl migration reaction to produce the corresponding 1,4-dicarbonyl compounds (2c–2e) in moderate to good yields. However, no desired reaction occurred when 1,1-diphenylbutan-1-ol was tested as the substrate under our standard conditions.
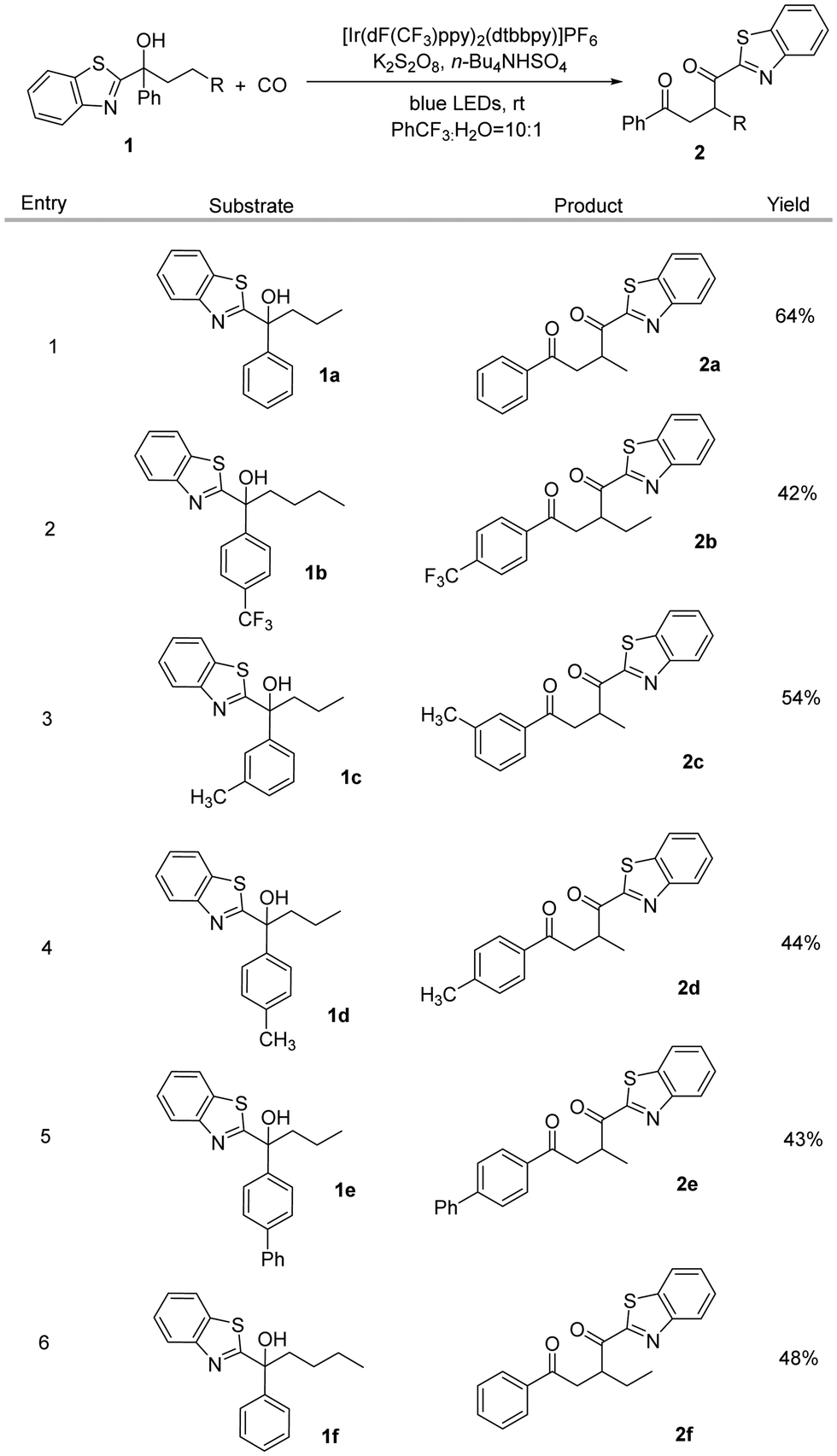 | ||
Scheme 1 Scope of substrates.a a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction conditions: 1 (0.1 mmol), photocatalyst (3 mol%), K2S2O8 (0.25 mmol), Bu4NHSO4 (0.05 mmol), PhCF3 (1.5 mL), H2O (150 μl) at rt for 36 h under CO (50 bar). bThe yield is calculated based on the isolated alcohol. Reaction conditions: 1 (0.1 mmol), photocatalyst (3 mol%), K2S2O8 (0.25 mmol), Bu4NHSO4 (0.05 mmol), PhCF3 (1.5 mL), H2O (150 μl) at rt for 36 h under CO (50 bar). bThe yield is calculated based on the isolated alcohol. | ||
Combined with the understanding based on previous work,24 we proposed a possible mechanism (Scheme 2). We chose tertiary alcohol containing butyl and benzothiazole to illustrate the reaction pathways and explain the selectivity. The photocatalysis of tertiary alcohols generates an alkoxide radical, which initiates hydrogen atom transfer. Generally, 1,5-HAT accounts for the majority of the product, but in the presence of carbon monoxide, 1,4-HAT accounts for the majority of the product as CO provides a carbon atom at the γ-C allowing benzothiazole to 1,4-migrate to form the more dominant product. In more detail, acyl radical intermediate II will be produced after capture of one molecule of CO once the radical intermediate I has been generated. Then, intermediate III will be formed after the 1,4-migration of benzothiazole, which can then be oxidized and gave cation intermediate IV. Finally, the targeted product can be produced after deprotonation. Benzothiazoles migrate to form the five-membered transition state, so the by-product (2f′) cannot be avoided, but the use of tertiary alcohols containing a propyl group instead of an n-butyl group can avoid the formation of the by-products.
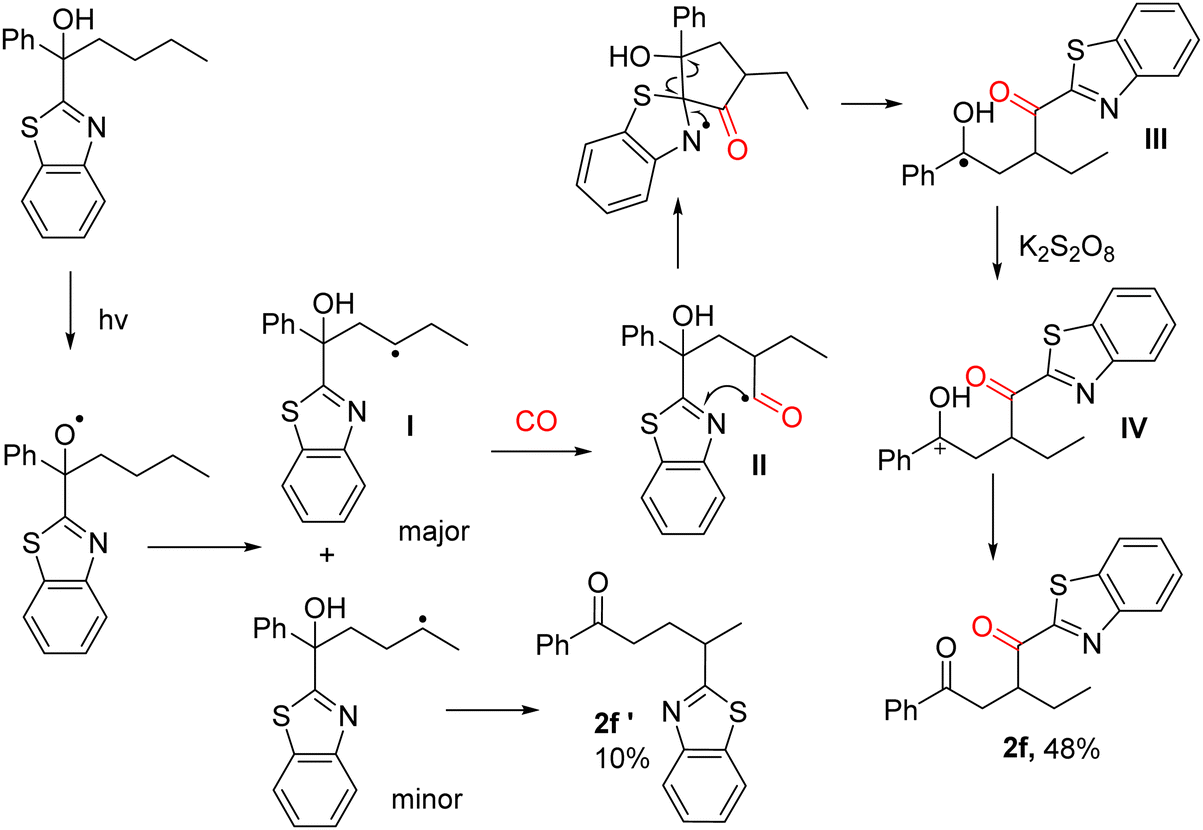 | ||
| Scheme 2 Proposed mechanism. | ||
In summary, we used tertiary alcohols containing benzothiazole moieties as the substrates studied γ-C(sp3)–H heteroarylative carbonylation through heteroaryl migration. The reaction proceeds via the formation of alkoxy radical via photocatalysis, extending the carbon chain using carbon monoxide to provide carbon sites for the migration of benzothiazoles, which made 1,4-HAT more advantageous, and we relied on this strategy to successfully achieve the synthesis of distal γ-functionalized 1,4-dicarbonyl compounds.
We are thankful for the financial support from the National Key R&D Program of China (2023YFA1507500) and DICP.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed.
- V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731–1770 CrossRef CAS PubMed.
- C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS PubMed.
- A. M. Messinis, J. C. A. Oliveira, A. C. Stückl and L. Ackermann, ACS Catal., 2022, 12, 4947–4960 CrossRef CAS.
- M. Albicker and N. Cramer, Angew. Chem., Int. Ed., 2009, 48, 9139–9142 CrossRef CAS PubMed.
- Z. Wu, C. Pi, X. Cui, J. Bai and Y. Wu, Adv. Synth. Catal., 2013, 355, 1971–1976 CrossRef CAS.
- Z. Li, R. Yu and H. Li, Angew. Chem., Int. Ed., 2008, 47, 7497–7500 CrossRef CAS PubMed.
- L. Yang, H. Xie, G. An and G. Li, J. Org. Chem., 2021, 86, 7872–7880 CrossRef CAS PubMed.
- M. Kapoor, A. Singh, K. Sharma and M. H. Hsu, Adv. Synth. Catal., 2020, 362, 4513 CrossRef CAS.
- Q. Qin and S. Yu, Org. Lett., 2015, 17, 1894–1897 CrossRef CAS PubMed.
- L. Wang, Y. Xia, K. Bergander and A. Studer, Org. Lett., 2018, 20, 5817–5820 CrossRef CAS PubMed.
- B. Xu and U. K. Tambar, ACS Catal., 2019, 9, 4627–4631 CrossRef CAS PubMed.
- X. Wu, H. Zhang, N. Tang, Z. Wu, D. Wang, M. Ji, Y. Xu, M. Wang and C. Zhu, Nat. Commun., 2018, 9, 3343 CrossRef PubMed.
- M. Wang, L. Huan and C. Zhu, Org. Lett., 2019, 21, 821–825 CrossRef CAS PubMed.
- Y. Zhu, K. Huang, J. Pan, X. Luo, Q. Qin, J. Wei, X. Wen, L. Zhang and N. Jiao, Nat. Commun., 2018, 9, 2625 CrossRef PubMed.
- J.-J. Guo, A. Hu, Y. Chen, J. Sun, H. Tang and Z. Zuo, Angew. Chem., Int. Ed., 2016, 55, 15319–15322 CrossRef CAS PubMed.
- A. Schoenberg, I. Bartoletti and R. F. Heck, J. Org. Chem., 1974, 39, 3318–3326 CrossRef CAS.
- L.-C. Wang, Y. Yuan, Y. Zhang and X.-F. Wu, Nat. Commun., 2023, 14, 7439 CrossRef PubMed.
- X.-F. Wu, X. Fang, L. Wu, R. Jackstell, H. Neumann and M. Beller, Acc. Chem. Res., 2014, 47, 1041–1053 CrossRef CAS PubMed.
- Z.-P. Bao, N.-X. Sun and X.-F. Wu, Chin. J. Catal., 2024, 60, 171–177 CrossRef CAS.
- A. Seayad, M. Ahmed, H. Klein, R. Jackstell, T. Gross and M. Beller, Science, 2002, 297, 1676–1678 CrossRef CAS PubMed.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- M. J. Howard, M. D. Jones, M. S. Roberts and S. A. Taylor, Catal. Today, 1993, 18, 325–354 CrossRef.
- Y. Wang, H. Yang, Y. Zheng, M. Hu, J. Zhu, Z.-P. Bao, Y. Zhao and X.-F. Wu, Nat. Catal., 2024, 7, 1065–1075 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc05361c |
| This journal is © The Royal Society of Chemistry 2024 |