DOI:
10.1039/D3CE01029E
(Paper)
CrystEngComm, 2024,
26, 604-619
Analysis of short contacts in crystals of halogenated amino acids: atom–atom interactions vs. energy frameworks†
Received
17th October 2023
, Accepted 8th December 2023
First published on 14th December 2023
Abstract
We investigated eight crystal structures of a series of chlorinated and iodinated alanine derivatives with different protective groups on carboxyl and amino functionalities. The crystal packing is determined by the H-bonding type interactions, primarily of the amide group, as well as of the acidic hydrogens of the stereogenic center and –CH2X (X = Cl or I) groups. These types of hydrogen bonding are similar to what is found in nature as seen in the secondary structures of proteins, i.e., α-helices and β-sheets, which are necessary to stabilize the three-dimensional structures of amino acid-based polymers. Two iodinated derivatives demonstrated either type II I⋯I halogen interactions or I⋯O multipolar interactions, whereas no indication of halogen bonding interactions was seen among the chlorinated derivatives. To a large extent, the packing is stabilized by dispersion forces, a conclusion drawn from the analysis of energy lattice networks performed with the help of Crystal Explorer.
Introduction
Amino acids serves as the fundamental building blocks for peptides, one of the three major biopolymers encountered in nature. Structures of the 20 canonical α-amino acids differ only by the residue attached to the stereogenic carbon. However, through functionalization of carboxylic and amino groups as well as the attachment of various residues to the stereogenic carbon, a vast array of amino acid derivatives can be synthesized. Utilizing X-ray diffraction, one may gain valuable insight into the structural aspects of these amino acids, their packing in the solid state and the subtle yet crucial intermolecular interactions. However, the overarching aim of the X-ray diffraction analysis is to elucidate the relationship between the molecular structure, the crystal packing adopted, and the consequent physical and chemical properties. The Cambridge Structural Database contains several thousand structures of α-amino acids and their derivatives1 and is updated by hundreds of new crystal structures yearly.
The majority of the research papers, which describe the X-ray structures of amino acids, have reported structures with non-modified carboxyl and amino groups.2 These reports give insight into the formation of supramolecular structures in the solid state3,4 and polymorphism,5 including that under high pressure,6 and describe the formation of solvates7,8 and metal complexes of amino acids.9
Far less X-ray structures of α-amino acids with protected carboxylic and amino groups were reported as described in a report in 2014,1 with a total number of less than 100 hits. In particular, they correspond to unnatural or non-canonical (not present amongst the 20 canonical amino acids) α-amino acids.10,11 This paper reports several structures of eight protected halogenated (chlorinated and iodinated) alanine derivatives. These compounds were prepared to serve as intermediates for the synthesis of non-canonical amino acids with donor and acceptor aromatic substituents using a Pd-catalyzed Negishi coupling reaction, a methodology developed by Jackson et al.12,13 Though most of these halogenated alanine derivatives have been reported before, only one has been investigated using X-ray crystallography.
The crystal packing of non-protected amino acids in the solid state is predominantly determined by the polar carboxylic and amine groups' weak interactions (electrostatic, H-bonding).2 Due to the presence of both hydrogen bond donors (primarily the HN-group of the amide) and acceptors, we expected that in our case the H-bonds should dominate the crystal packing.14,15 The presence of halogens suggested that the interactions between the protected carboxyl and amino groups could be complemented by halogen bonds16–19 (XB) of different types and geometries.
Halogens are not very common in natural compounds, still, they play a role in several important molecular recognition processes, for example, in the binding of iodinated thyroid hormones and inhibitors against cancer targets, such as protein kinases.20 In the last decade, it was demonstrated that halogen bonds can also find use in peptide systems.21,22 Using synthetic halogenated amino acids, in particular halogenated phenylalanine derivatives, it is possible to modulate peptide conformation and folding by formation of halogen bonds.23–28 Halogenated amino acids were also tested as building blocks in antimicrobial peptides and peptidomimetics.29
In the crystal packing of the halogenated alanine derivatives, our analysis revealed that N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of the carbamate H-bonding interaction was present in most of the structures. Additionally, these interactions were often complemented by C–H⋯OC weak H-bonds,30 in which hydrogen belonged either to the stereogenic carbon center or to the –CH2X group. One iodinated amino acid demonstrated I⋯I halogen bonds31 of type II,32,33 and in another one, short I⋯O contacts were observed. In contrast, none of the chlorinated derivatives' chlorine atoms participated in halogen bonding interactions. We also conducted a Hirshfeld surface and energy framework analysis of the crystal structures reported here, allowing us to estimate the contribution of different types of weak intermolecular interactions to the total stabilization energy of crystal structures.
C of the carbamate H-bonding interaction was present in most of the structures. Additionally, these interactions were often complemented by C–H⋯OC weak H-bonds,30 in which hydrogen belonged either to the stereogenic carbon center or to the –CH2X group. One iodinated amino acid demonstrated I⋯I halogen bonds31 of type II,32,33 and in another one, short I⋯O contacts were observed. In contrast, none of the chlorinated derivatives' chlorine atoms participated in halogen bonding interactions. We also conducted a Hirshfeld surface and energy framework analysis of the crystal structures reported here, allowing us to estimate the contribution of different types of weak intermolecular interactions to the total stabilization energy of crystal structures.
Results and discussion
Structures of the halogenated alanine derivatives discussed in this study are shown in Fig. 1. They differ by the halogen atom (chlorine or iodine), thus making two distinct groups based on it. Each of the two sets includes amino acid derivatives with different protective groups on the carboxyl functionality (Me, tBu, and Bn) and amino functionality (either Boc or Fmoc).
 |
| | Fig. 1 Structures of halogenated L-alanine derivatives described in this study. | |
The characteristic features of the packing of these amino acid derivatives in the crystal state are described below. Analysis of the crystal structures is performed at the molecular level (molecular geometry) and supramolecular level (packing of the molecules in the crystal structures), and, therefore, the manuscript is subdivided into the two corresponding sections. At the molecular level, all analyzed molecules did not feature any abnormalities regarding bond lengths and angles.34 Ester groups and carbamate linkages (both including the corresponding carbonyl oxygen atoms) were almost planar for most of the amino acid derivatives, and the angle between these two planes served as a characteristic feature of the overall arrangement of the molecular backbone. Establishing the absolute configurations of all enantiopure amino acid derivatives did not present any problems because of the significant anomalous scattering of chlorine and iodine. The bonding interactions and short contacts were analysed using Platon35 outputs and by visualizing the molecules using the Mercury36 graphical interface. For the in-depth analysis of weak interactions and their contribution to the stability of the crystal lattices, we used energy framework analysis37 using Crystal Explorer38 software.
Molecular geometry
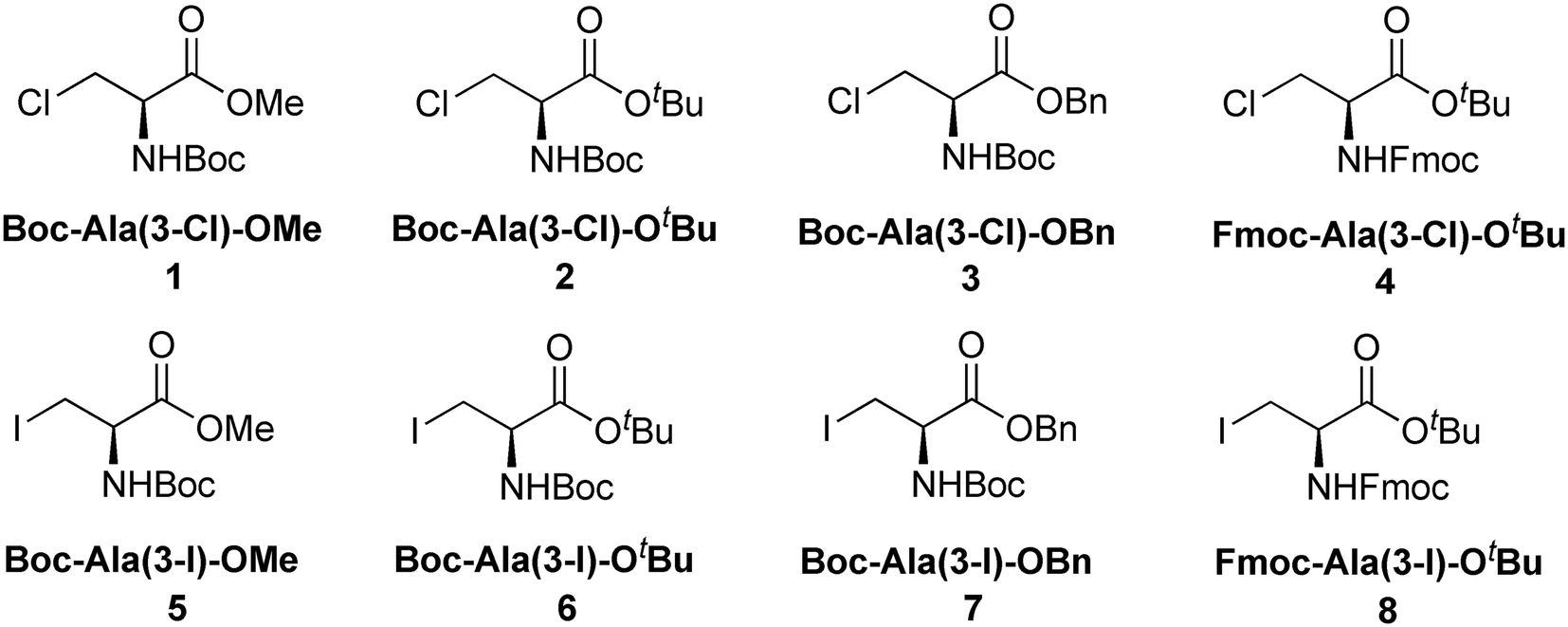
Boc-Ala(3-Cl)-OMe (1) (Fig. 2)
The carbamate linkage (from C2 to C6, including O3) and ester linkage (C2 to C5, including O1) are almost planar (deviation of 0.038(3) Å and 0.011 Å, respectively). The dihedral angle between these two groups amounts to 76.41(4)°. The backbone of the molecule (from C5 to Cl1) features an all-stretched conformation (all bonds in the chain in the anti-conformation), with all atoms (including O1) residing close to one plane.
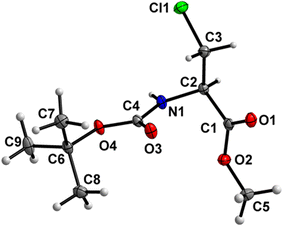 |
| | Fig. 2 Molecular structure of 1 with atom labels. Atom labels for hydrogen atoms are omitted for clarity. | |
Boc-Ala(3-Cl)-O
t
Bu (2) (Fig. 3)
Both the carbamate linkage (C2 to C9, including O3) and ester linkage (C2 to C5, including O1) are almost planar (deviation of 0.022 Å and 0.008 Å, respectively), with a dihedral angle of 37.5(2)° between these two groups. The conformation along the C2–C3 bond is staggered, and, interestingly, the bulky chlorine substituent is gauche to both ester and amido groups and not trans to any one of them. This is notable since, in protein structures, anti-parallel β-sheets with amino acids interacting via hydrogen interactions have their side chains oriented in the same direction, thus strengthening the notion that such non-natural amino acids retain some canonical characteristics.39
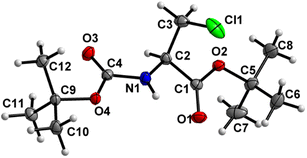 |
| | Fig. 3 Molecular structure of 2 with atom labels. Atom labels for hydrogen atoms are omitted for clarity. | |
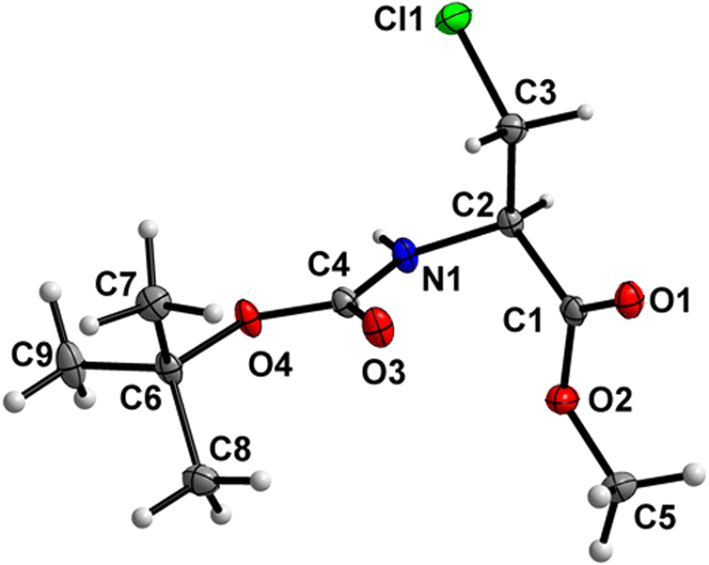
Boc-Ala(3-Cl)-OBn (3)
The orientation of the Bn ester group is distorted between two positions in a 54/46 ratio (Fig. 4). Both the carbamate linkage (C2 to C12, including O3) and ester linkage (C2 to C5, including O1) are almost planar (deviation of 0.017 Å and 0.026 Å, respectively), with a dihedral angle of 79.9(5)° between the planes of these two groups (ester group taken in its major orientation). The backbone of the molecule (from C5 to Cl1) features an all-stretched conformation, with all atoms (including O1) residing close to one plane, whereas the phenyl ring is rotated to the side out of this plane.
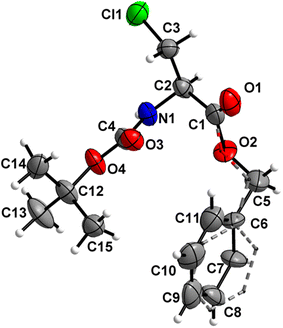 |
| | Fig. 4 Molecular structure of 3 with atom labels. The Bn group is disordered with 54/46 occupancies; the minor 46% occupancy is indicated using dashed lines. Atom labels for hydrogen atoms are omitted for clarity. | |
Fmoc-Ala(3-Cl)-O
t
Bu (4)
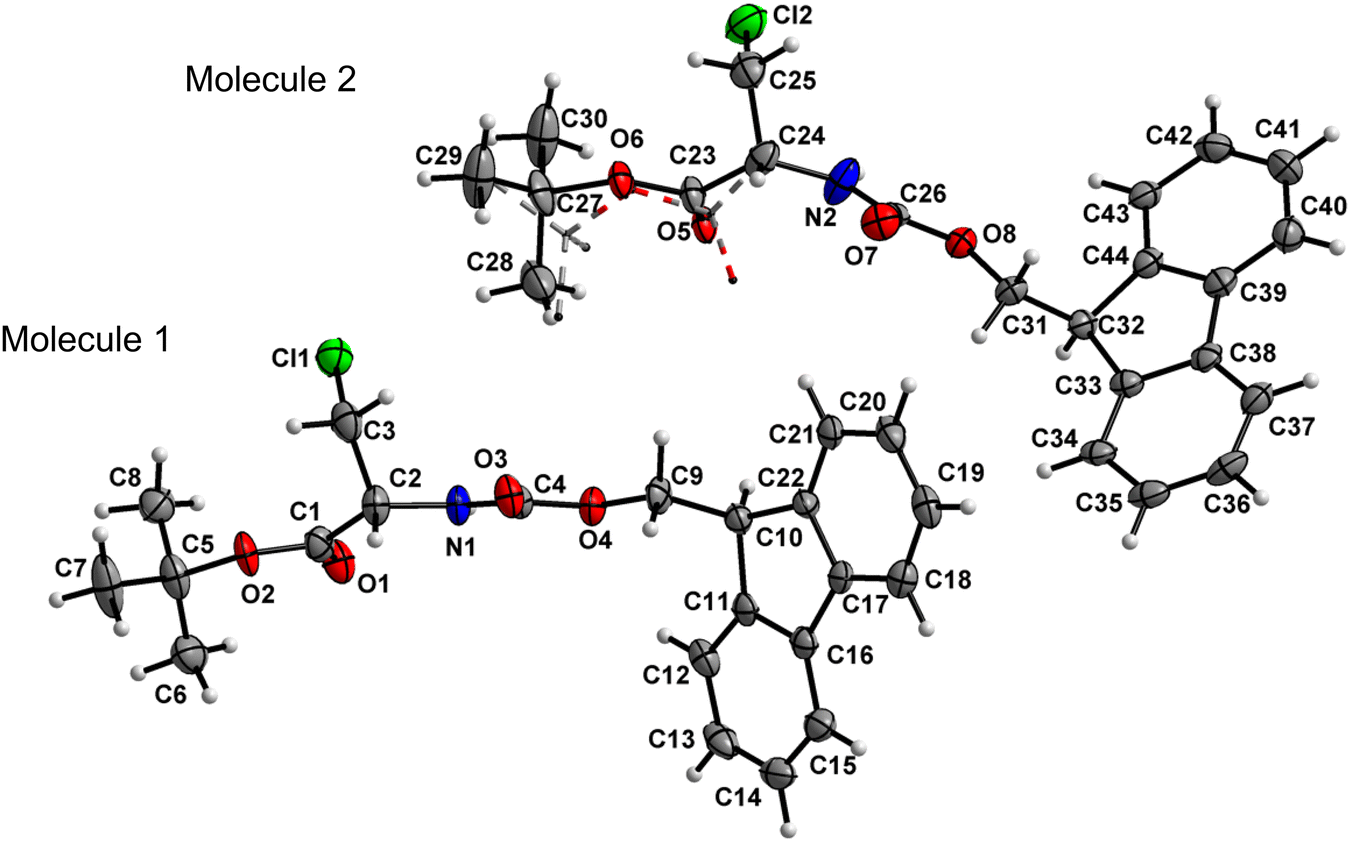
Amino acid derivative 4 features two molecules in the asymmetric unit cell (ASU), in one of which the tert-butyl group is disordered with 56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :44 occupancies. The other molecule within the ASU crystallized without a disorder (Fig. 5). Both the carbamate linkage (C2 to C9, including O3) and ester linkage (C2 to C5, including O1) of molecule 1 in the asymmetric unit are almost planar (deviation of 0.028 Å and 0.058 Å, respectively), with a dihedral angle of 18.2(1)° between the planes of these two groups. The carbamate linkage of molecule 2 (C24 to C31, including O7) is slightly bent out of planarity (r.m.s. deviation of 0.096 Å). In contrast, the ester linkage (C24 to C27, including O5) is almost planar (deviation of 0.058 Å), and the dihedral angle between these two planes amounts to 44.8(3)°. The conformations along the C2–C3 (molecule 1) and C24–C25 (molecule 2) bonds are staggered, and, in both cases, the bulky chlorine substituent is gauche to both ester and amido groups, in a similar manner to compound 2.
:44 occupancies. The other molecule within the ASU crystallized without a disorder (Fig. 5). Both the carbamate linkage (C2 to C9, including O3) and ester linkage (C2 to C5, including O1) of molecule 1 in the asymmetric unit are almost planar (deviation of 0.028 Å and 0.058 Å, respectively), with a dihedral angle of 18.2(1)° between the planes of these two groups. The carbamate linkage of molecule 2 (C24 to C31, including O7) is slightly bent out of planarity (r.m.s. deviation of 0.096 Å). In contrast, the ester linkage (C24 to C27, including O5) is almost planar (deviation of 0.058 Å), and the dihedral angle between these two planes amounts to 44.8(3)°. The conformations along the C2–C3 (molecule 1) and C24–C25 (molecule 2) bonds are staggered, and, in both cases, the bulky chlorine substituent is gauche to both ester and amido groups, in a similar manner to compound 2.
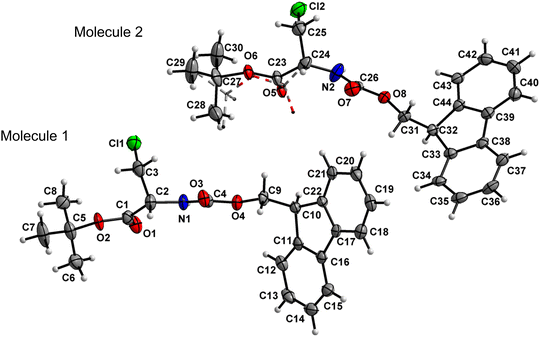 |
| | Fig. 5 Molecular structure of 4 with atom labels. Both molecules of the asymmetric unit are shown. One tBu group of molecule 2 is disordered with 56:44 occupancies; the minor 44% occupancy is illustrated in dashed lines. Atom labels for hydrogen atoms are omitted for clarity. | |
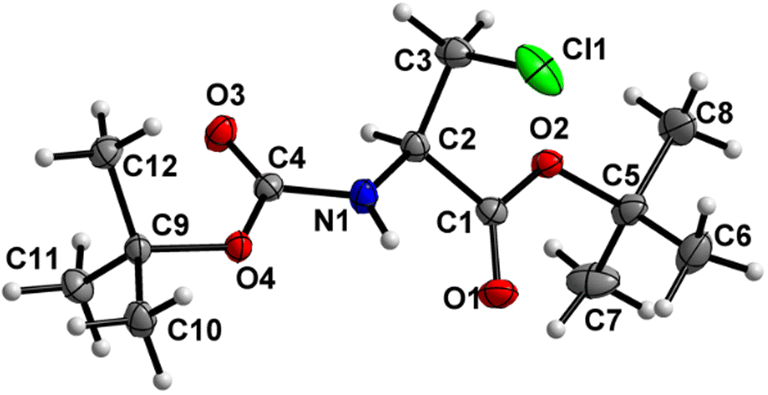
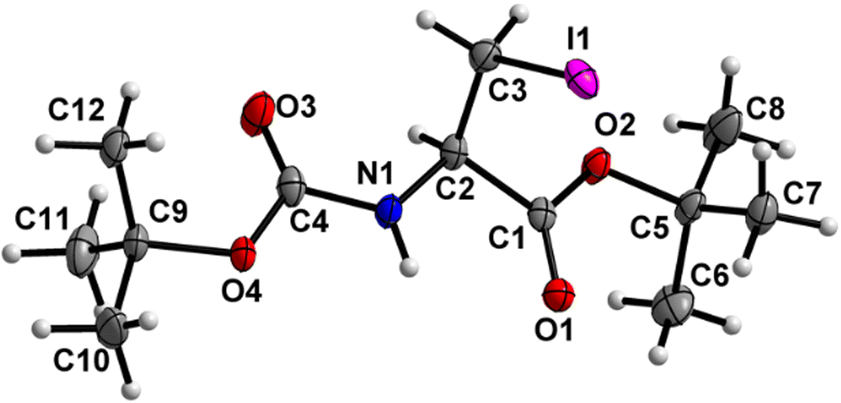
Boc-Ala(3-I)-OMe (5) (Fig. 6)
The carbamate linkage (C2 to C6, including O3) demonstrates noticeable bending out of planarity (deviation of 0.085 Å), whereas the ester linkage (C2 to C5, including O1) is almost planar (deviation of 0.011 Å). The dihedral angle between these two planes is 78.7(5)°. The backbone of the molecule (from C5 to I1) features an all-stretched conformation, with all atoms (including O1) residing close to one plane.
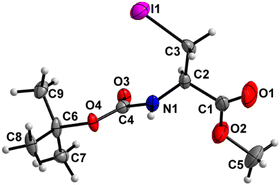 |
| | Fig. 6 Molecular structure of 5 with atom labels. Atom labels for hydrogen atoms are omitted for clarity. | |
Boc-Ala(3-I)-O
t
Bu (6) (Fig. 7)
Both the carbamate linkage (C2 to C9, including O3) and ester linkage (C2 to C5, including O1) are close to planarity (deviation of 0.023 Å and 0.021 Å, respectively), with a dihedral angle of 20.79(9)° between these two planes. The conformation along the C2–C3 bond is staggered, and the bulky iodine substituent is gauche to both ester and amido groups and not trans to any of them.
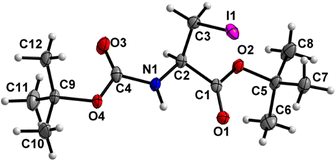 |
| | Fig. 7 Molecular structure of 6 with atom labels. Atom labels for some hydrogen atoms are omitted for clarity. | |
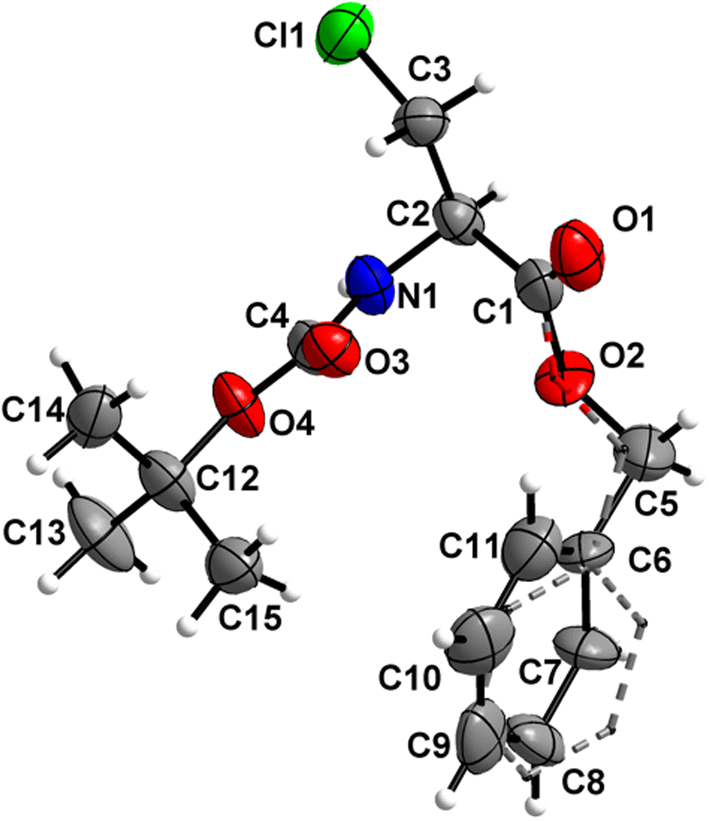
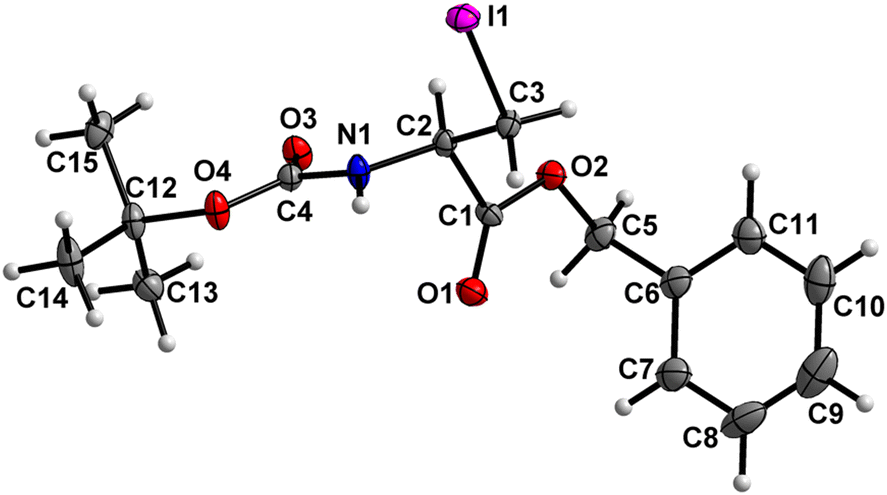
Boc-Ala(3-I)-OBn (7) (Fig. 8)
The carbamate linkage (C2 to C12, including O3) and the ester linkage (C2 to C5, including O1) are almost planar (deviation of 0.022 Å and 0.002 Å, respectively), with a dihedral angle of 81.7(2)° between these two planes.
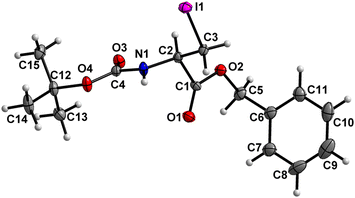 |
| | Fig. 8 Molecular structure of 7 with atom labels. Atom labels for hydrogen atoms are omitted for clarity. | |
Fmoc-Ala(3-I)-O
t
Bu (8)
The crystal structure has been reported previously40 and also belongs to the same series of compounds based on its structure, and, therefore, we add it to the discussion. For the details of its molecular structure, we refer the reader to the original publication.40
Thus, from the perspective of the molecular conformation, amino acid derivatives with less bulky ester groups (Me, Bn) prefer the all-stretched conformation (type 1) from the ester to the terminal methylene group of the alanine backbone (structures 1, 3, 5, 7), which is indicated by the dihedral angle of about 75–85° between the planes of the ester and carbamate groups (see Table 1). On the other hand, the molecules with bulky tBu esters prefer the conformation that features the ester group and the residue of carbamate protective groups maximally away from each other (structures 2, 4, 6, 8), which is accompanied by a dihedral angle of 45° or less between the planes of the ester and carbamate groups (type 2).
Table 1 Dihedral angles and molecular geometry of 1–8
| Compound |
Conformation |
Angles |
Type |
PG1 |
PG2 |
| PG1: carboxylate protective group; PG2: amine protective group. |
|
1
|
Stretched |
76.4 |
1 |
OMe, |
Boc |
|
2
|
Bent |
38.2 |
2 |
OtBu |
Boc |
|
3
|
Stretched |
80.62 |
1 |
OBn |
Boc |
|
4
|
Bent |
18.3; 43.7 |
2 |
OtBu |
Fmoc |
|
5
|
Stretched |
78.3 |
1 |
OMe |
Boc |
|
6
|
Bent |
20.7 |
2 |
OtBu |
Boc |
|
7
|
Stretched |
81.6 |
1 |
OBn |
Boc |
|
8
|
Bent |
33.69 |
2 |
OtBu |
Fmoc |
Crystal packing
Historically, analysis of molecular packing was performed by the manual analysis of short intermolecular contacts, using either Platon35 outputs or graphical interfaces, e.g. using Mercury36 software. This approach allowed us to identify the principal short contacts in the crystal packing of amino acid derivatives 1–8. Not surprisingly, most of these interactions can be classified as H-bonds, due to the presence of several H-bonding interactions in all molecular structures. All H-bond type interactions of compounds 1–8 are summarized in Table 2. In addition to H-bonds, two iodinated amino acid derivatives demonstrated halogen bond-type interactions, and in one of the compounds with the Fmoc protective group, a π–π stacking interaction between two aromatic ring systems was observed.
Table 2 H-bond type close contacts of amino acid derivatives 1–8
|
|
Donor (D) |
H-atom |
Acceptor (A) |
Symmetry operator |
Distance, Å |
Angle, ° |
| D–H |
H⋯A |
D⋯A |
D–H⋯A |
| Compound 1 |
| (i) |
N1 |
H1 |
O3 |
x, −1 + y, z |
0.82(2) |
2.15(2) |
2.954(2) |
169.3(2) |
| (ii) |
C2 |
H2 |
O1 |
x, −1 + y, z |
1.00 |
2.44 |
3.146(2) |
165 |
| (iii) |
C2 |
H2 |
O1 |
2 − x, −1/2 + y, 1 − z |
1.00 |
2.66 |
3.192(2) |
113 |
| (iv) |
C3 |
H3A |
O1 |
2 − x, −1/2 + y, 1 − z |
0.99 |
2.52 |
3.174(2) |
123 |
| Compound 2 |
| (i) |
C3 |
H3B |
O1 |
−1 + x, y, z |
0.99 |
2.62 |
3.435(2) |
139 |
| (ii) |
C3 |
H3A |
O4 |
−1 + x, y, z |
0.99 |
2.64 |
3.497(2) |
146 |
| Compound 3 |
| (i) |
N1 |
H1 |
O3 |
−1 + x, y, z |
0.99(3) |
2.01(4) |
2.98(1) |
166(8) |
| (ii) |
C2 |
H2 |
O1 |
−1 + x, y, z |
1.00 |
2.57 |
3.54(1) |
163 |
| (iii) |
C2 |
H2 |
O1 |
1/2 + x, 3/2 − y, 1 − z |
1.00 |
2.56 |
3.12(1) |
115 |
| (iv) |
C3 |
H3A |
O1 |
1/2 + x, 3/2 − y, 1 − z |
1.00 |
2.69 |
3.29(1) |
118 |
| Compound 4 |
| (i) |
N1 |
H1 |
O3 |
x, 1 + y, z |
0.88 |
2.24 |
3.083(5) |
161.4 |
| (ii) |
N2 |
H2 |
O7 |
x, 1 + y, z |
0.88 |
2.21 |
3.035(6) |
157 |
| (iii) |
C2 |
H2 |
O1 |
x, 1 + y, z |
1.00 |
2.28 |
3.252(6) |
164 |
| (iv) |
C24 |
H24 |
O5 |
x, 1 + y, z |
0.99 |
2. 49 |
3. 466(2) |
169 |
| (v) |
C41 |
H41 |
O7 |
−x, 1/2 + y, 2 − z |
0.95 |
2.60 |
3.423(6) |
147 |
| Compound 5 |
| (i) |
N1 |
H1 |
O3 |
1 + x, y, z |
0.69(10) |
2.39(10) |
2.917(1) |
135(1) |
| (ii) |
C3 |
H3B |
O3 |
1 + x, y, z |
0.99 |
2.57 |
3.331(1) |
133 |
| (iii) |
C3 |
H3A |
O1 |
−1/2 + x, −3/2 − y, −1 − z |
0.99 |
2.39 |
3.066(1) |
125 |
| (iv) |
C2 |
H2 |
O1 |
−1/2 + x, −3/2 − y, −1 − z |
0.99 |
2.68 |
3.201(1) |
112 |
| Compound 6 |
| (i) |
N1 |
H1 |
O3 |
1 + x, y, z |
0.89(2) |
2.69(2) |
3.568(3) |
167(2) |
| (ii) |
C2 |
H2 |
O1 |
1 + x, y, z |
1.00 |
2.62 |
3.374(3) |
132 |
| (iii) |
C8 |
H8B |
O1 |
2 − x, 1/2 + y, 1/2 − z |
0.98 |
2.52 |
3.498(4) |
177 |
| Compound 7 |
| (i) |
N1 |
H1 |
O3 |
x, y, −1 + z |
1.00(6) |
2.01(6) |
2.986(5) |
164(5) |
| (ii) |
C3 |
H3B |
O3 |
x, y, −1 + z |
0.99 |
2.54 |
3.229(6) |
126 |
| Compound 8 |
| (i) |
N1 |
H1 |
O3 |
1/2 + x, 1/2 − y, 1 − z |
0.881(2) |
2.489(2) |
3.309(3) |
155(1) |
To achieve the holistic view of the intermolecular interactions contributing to the stabilization of the crystal structure, it is also necessary to take into account the interactions that cannot be extracted from the Platon output or from the visual analysis of short atom–atom contacts. In particular, they refer to London dispersion forces, which are weak but additive over the whole molecular surface and, therefore, may be the major contributing factor for the attractive intermolecular interactions of the non-polar molecules with large surfaces. The last versions of Crystal Explorer38 software offer a computational approach combining calculations of intermolecular interaction energies with their graphical representation. This approach offers accurate computation of intermolecular interaction energies between molecular pairs in crystal structures and their visualization using colored cylinders,37,41 and has been shown to be particularly useful in the analysis of weak interactions, such as hydrogen, halogen and chalcogen bonds.42–47 These cylinders represent the energies between molecular pairs in crystals; the thicker the cylinder, the higher the interaction energy. Contributions of different types of intermolecular interactions (electrostatic, dispersion, polarization, repulsion) can be determined for each interacting molecular pair. Though the CLP PIXEL method48–50 developed before by Gavezzotti offers the possibility for the same type of calculation, the graphical interface of Crystal Explorer and approximately the same accuracy of calculations51 make it the method of choice for the calculation of intermolecular interaction in crystal structures. A summary of all interaction terms for the closest intermolecular interactions is presented in Table 3 below, followed by the descriptions of crystal packing by each of the amino acid derivatives 1–8.
Table 3 Interaction energies (kJ mol−1) for the closest intermolecular interactions
| Compound |
Electron density |
R
|
E
ele
|
E
pol
|
E
dis
|
E
rep
|
E
tol
|
|
R = distance in Å between molecular centroids, Eele = electrostatic component, Epol = polarisation component, Edis = dispersion component, Erep = repulsion component. |
|
1
|
B3LYP/6-31G(d,p) |
5.08 |
−38.2 |
−7.3 |
−38.9 |
49.3 |
−49.2 |
|
2
|
B3LYP/6-31G(d,p) |
5.97 |
−10.6 |
−2.6 |
−40.7 |
27.3 |
−31.8 |
|
3
|
B3LYP/6-31G(d,p) |
5.11 |
−44.3 |
−8.4 |
−67.5 |
71.7 |
−67.5 |
|
4
|
B3LYP/6-31G(d,p) |
5.11 |
−35.4 |
−9.0 |
−84.4 |
90.1 |
−61.8 |
|
5
|
B3LYP/DGDZVP |
5.13 |
−37.4 |
−10.1 |
−42.8 |
50.2 |
−53.3 |
|
6
|
B3LYP/DGDZVP |
5.45 |
−19.0 |
−3.6 |
−47.3 |
36.1 |
−41.6 |
|
7
|
B3LYP/DGDZVP |
5.17 |
−54.4 |
−9.8 |
−69.4 |
91.0 |
−69.0 |
|
8
|
B3LYP/DGDZVP |
4.82 |
−48.7 |
−7.0 |
−101.1 |
97.9 |
−84.2 |
Boc-Ala(3-Cl)-OMe (1)
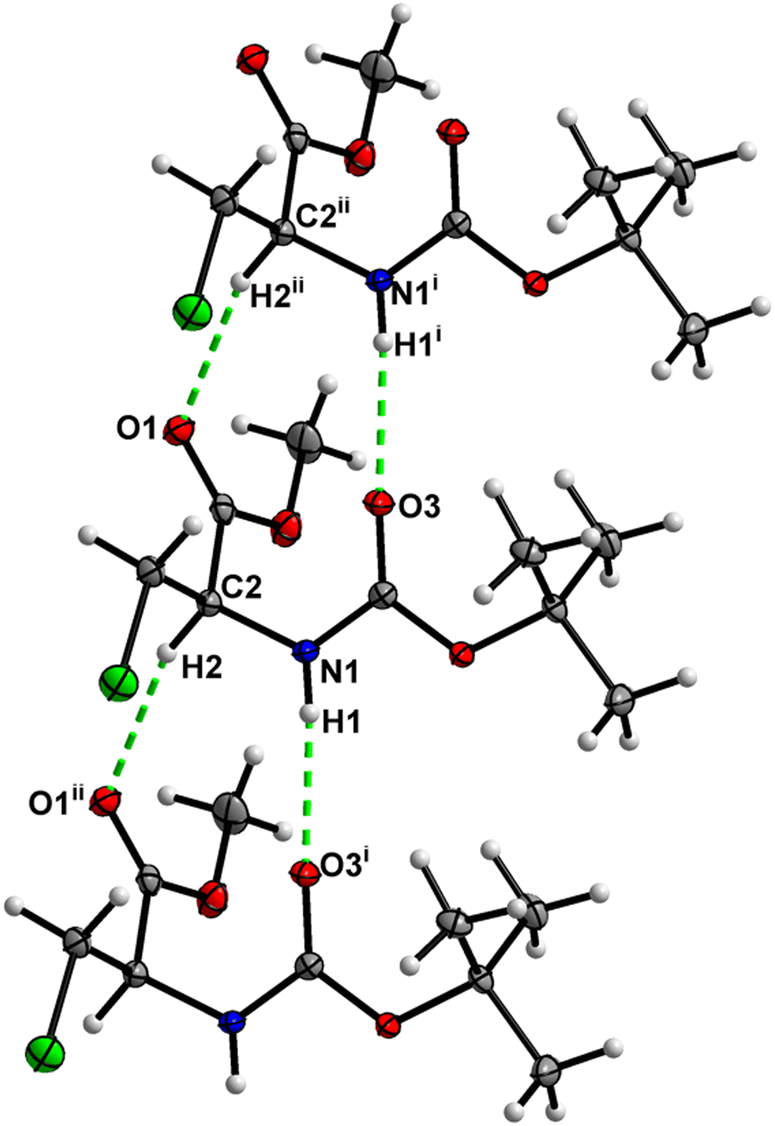
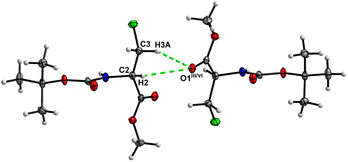
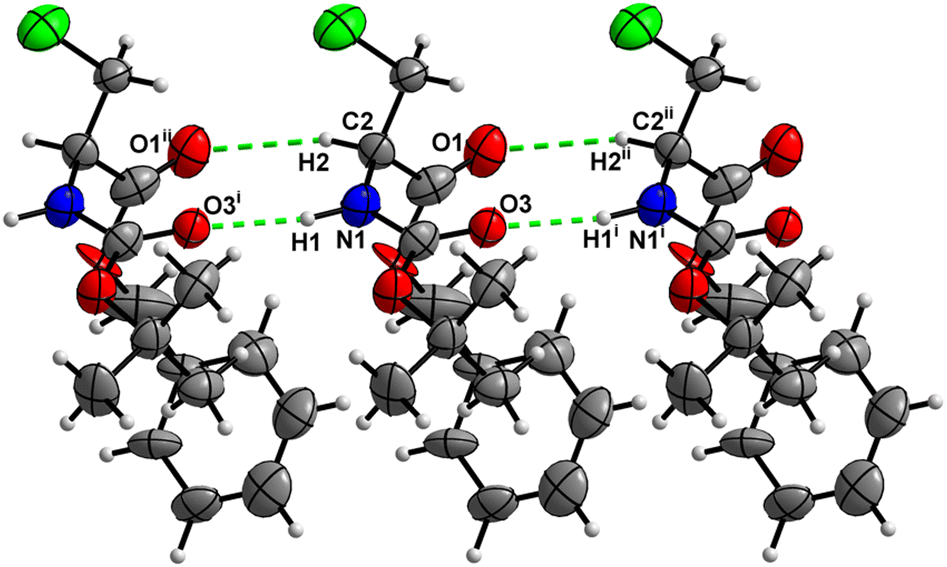
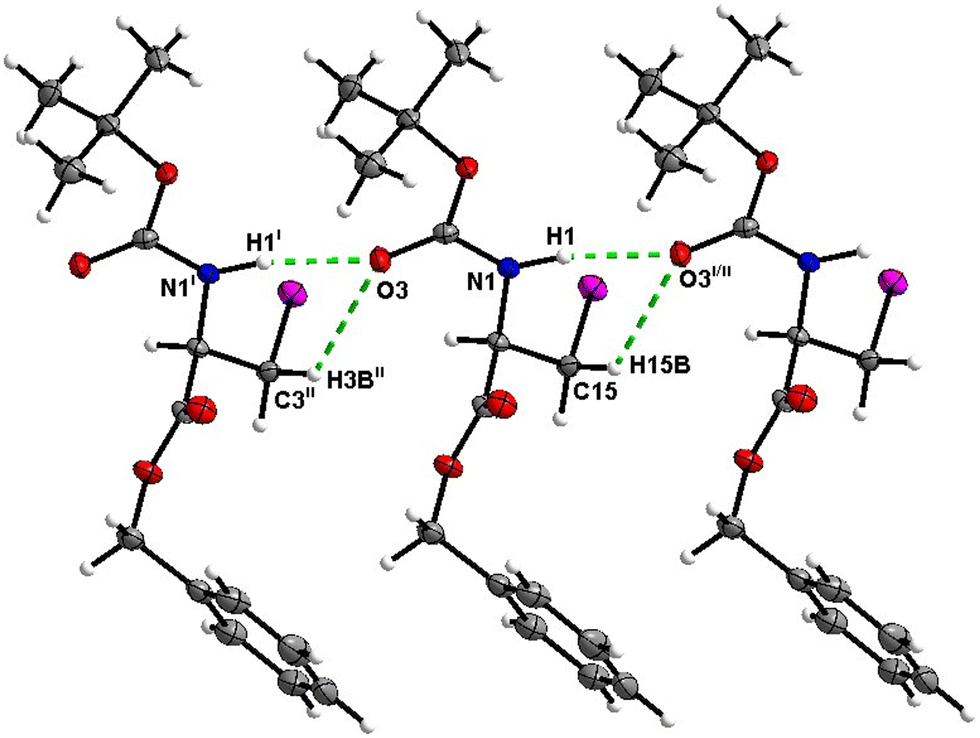
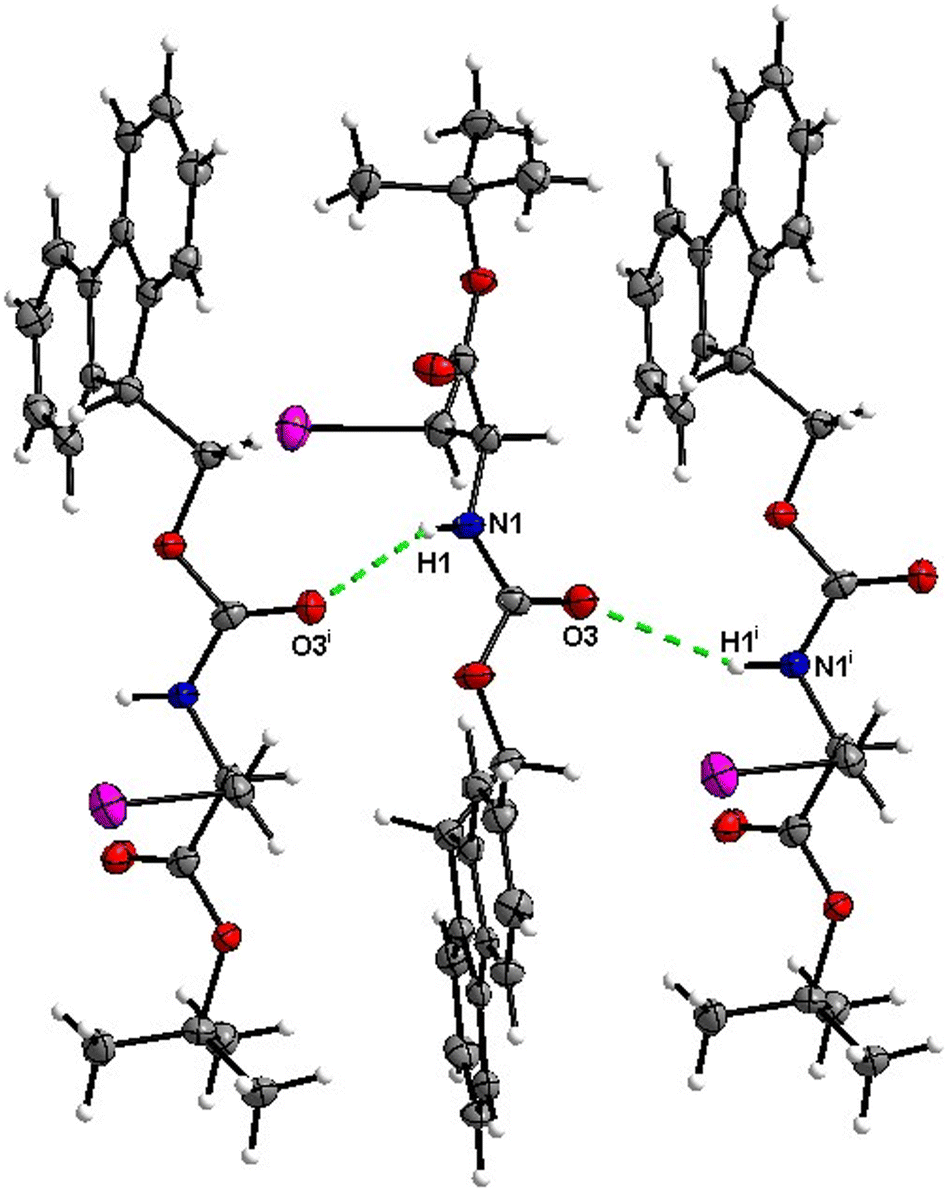
In the crystal, molecules of 1 are linked together into infinite chains by CO⋯H–N hydrogen bonds, with both H-bond donors and acceptors belonging to the carbamate groups of the interacting molecules (Fig. 9) This bond is complemented by the weak H-bond type CO⋯H–C interaction, in which the C–H of the stereocenter serves as the H-bond donor, and the carbonyl of the ester group serves as the H-bond acceptor. Two other H-bond type interactions of the ester carbonyl link the molecule to the C–H of the stereocenter and the C–H of the –CH2Cl residue of molecule 2 (Fig. 10). In combination, these interactions organize the molecule into dimeric ribbons along [010].
 |
| | Fig. 9 View of the H-bond interaction (i) and close C–H⋯O contacts (ii) parallel to [010] in the crystal structure of 1. | |
 |
| | Fig. 10 View of the short C–H⋯O contacts (iii) and (iv) in the crystal structure of 1. | |
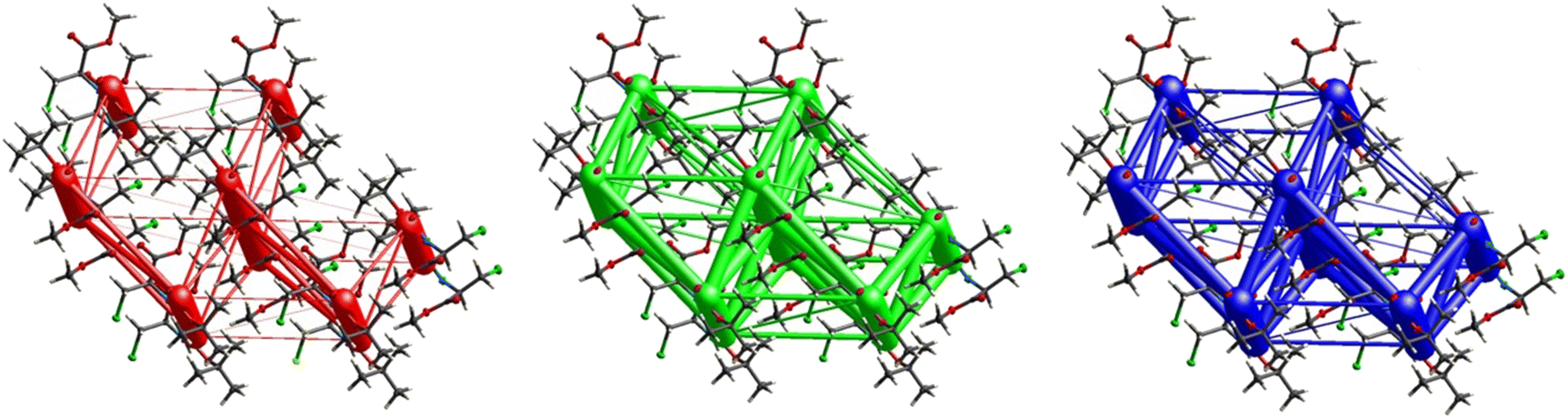
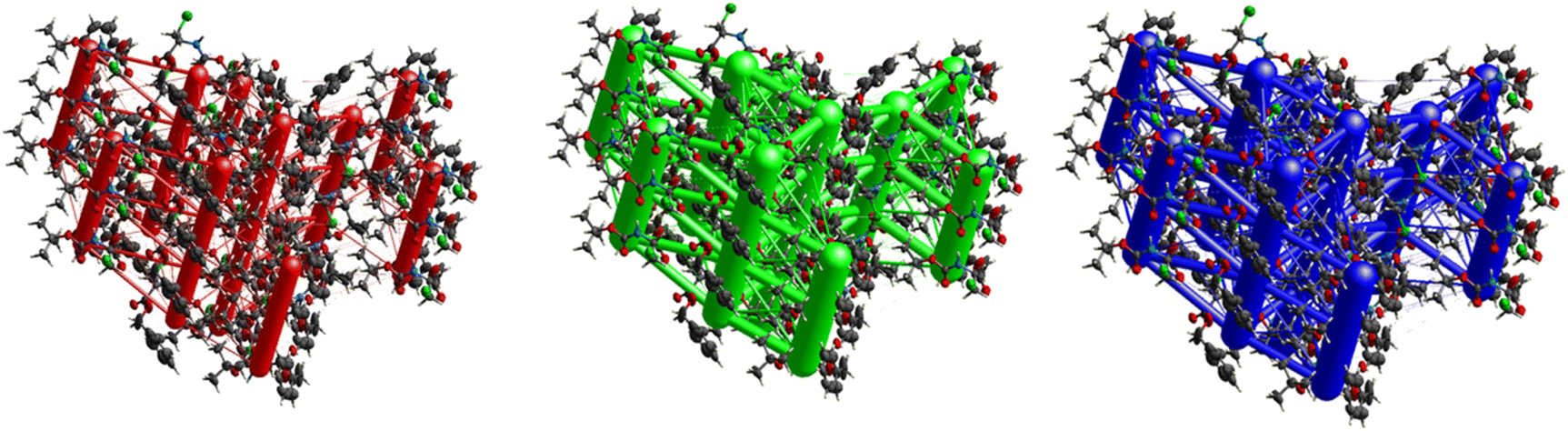
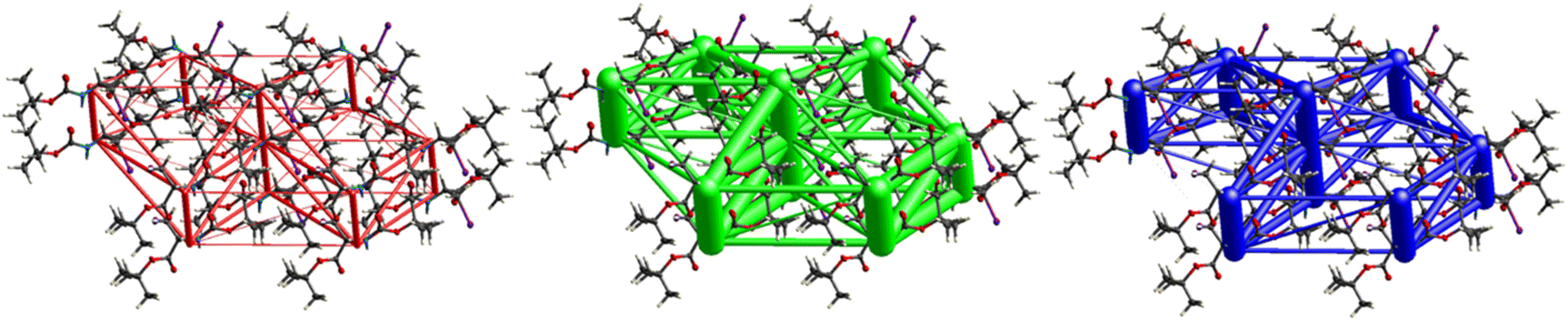
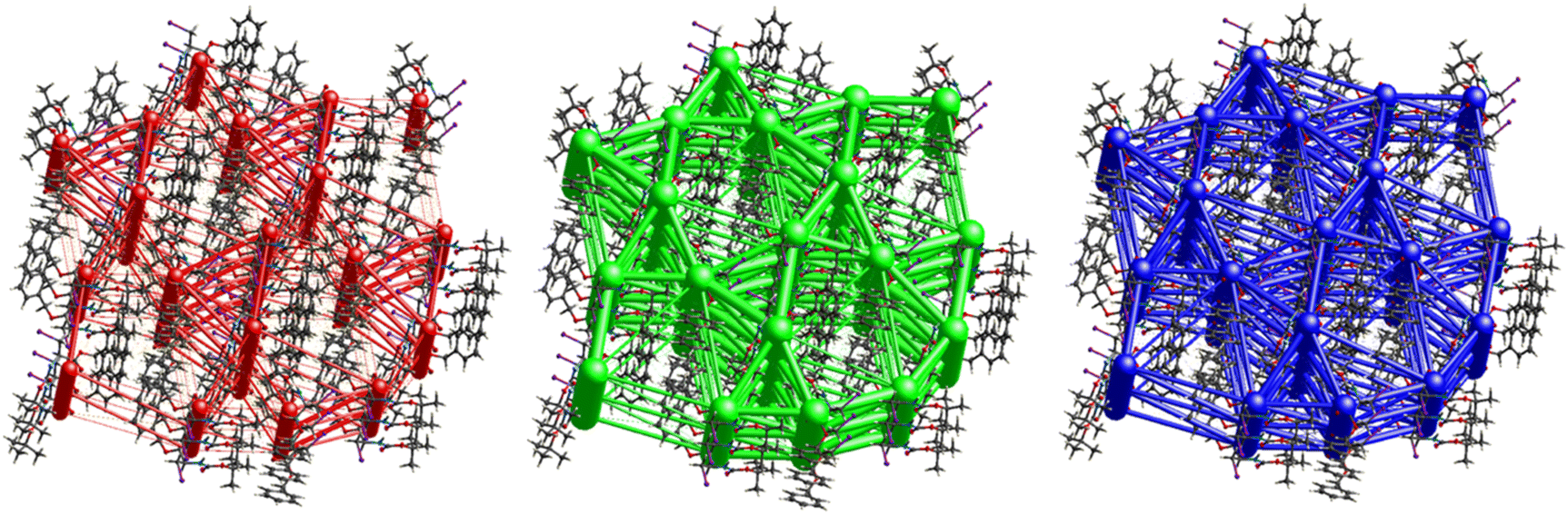
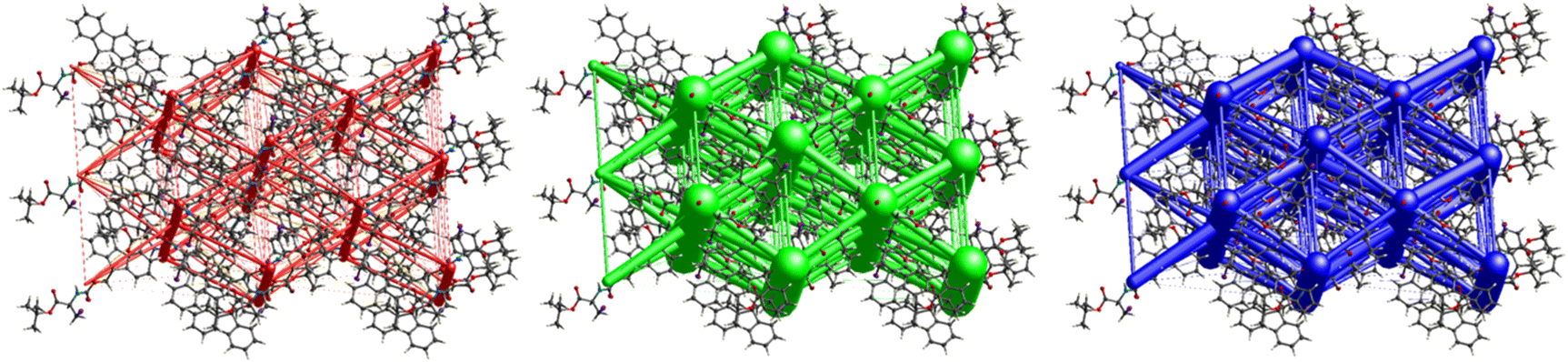
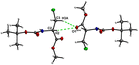
Fig. 11 demonstrates the energy frameworks obtained for 1 using Crystal Explorer. It shows an isotropic hexagonal packing topology with one extra cylinder at the centroid. The crystal packing is stabilized by strong electrostatic attractive interactions running along [010] and corresponds to the CO⋯H–N and the CO⋯H–C hydrogen bonds (Fig. 9), and the dispersion energy cylinders also adopt this direction. The similarity of the magnitude of the two energies is visible in Fig. 11; this similarity is also revealed by the values indicated in Table 1 for Eele and Edis. However, the magnitude of the total energies mirrors that of the dispersion energy, suggesting that the repulsion terms largely cancel the electrostatic energies. Therefore, the total stabilization energy of the crystal amounting to −49.2 kJ mol−1 is dominated by the dispersion energies.
 |
| | Fig. 11 Energy frameworks for electrostatic (red), dispersion (green) and total interaction energies (blue) for a cluster of nearest-neighbour molecules in Boc-Ala(3-Cl)-OMe (1). The energy tube size is 200, and the energy threshold is 0 kJ mol−1. | |
Boc-Ala(3-Cl)-O
t
Bu (2)
The number of short contacts classified as weak hydrogen bonds is surprisingly low. This can be traced to two short contacts of each of the H-atoms of the –CH2Cl group, one pointing to the carbonyl of the ester group and the second one to the alkylated O-atom of the ester group (Fig. 12). This interaction organizes the molecules into infinite chains along [100].
 |
| | Fig. 12 Short C–H⋯O contacts (i) and (ii) along [100] in the crystal structure of 2. | |
Electrostatic energy contribution in the crystal structure of 2 is rather insignificant due to the absence of strong H-bonds of the CO⋯H–N type, as corroborated by Fig. 12 and 13 and Table 3. However, the total topology adopted is that of corrugated slabs running anti-parallel to each other along [100]. Even more pronounced than in compound 1, the interactions are predominantly dispersion-based as seen in Fig. 13, with a total energy of −31.8 kJ mol−1, which is the lowest one among all the crystal lattices in this study.
 |
| | Fig. 13 Energy frameworks for electrostatic (red), dispersion (green) and total interaction energies (blue) for a cluster of nearest-neighbour molecules in Boc-Ala(3-Cl)-OtBu (2). The energy tube size is 200, and the energy threshold is 0 kJ mol−1. | |
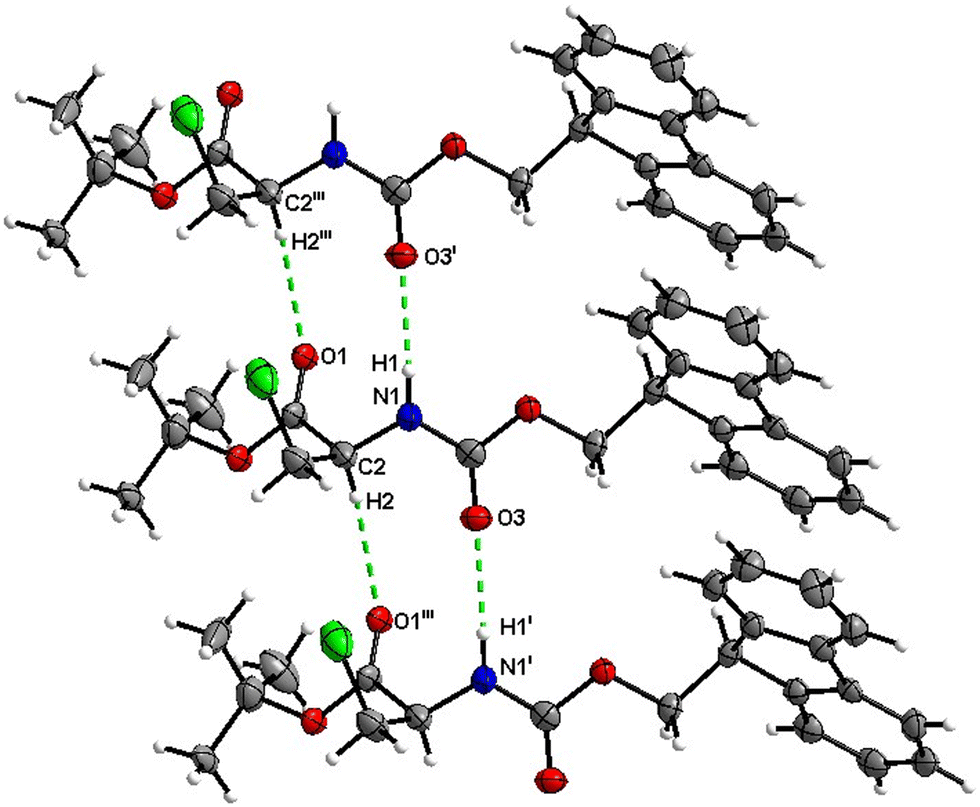
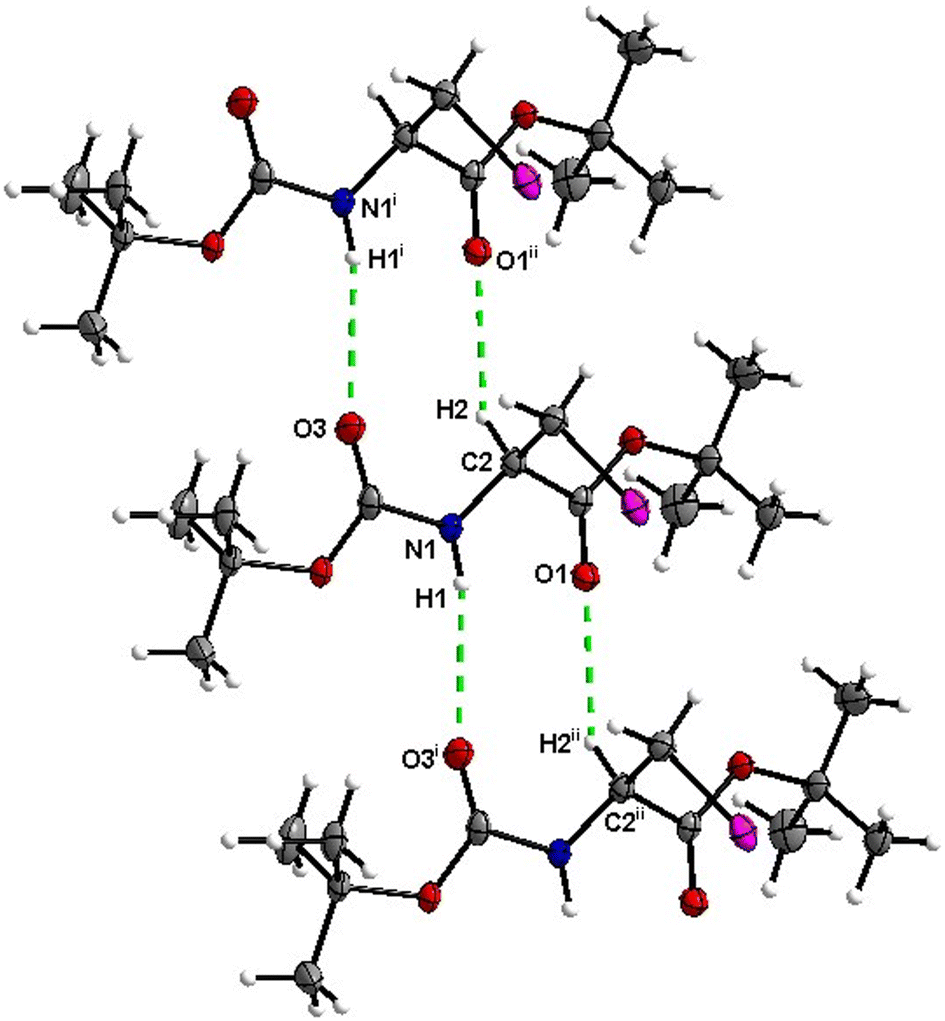
Boc-Ala(3-Cl)-OBn (3)
Molecules of 3 are interconnected into infinite chains by CO⋯H–N hydrogen bonds, in which both H-bond donors and acceptors belong to the carbamate groups of the interacting molecules (Fig. 14). This bond is complemented by the weak H-bond type CO⋯H–C interaction, in which the C–H of the stereocenter serves as the H-bond donor, and the carbonyl of the ester group serves as the H-bond acceptor. Two other H-bond type interactions of the ester carbonyl link the molecule to the C–H of the stereocenter (forming a bifurcated H-bond) and the C–H of the –CH2Cl residue of molecule 2 (Fig. 15). Altogether, these H-bond type interactions organize the molecule into dimeric ribbons along [100].
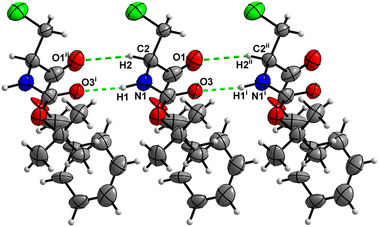 |
| | Fig. 14 View of the H-bond interaction (i) and close C–H⋯O contacts (ii) parallel to [100] in the crystal structure of 3. Minor occupancy of the disorder is omitted for clarity. | |
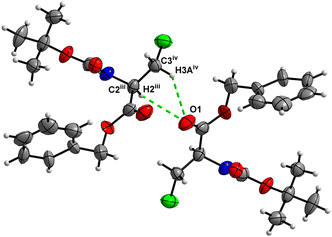 |
| | Fig. 15 View of the close contacts (iii) and (iv) in the crystal structure of 3. Minor occupancy of the disorder is omitted for clarity. | |
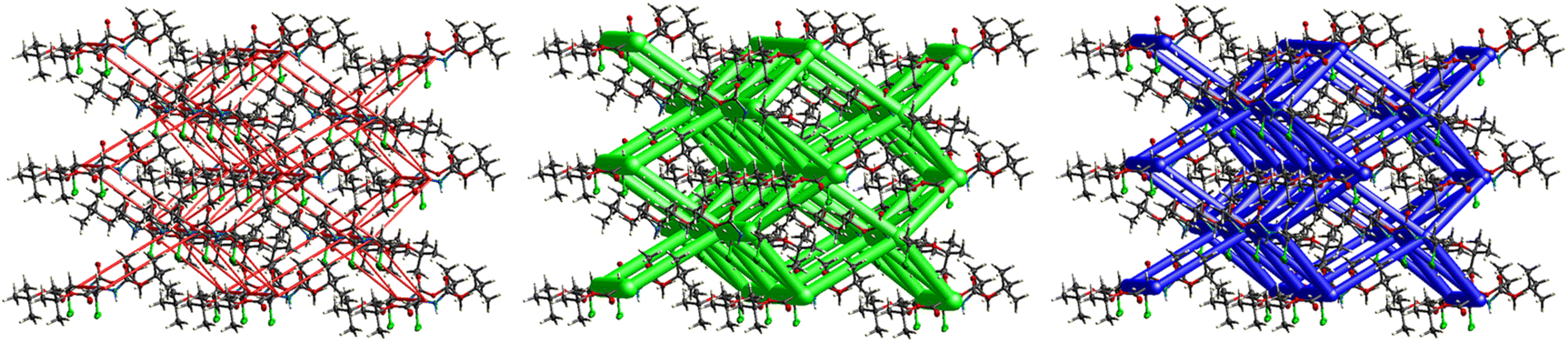
A strong electrostatic energy framework, running along the ab plane in the packing framework of 3, is due to the combination of CO⋯H–N and CO⋯H–C H-bonding interactions (Fig. 16). These electrostatic cylinders are accompanied by even thicker dispersion energy cylinders running along the same [100] direction. In the dispersion framework, each cylinder is linked to three additional cylinders of the same magnitude by at least six less dispersion terms. A high lattice energy of −67.5 kJ mol−1 is indicative of strong dispersion forces combined with the energy of the CO⋯H–N and the CO⋯H–C hydrogen bonds.
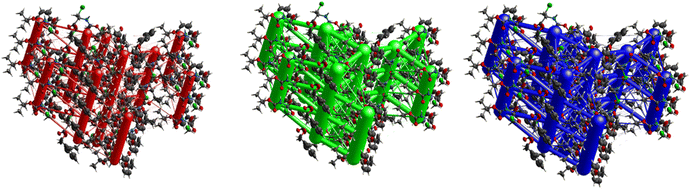 |
| | Fig. 16 Energy frameworks for electrostatic (red), dispersion (green) and total interaction energies (blue) for a cluster of nearest-neighbour molecules in Boc-Ala(3-Cl)-OBn (3). The energy tube size is 200, and the energy threshold is 0 kJ mol−1. | |
Fmoc-Ala(3-Cl)-O
t
Bu (4)
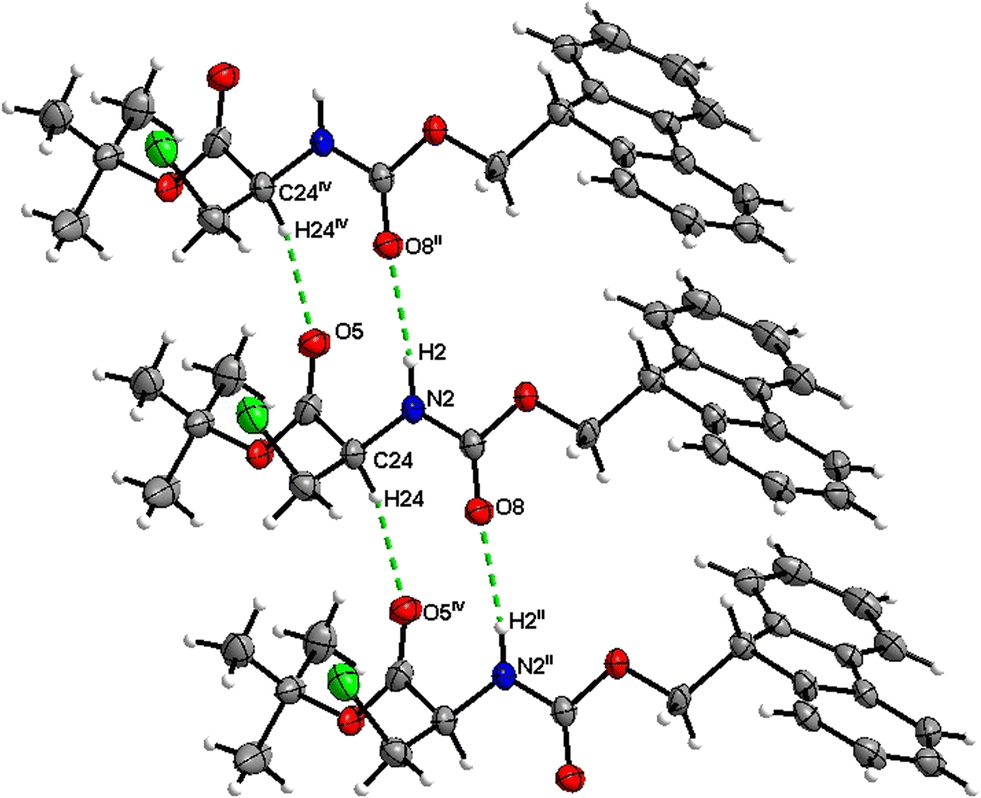
The number of H-bond type close contacts is relatively low. Populations of both types of molecules in the asymmetric unit are interconnected into infinite chains by the two almost parallel interactions along [010]. One of these interactions is a CO⋯H–N hydrogen bond, with the carbonyl belonging to the carbamate group, and one is a CO⋯H–C close contact, with the carbonyl belonging to the ester group and C–H to the stereocenter of the amino acid (Fig. 17 and 18).
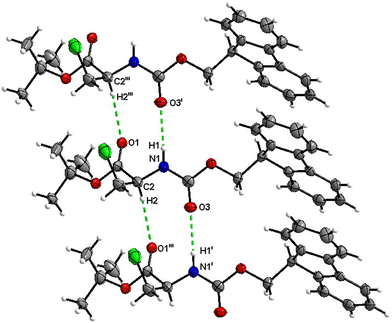 |
| | Fig. 17 View of the H-bond interaction (i) and close C–H⋯O contacts (iii) parallel to [010] in the crystal structure of 4. | |
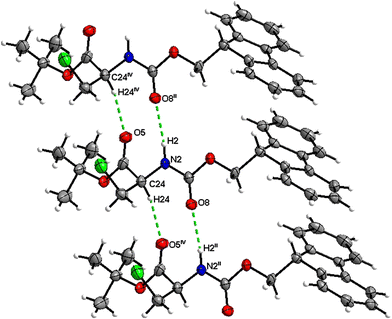 |
| | Fig. 18 View of the H-bond interaction (ii) and close C–H⋯O contacts (iv) parallel to [010] in the crystal structure of 4. Only the major disorder of the tBu group is shown for clarity. | |
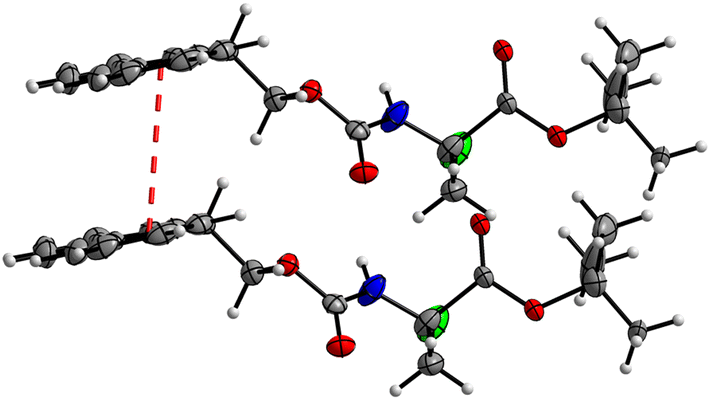
In addition, crystal packing of 4 features a π–π stacking interaction with an interplanar Cg1–Cg2 (Cg1 = C32–C33–C38–C39–C44, Cg2 = C33–C34–C35–C36–C37–C38) distance of 3.943(3) Å (Fig. 19). Interestingly, this short contact exists between only one set of the molecules in the asymmetric unit; the other set does not feature any close π–π contacts. The large lateral displacement of aromatic rings implies the relatively low significance of this interaction.
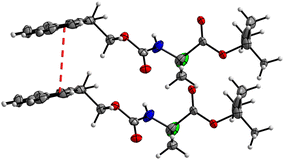 |
| | Fig. 19 View of the π–π stacking interaction between two aromatic ring systems, Cg1 and Cg2, in the crystal structure of 4 with a distance of 3.943(3) Å (symmetry operator: x, 1 + y, z). | |
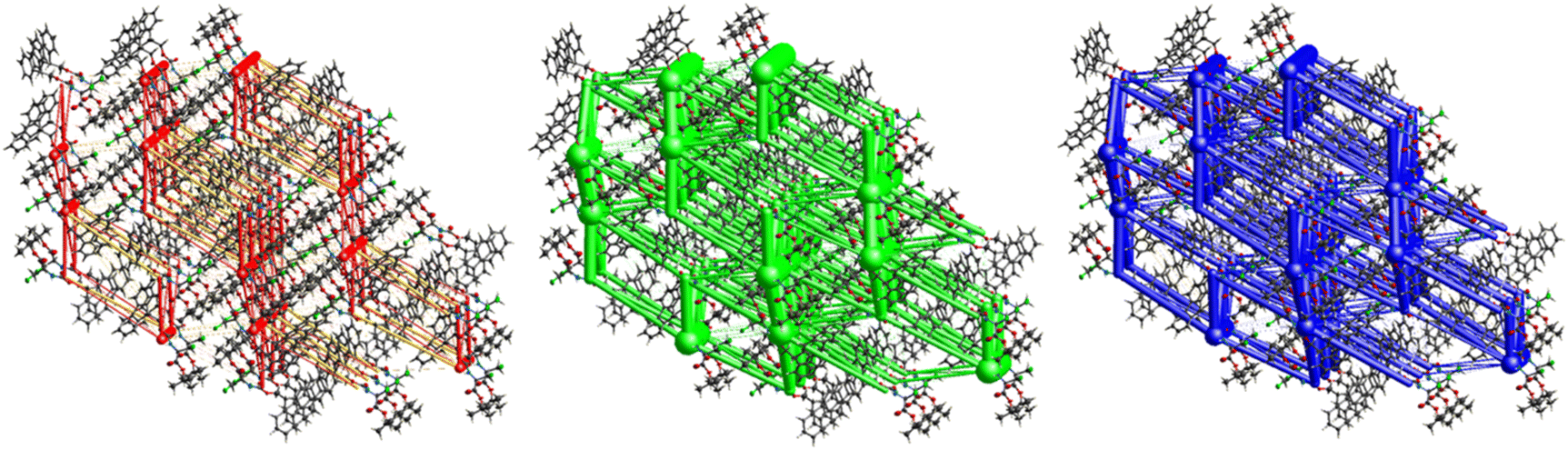
The interactions in 4 (Fig. 20) are dominated by dispersion energies moving along [010] parallel to the ab plane, and these cylinders are linked by weaker dispersion terms resulting in a more isotropic topology. Although very weak, the electrostatic terms seem to be accompanied by weaker destabilizing electrostatic energies of 10.1 kJ mol−1, yellow columns as seen in Fig. 20, but they are compensated by the attractive dispersion interactions. Destabilizing interactions such as these may remain unnoticed if one only focuses on the analysis of typical bonding patterns, such as the direct atom–atom interactions discussed in the first section.
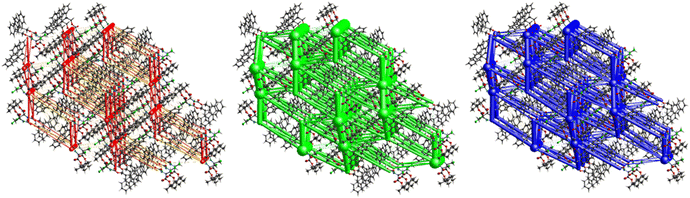 |
| | Fig. 20 Energy frameworks for electrostatic (red), dispersion (green) and total interaction energies (blue) for a cluster of nearest-neighbour molecules in Fmoc-Ala(3-Cl)-OtBu (4). The energy tube size is 200, and the energy threshold is 0 kJ mol−1. | |
Boc-Ala(3-I)-OMe (5)
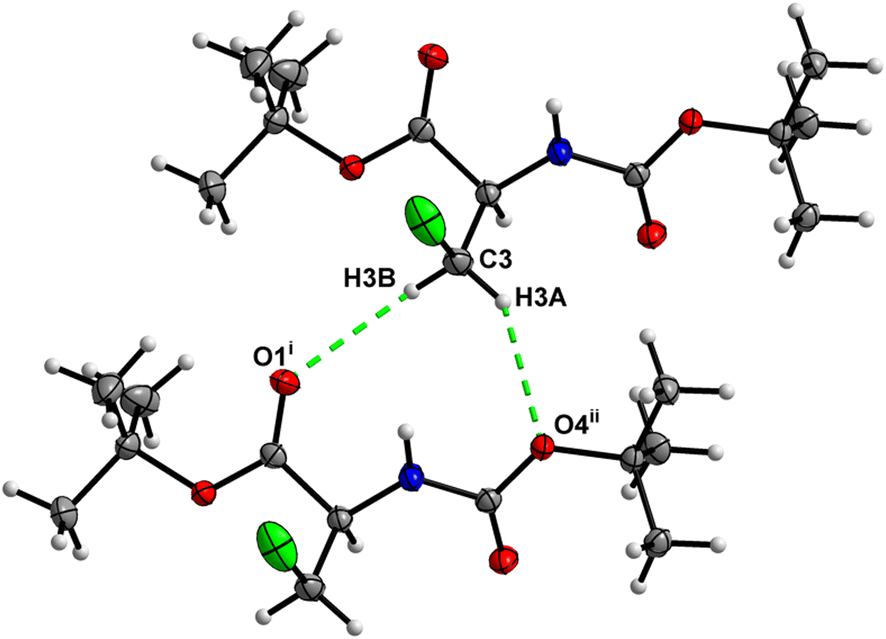
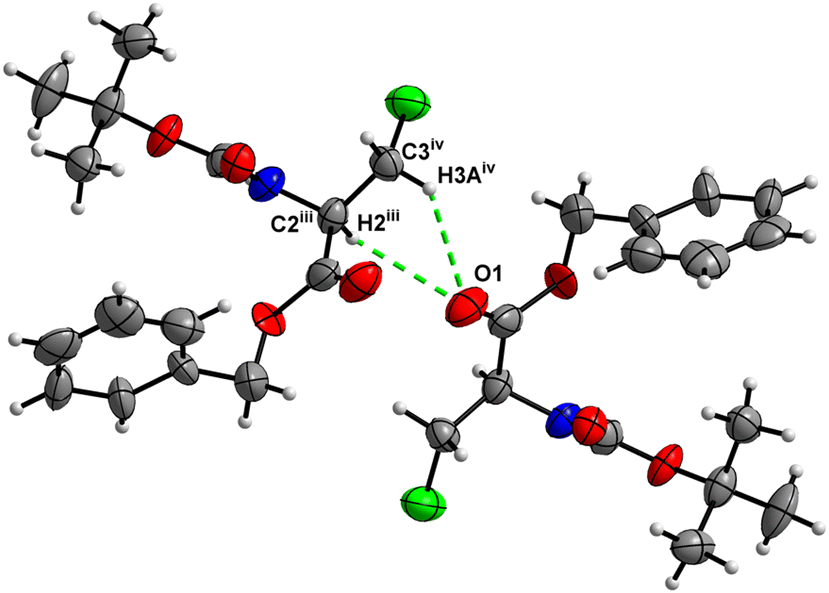
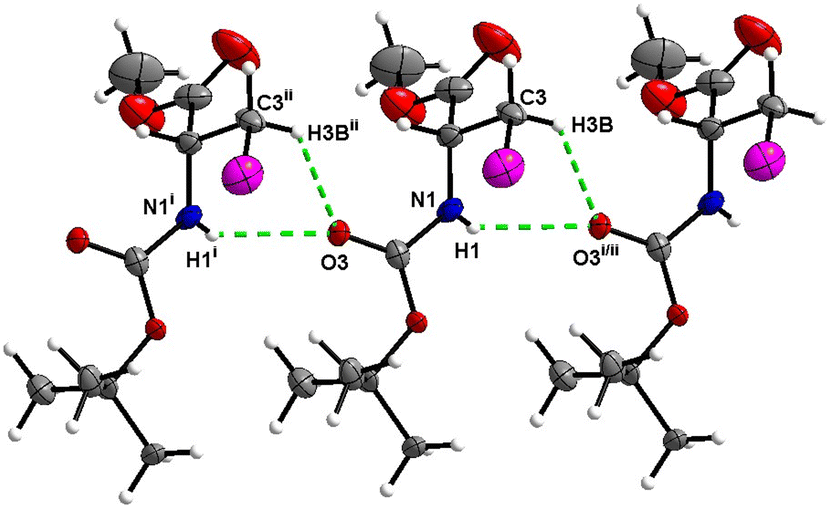
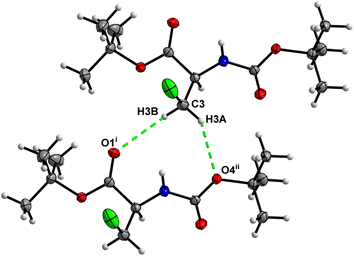
In the crystal, molecules of 5 are linked together into infinite chains by CO⋯H–N hydrogen bonds, with both H-bond donors and acceptors belonging to the carbamate groups of the interacting molecules (Fig. 21). This bond is complemented by the weak H-bond type CO⋯H–C interaction, in which the C–H of the stereocenter serves as the H-bond donor and the carbonyl of the carbamate group serves as the H-bond acceptor, thus forming a bifurcated H-bond (Fig. 21). Two other H-bond type interactions of the ester carbonyl link the molecule to the C–H of the stereocenter and the C–H of the –CH2I residue of molecule 2 (Fig. 22). Combined, these interactions arrange the molecule into dimeric ribbons along [100].
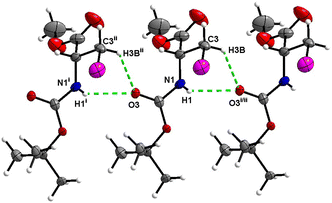 |
| | Fig. 21 View of the H-bond interaction (i) and close C–H⋯O contacts (ii) parallel to [100], with O2 being a bifurcated acceptor in the crystal structure of 5. | |
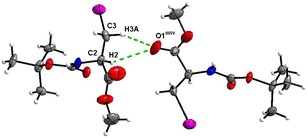 |
| | Fig. 22 View of the close C–H⋯O contacts (iii) and (iv) in the crystal structure of 5. | |
The crystal packing of 5 (Fig. 23) revealed a rather interesting zigzag packing fashion linked by strong CO⋯H–N and CO⋯H–C H-bonding interactions. Anti-parallel frameworks of weak electrostatic energies stabilize this compound due to these CO⋯H–C and CO⋯H–N hydrogen bonding interactions. Compound 4 also gave a corrugated-slab packing mode similar to 5, but with a higher lattice energy of −61.8 kJ mol−1, whereas for 5, calculations predicted −53.3 kJ mol−1 lattice energy.
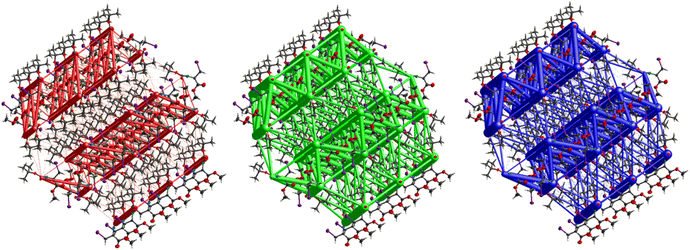 |
| | Fig. 23 Energy frameworks for electrostatic (red), dispersion (green) and total interaction energies (blue) for a cluster of nearest-neighbour molecules in Boc-Ala(3-I)-OMe (5). The energy tube size is 200, and the energy threshold is 0 kJ mol−1. | |
Boc-Ala(3-I)-O
t
Bu (6)
Hydrogen bond type close contacts are not abundant in the crystal packing of 6 (Fig. 24). Molecules are interconnected into infinite chains by the two almost parallel interactions along [100]. One of these interactions is the CO⋯H–N hydrogen bond, with carbonyl belonging to the carbamate group, and another one is the CO⋯H–C close contact, with carbonyl belonging to the ester group and C–H to the stereocenter of the amino acid. The structure of Boc-Ala(3-Cl)-OtBu2 differs from that of 6 only by the halogen atom (chlorine versus iodine), and it is noteworthy that in the crystal structure of 2, no CO⋯H–N hydrogen interaction is observed.
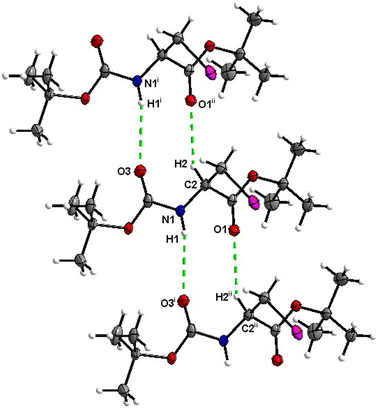 |
| | Fig. 24 View of the H-bond interaction (i) and close C–H⋯O contacts (ii) parallel to [100] in the crystal structure of 6. | |
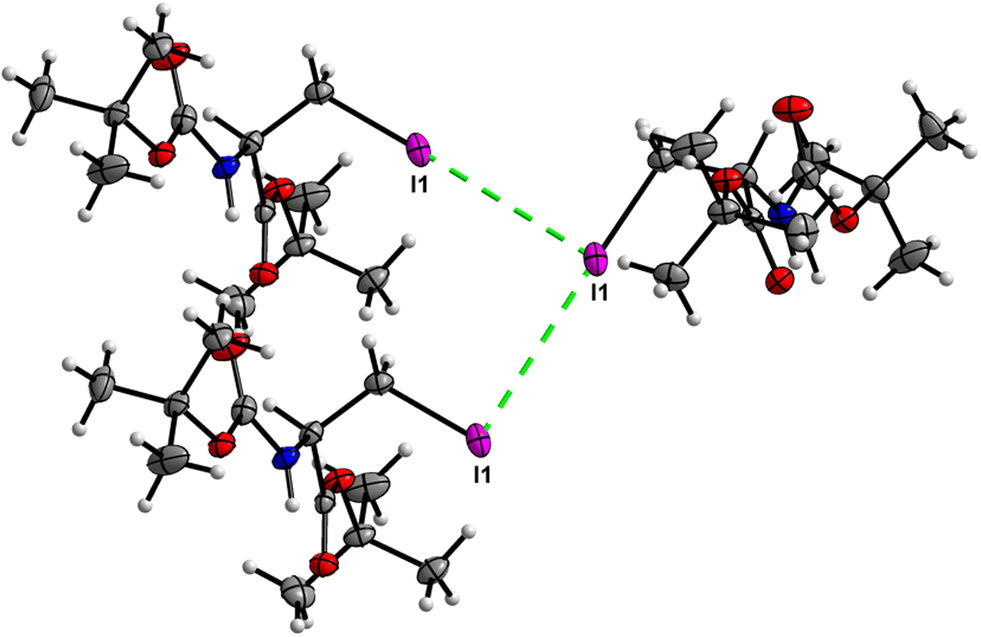
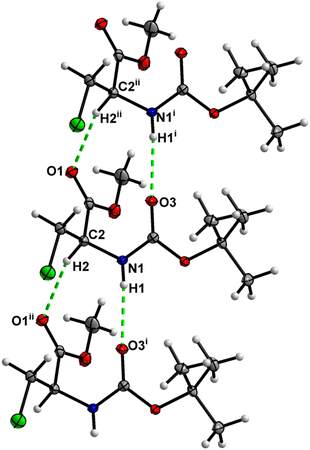
Crystal packing of 6 is the only one featuring halogen interactions between the iodine atoms of the neighbouring molecules (Fig. 25, symmetry operator −1/2 + x, 3/2 − y, 1 − z). It is a typical type II halogen interaction32,33 featuring a typical zigzag pattern. The electrophilic region of one iodine atom (σ-hole, noticeable as a blue spot on the ESP surface, see Fig. S11†) points to the nucleophilic region of the other halogen atom with an I⋯I distance of 3.8822(6) Å and an I⋯I⋯I angle of 89.094(7)°. This zigzag pattern of this interaction stretches along [100].
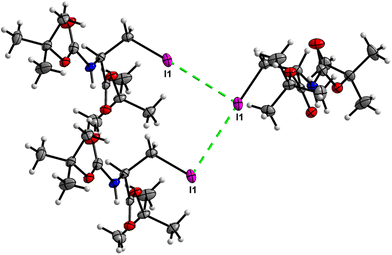 |
| | Fig. 25 View of the zigzag short I⋯I contact of type II in the crystal structure of 6. | |
The crystal structure of compound 6, as seen in Fig. 26, gave a similar hexagonal energy framework to that of 1. However, unlike 1, the crystal packing is dominantly stabilized by dispersion energies linked by weaker dispersion cylinders to each other and to the centroid cylinder. The highest total pairwise energy is also lower in 6 by a slight difference of ∼8 kJ mol−1. The absence of links between molecules associated with I⋯I⋯I interactions should not be taken as an indication of no halogen–halogen bonding but rather as an indication of low I⋯I⋯I energy magnitude. This observation implies that the I⋯I⋯I contacts do not play a significant stabilizing role in the packing mode of this compound.
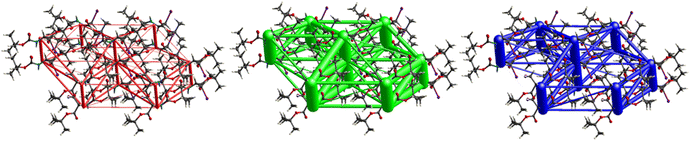 |
| | Fig. 26 Energy frameworks for electrostatic (red), dispersion (green) and total interaction energies (blue) for a cluster of nearest-neighbour molecules in Boc-Ala(3-I)-OtBu (6). The energy tube size is 200, and the energy threshold is 0 kJ mol−1. | |
Boc-Ala(3-I)-OBn (7)
Molecules of 7 are linked together into infinite chains by CO⋯H–N hydrogen bonds in the crystal, with both H-bond donors and acceptors belonging to the carbamate groups of the interacting molecules (Fig. 27). This bond is complemented by the weak H-bond type CO⋯H–C interaction, in which the C–H of the stereocenter serves as the H-bond donor, and the carbonyl of the carbamate group serves as the bifurcated H-bond acceptor for both H-bond type interactions (Fig. 27).
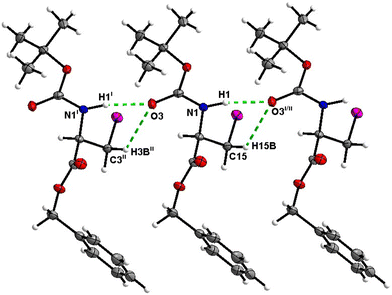 |
| | Fig. 27 View of the H-bond interactions (i) and (ii) parallel to the [001] axis in the crystal structure of 7. | |
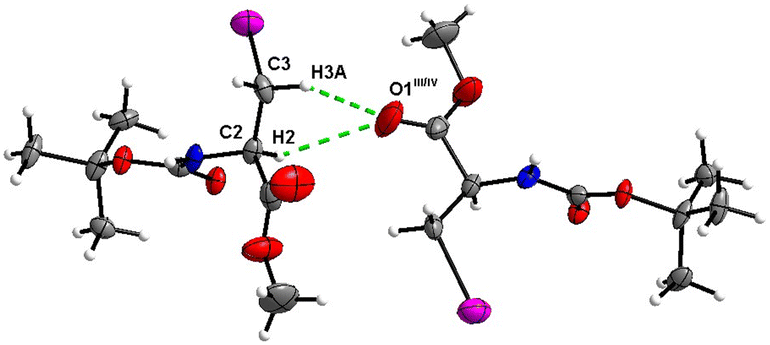
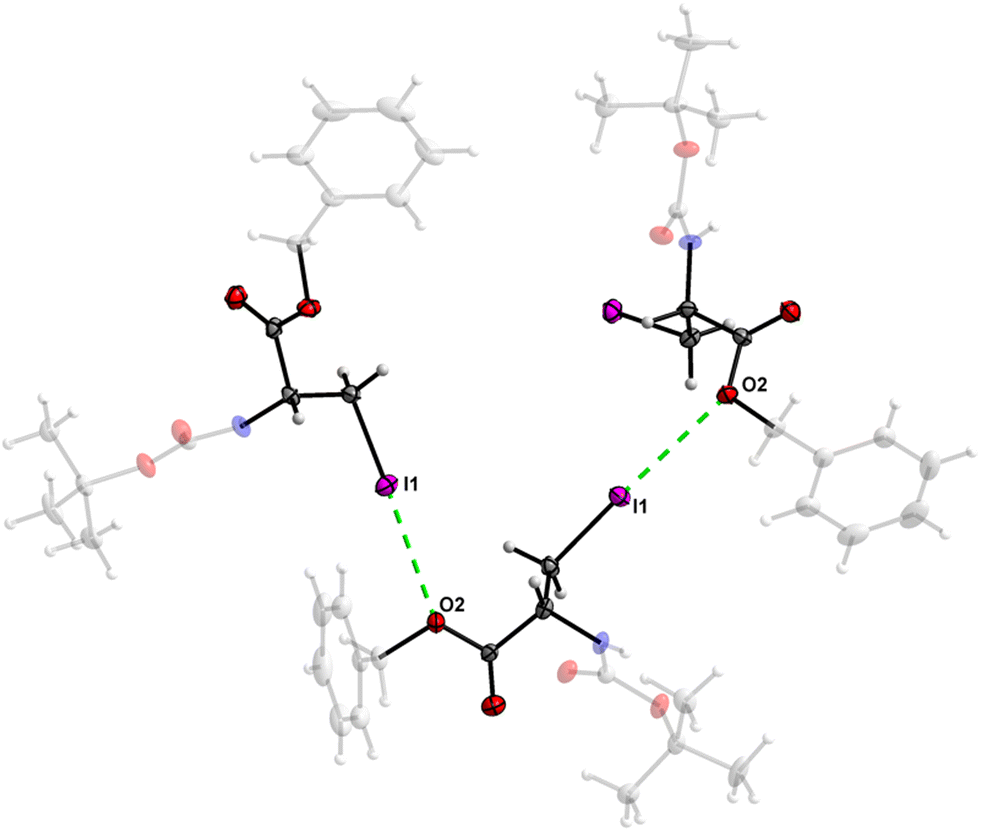
The crystal structure of 7 features an interesting type of iodine oxygen interaction, in which the I1 atom points with its electron-deficient σ-hole21 (sigma hole) to the electron-rich domain of the O4 atom of the ester group with a contact distance of 3.193(3) Å with a C3–I1–O2 angle of 177.9(2)°, indicating an almost linear arrangement of these three atoms. This I⋯O interaction (Fig. 28, symmetry operator −y, x − y, −2/3 + z) forms infinite spiral chains along [001].
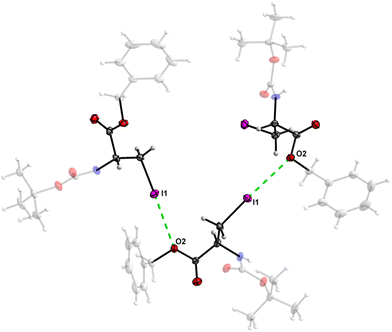 |
| | Fig. 28 The I⋯O interaction forms an infinite spiral chain along the [001] axis in the crystal structure of 7. | |
The crystal packing of 7 is demonstrated in Fig. 29 and shows a somewhat complex isotropic packing mode with total interaction energies as high as −69.0 kJ mol−1. Weak electrostatic energies corresponding to the C–H⋯O and N–H⋯O interactions can be seen clearly when one views the packing along [001]. Each electrostatic cylinder is linked to two neighbouring cylinders in a triangular prism fashion by cylinders corresponding to the C–I⋯O. Thick dispersion cylinders can also be seen along [001] running parallel to the ac plane also in a triangular prism fashion but with an extra low energy cylinder linking each neighbouring triangle to one another. The magnitude of the total energies mirrors that of the dispersion ones indicating the dominance of the dispersion terms due to the cancellation of the electrostatic terms by the repulsion ones. A more isotropic topology is evident, as seen in Fig. 15.
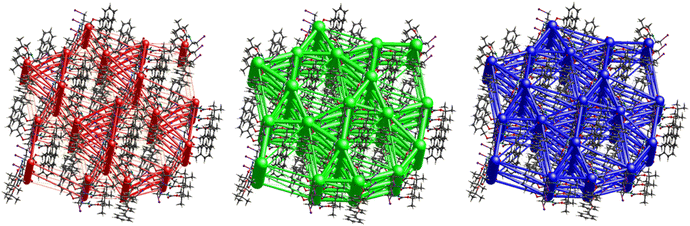 |
| | Fig. 29 Energy frameworks for electrostatic (red), dispersion (green) and total interaction energies (blue) for a cluster of nearest-neighbour molecules in Boc-Ala(3-I)-OBn (7). The energy tube size is 200, and the energy threshold is 0 kJ mol−1. | |
Fmoc-Ala(3-I)-O
t
Bu (8)
The molecular packing of 8 is not rich with H-bonds, with the major interaction being the CO⋯H–N hydrogen bond with the H-bond donor and acceptor belonging to the carbamate groups of the interacting molecules (Fig. 30).
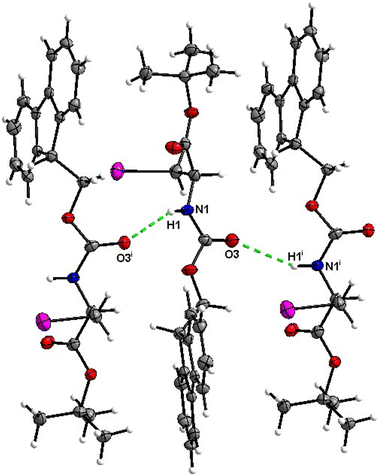 |
| | Fig. 30 View of the H-bond interactions (i) parallel to the [100] axis in the crystal structure of 8. | |
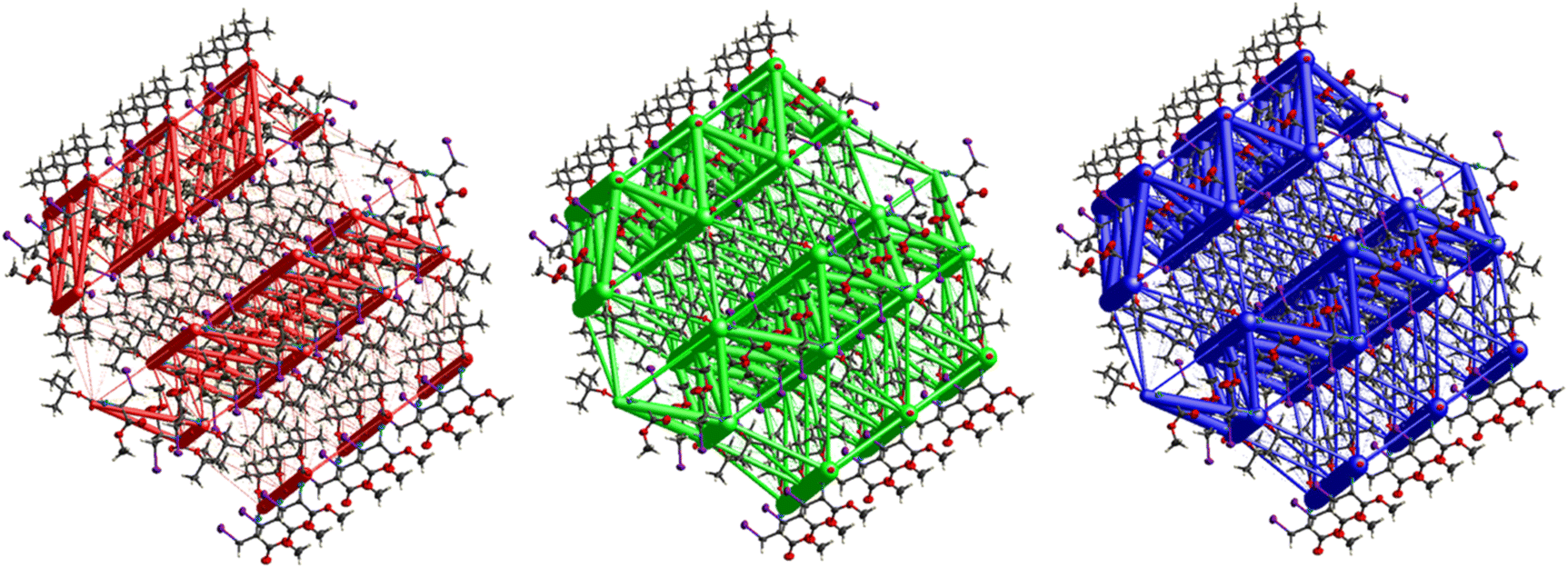
The hexagonal isotropic crystal topology of compound 8 (Fig. 31) is well pronounced and very similar to the topologies of compounds 1 and 6. The crystal stabilization of 8 is also dominated by strong dispersion terms running along [100] parallel to the ac plane. The weak electrostatic energies running along [100] seem to be cancelled by the repulsion energies. Compound 8 gave the highest lattice energy of −84.2 kJ mol−1, resulting in a well-pronounced energy framework as seen in Fig. 31.
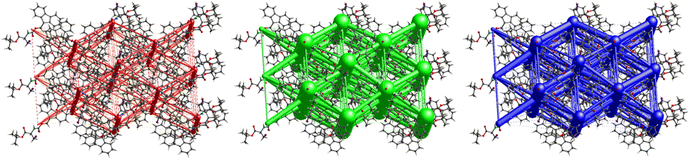 |
| | Fig. 31 Energy frameworks for electrostatic (red), dispersion (green) and total interaction energies (blue) for a cluster of nearest-neighbour molecules in Fmoc-Ala(3-I)-OtBu (8). The energy tube size is 200, and the energy threshold is 0 kJ mol−1. | |
The Hirshfeld surfaces and fingerprint plots,52,53 generated for compounds 1–8 (see the ESI,† Fig. S2, S4, S6, S8, S10, S12, S14 and S16) using Crystal Explorer, may be used to complement the classification of intermolecular interactions based on the analysis of individual short contacts. The dominant CO⋯H–N hydrogen bonds manifest themselves as sharp protrusions in the fingerprint plots of compounds 1 and 3–8, and are absent for the fingerprint plot of 2 lacking this type of interaction. Intermolecular I⋯I contacts are well observable in the fingerprint plot of 6, whereas the I⋯O contact for 7 is not clearly seen, being “hidden” under the signatures of other contacts. Interestingly, fingerprint plots for compounds 1, 2, 4–6 and 8 demonstrate features that can be classified as weak hydrogen bonds with halogen atoms, but closer analysis of the crystal structures indicated that all these contacts were either slightly above the sum of vdW radii, or/and their geometry did not allow them to be classified as weak H-bonds. Therefore, it can be concluded that, though Hirshfeld fingerprint plots may serve as a useful visualization tool of short intermolecular contacts, they lack the predictive power regarding the energy contributions of such interactions.
Conclusions
Our study compared four chlorinated and four iodinated protected alanine derivatives. The prominent N–H⋯OC H-bonding interaction of the carbamate was present in all but one structure and, likely, played the key role in the organization of molecular packing. This type of hydrogen bonding is known to be dominant in the folding of α-helices and β-sheets found in protein structures.39 Interestingly, these H-bonding interactions are conserved throughout the crystal structures reported in this study and suggest that even in protected non-canonical amino acids, they are at the core of the properties that determine the nature of intermolecular contacts of an amino acid in the solid state. Additional stabilization can be brought by the H-bond type short contacts to the acidic C–H of the stereogenic centre and, in a couple of instances, to the hydrogens of the –CH2X group. Among the iodinated alanine derivatives, compound 6 demonstrated I⋯I halogen bonds of type II,32 whereas I⋯O attractive interactions were observed in the crystal structure of compound 7. None of the chlorinated compounds demonstrated any type of halogen bonding interaction, which can be explained by the low polarizability of chlorine. Therefore, the role of halogen bonding in the crystal packing of the studied halogenated amino alanine derivatives was found to be almost negligible. The plausible explanation for this observation is that halogen interactions are common in the compounds with halogens attached to more electronegative atoms or moieties, such as a phenyl ring or an sp-hybridized carbon, whereas in the reported halogenated amino acid derivatives, there are no electron withdrawing groups in the vicinity of halogen atoms. A plethora of available literature reports support this assumption: for example, multiple articles published in several recent special issues on halogen bonds.54–56 Still, London dispersion forces were found to be the main contributing factor to the stabilization of crystal packing. It is not surprising due to the presence of large alkyl or aromatic moieties with extended molecular surfaces in all eight studied amino acid derivatives. Although dispersion interactions are impossible to determine from the list of short contacts, energy lattice diagrams obtained using the Crystal Explorer offer a convenient method to decipher the energy contributions of the different types of interactions to the total packing energy of a crystal. To sum up, in the series of these protected halogenated amino acid derivatives, H-bonding interactions are the interactions that, supposedly, define the relative orientation of the molecules in the crystal lattice, whereas the London dispersion forces are the interactions that contribute most to the crystal lattice energy.
Experimental part
Synthesis
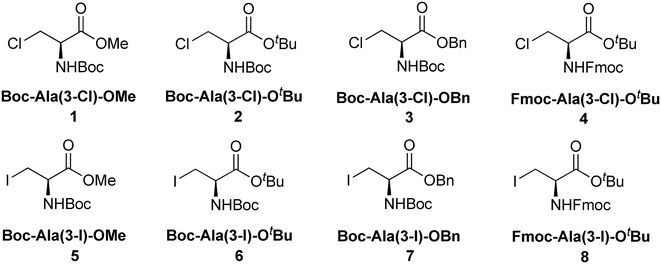
Halogenated amino acid derivatives were prepared starting from L-serine (Scheme 1). Chlorinated alanine derivatives Boc-Ala(3-Cl)-OMe (1),57Boc-Ala(3-Cl)-OtBu (2), Boc-Ala(3-Cl)-OBn (3), and Fmoc-Ala(3-Cl)-OtBu (4)58 were prepared from the corresponding protected L-serine derivatives by chlorination using Appel reaction conditions. Iodinated alanine derivatives Boc-Ala(3-I)-OMe (5),59Boc-Ala(3-I)-OtBu (6),60 and Boc-Ala(3-I)-OBn (7)61 were prepared by iodination of the corresponding protected L-serine derivatives with I2/Ph3P/imidazole, whereas Fmoc-Ala(3-I)-OtBu (8)40 was prepared from Fmoc-Ala(3-Cl)-OtBu (4) by nucleophilic substitution with iodide. All halogenated alanine derivatives were colorless crystalline solids readily affording crystals suitable for X-ray diffractometry.
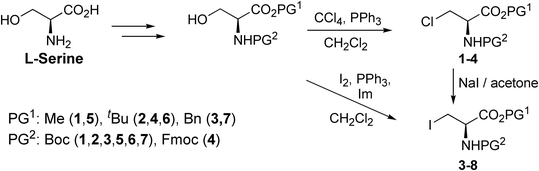 |
| | Scheme 1 The general synthetic pathway of producing halogenated alanine derivatives. | |
3-Chloro-N-[tert-butoxycarbonyl]-L-alanine tert-butyl ester (2).
Boc-L-Ser-OtBu (0.110 g, 0.42 mmol, 1 eq.) and triphenylphosphine (0.221 g, 0.876 mmol, 2 eq.) were dissolved in DCM (5 mL) and cooled to 0 °C while stirring. Carbon tetrachloride (0.165 mL, 1.682 mmol, 4 eq.) was then added to the solution dropwise, after which the solution was allowed to warm to room temperature, where stirring commenced until completion of the reaction tracked by TLC. Methanol was then added to quench the reaction mixture and stirred until TLC observed no triphenylphosphine. The solvent was evaporated under reduced pressure, and the resulting crude was purified by column chromatography with DCM as an eluent, yielding the product compound as colourless crystals (0.089 g; 0.318 mmol; 76%). Large transparent X-ray quality crystals were grown by slow evaporation from hexane/DCM at 5 °C. Rf 0.5 (DCM); Mp 70.6 °C; 1H NMR (400 MHz, CDCl3): δ 5.41 (1H, bd, J = 7.8 Hz, NH), 4.55–4.58 (1H, m, αCH), 3.94 (1H, dd, J = 11.1, 3.1 Hz, βCH2), 3.84 (1H, dd, J = 11.1, 3.3 Hz, βCH2), 1.49 (9H, s, OtBu), 1.46 (9H, s, OtBu); 13C NMR (101 MHz, CDCl3): δ 168.0, 155.1, 83.1, 80.2, 54.7, 46.0, 28.3, 27.9; HRMS (ESI-TOF) m/z calcd for C12H23NO4Cl [M + H]+: 280.1316, found: 280.1314.
3-Chloro-N-[tert-butoxycarbonyl]-L-alanine phenylmethyl ester (3).
Boc-L-Ser-OBn (0.141 g; 0.48 mmol; 1 eq.) and triphenylphosphine (0.250 g; 0.95 mmol; 2 eq.) were dissolved in DCM (5 mL) and cooled to 0 °C while stirring. Carbon tetrachloride (0.184 mL; 1.90 mmol; 4 eq.) was then added to the solution dropwise, after which the solution was allowed to warm to room temperature, where stirring commenced until completion of the reaction tracked by TLC. Methanol was then added to quench the reaction and stirred until TLC observed no triphenylphosphine. The solvent was evaporated under reduced pressure, and the resulting crude was purified by column chromatography with DCM as an eluent, yielding the product compound as thin transparent needles (0.082 g; 0.261 mmol; 55%). X-ray quality crystals were grown by slow evaporation from pentane at 5 °C. Rf 0.48 (DCM); Mp 65.2 °C; 1H NMR (400 MHz, CDCl3): δ 7.33–7.40 (5H, m, Ar–H), 5.44 (1H, bd, J = 8.0 Hz, NH), 5.24 (1H, d, J = 12.2 Hz, CH2Ph), 5.21 (1H, d, J = 12.2 Hz, CH2Ph), 4.73–4.76 (1H, m, αCH), 4.00 (1H, dd, J = 11.3, 3.2 Hz, βCH2), 3.85 (1H, dd, J = 11.3, 3.5 Hz, βCH2), 1.45 (9H, s, Boc); 13C NMR (101 MHz, CDCl3): δ 169.0, 155.0, 134.9, 128.7, 128.6, 128.4, 80.5, 67.8, 54.5, 45.6, 28.3; HR-MS (ESI-TOF) m/z calcd for C15H20ClNO4Na [M + Na]+: 336.0979, found: 336.0972.
X-ray crystallography
X-ray diffraction analyses were performed using either a Bruker Venture D8 4K Kappa Photon III C28 equipped with a graphite monochromator using either a Mo-Kα and Cu-Kα X-ray generator operating at a wavelength of λ = 0.71073 Å and λ = 1.54178 Å, respectively, or a Bruker D8 Quest diffractometer equipped with a graphite monochromator utilizing a Mo-Kα X-ray generator operating at a wavelength of λ = 0.71073 Å.
Refinement
Bruker SAINT-Plus and XPREP software packages were used for frame integration and data reduction, respectively. SADABS was used for absorption correction through the multi-scan method while SHELXS, SHELXL-2018/3,62 WinGX63 and Olex264 software programs were used to solve and refine the crystal structures. DIAMOND 3.0 software was used for generating molecular images. All hydrogen atom labels were omitted for clarity unless indicated to illustrate interactions. All thermal ellipsoids were drawn with 50% probability, and all non-hydrogen atoms were refined anisotropically. Methyl, methylene, and aromatic hydrogens were placed in geometrically idealized positions riding them on the parent atoms (C–H = 0.950–0.980 Å, Uiso(H) = 1.5Ueq(C) and 1.2Ueq(C)). Hydrogen atoms bound to non-carbon atoms were placed and refined according to the Fourier electron density difference map. Crystal data collection and structure refinement parameters of 1–7 are summarized in Table 4.
Table 4 Crystal data collection and structure refinement parameters of acid derivatives 1–7
| Crystal data |
1
|
2
|
3
|
4
|
5
|
6
|
7
|
| Computer programs: APEX3 (Bruker, 2016), SAINT (Bruker, 2009, 2016), SHELXT, SHELXL, SADABS (Bruker, 2016) and OLEX2. |
| Chemical formula |
C9H16ClNO4 |
C12H22ClNO4 |
C15H20ClNO4 |
C22H24ClNO4 |
C9H16INO4 |
C12H22INO4 |
C15H20INO4 |
| Crystallizing solvent |
CH2Cl2/hexane |
CH2Cl2/hexane |
pentane |
CHCl3/PhMe |
CHCl3/hexane |
CHCl3/hexane |
CHCl3/hexane |
|
M
r
|
237.68 |
279.75 |
313.77 |
401.87 |
329.13 |
371.20 |
405.22 |
| Crystal system, space group |
Monoclinic, P21 |
Monoclinic, P21 |
Orthorhombic, P212121 |
Monoclinic, P21 |
Orthorhombic, P212121 |
Orthorhombic, P212121 |
Trigonal, P31 |
| Temperature (K) |
100 |
100 |
100 |
100 |
100 |
100 |
100 |
|
a, b, c (Å) |
9.2240(6), 5.0773(3), 13.2741(9) |
5.9666(5), 9.8723(9), 12.6022(11) |
5.1069(15), 11.791(3), 26.327(8) |
15.207(2), 5.1052(7), 26.456(4) |
5.1261(9), 8.7280(14), 29.053(5) |
5.4467(8), 11.4173(18), 25.107(4) |
16.6588(11), 16.6588(11), 5.1740(5) |
|
α, β, γ (°) |
90, 107.467(2), 90 |
90, 93.118(3), 90 |
90, 90, 90 |
90, 99.547(5), 90 |
90, 90, 90 |
90, 90, 90 |
90, 90, 120 |
| Volume (A3) |
593.00(7) |
741.22(11) |
1585.3(8) |
2025.4(5) |
1299.9(4) |
1561.3(4) |
1243.5(2) |
|
Z
|
2 |
2 |
4 |
4 |
4 |
4 |
3 |
| Radiation type |
MoKα |
MoKα |
CuKα |
CuKα |
CuKα |
MoKα |
MoKα |
|
μ (mm−1) |
0.317 |
0.264 |
2.268 |
1.900 |
19.359 |
2.058 |
1.946 |
| Crystal size (mm) |
0.485 × 0.173 × 0.107 |
0.408 × 0.351 × 0.227 |
0.217 × 0.021 × 0.02 |
0.396 × 0.077 × 0.02 |
0.083 × 0.077 × 0.038 |
0.169 × 0.094 × 0.032 |
0.373 × 0.043 × 0.041 |
| Data collection |
|
|
|
|
|
|
|
| Diffractometer |
Bruker D8 Quest |
Bruker Venture X8 |
Bruker Venture X8 |
Bruker Venture X8 |
Bruker Venture X8 |
Bruker D8 Quest |
Bruker Venture X8 |
| Absorption correction |
Multi-scan |
Multi-scan |
Multi-scan |
Multi-scan |
Multi-scan |
Multi-scan |
Multi-scan |
|
T
min, Tmax |
0.7042, 0.7457 |
0.6535, 0.7457 |
0.5166, 0.7528 |
0.5923, 0.7505 |
0.3989, 0.7516 |
0.6306, 0.7457 |
0.6057, 0.7457 |
| Measured and independent reflections |
12442, 2911 |
12661, 3606 |
4985, 2156 |
19172, 4721 |
19737, 1873 |
26043, 3883 |
16446, 4125 |
|
R
int
|
0.0189 |
0.0294 |
0.0667 |
0.0677 |
0.1024 |
0.0408 |
0.0647 |
| Refinement |
|
|
|
|
|
|
|
|
R[F2 > 2σ(F2)], wR(F2), S |
0.0212, 0.0530, 1.074 |
0.0231, 0.0617, 1.046 |
0.0802, 0.2465, 1.061 |
0.0380, 0.1039, 1.036 |
0.0413, 0.1071, 1.038 |
0.0194, 0.0389, 1.131 |
0.0273, 0.0656, 1.036 |
| No. of parameters |
144 |
173 |
247 |
541 |
145 |
169 |
194 |
| No. of restraints |
1 |
1 |
97 |
3 |
0 |
0 |
1 |
| Δpmax, Δpmin (e Å−3) |
0.22, −0.17 |
0.21, −0.14 |
0.40, −0.53 |
0.21, −0.29 |
1.05, −0.48 |
0.65, −0.45 |
0.34, −0.74 |
Calculations of energy frameworks
Calculations of energy frameworks were performed using Crystal Explorer 17 at the B3LYP/6-31G(d,p) and B3LYP/DGDZVP level of theory for the chlorine- and iodine-containing derivatives, respectively, calculated at the crystal geometry. The disorders were removed and refined for each structure, and then Hirshfeld surfaces were calculated and defined by electron density at high resolution. Molecules were generated by the expansion of the unit cell for energy framework calculations. Lastly, the energy threshold was kept at 0.00 kJ mol−1 and tube size at 200 in all cases.
Author contributions
FDB: chemical synthesis and growth of crystals for most of the compounds, FJFJ: data collection and refinement, calculations using Crystal Explorer, generation of images; AN: chemical synthesis and growth of crystals for one of the compounds; DVK: conceptualization, data collection and refinement, analysis of the data, writing of the paper; VAA: conceptualization, growth of crystals, analysis of the data, writing of the paper.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This project was supported by the South African National Research Foundation, SA-NRF (Grant No. 120464). Funding for crystal data collection from the SA-NRF (Grant No. 116180 and 137759) is gratefully acknowledged. FJDB and FJFJ are grateful for their PhD bursaries from the SA-NRF.
References
- C. H. Görbitz, Crystallogr. Rev., 2015, 21, 160–212, DOI:10.1080/0889311X.2014.964229.
-
E. Boldyreva, in Models, Mysteries and Magic of Molecules, Springer, Dordrecht, 2008, pp. 167–192 Search PubMed.
- B. Schade and J. H. Fuhrhop, New J. Chem., 1998, 22, 97–104, 10.1039/A708317C.
- S. Bera, S. Mondal, S. Rencus-Lazar and E. Gazit, Acc. Chem. Res., 2018, 51, 2187–2197, DOI:10.1021/acs.accounts.8b00131.
- F. S. Ihlefeldt, F. B. Pettersen, A. Von Bonin, M. Zawadzka and C. H. Görbitz, Angew. Chem., Int. Ed., 2014, 53, 13600–13604, DOI:10.1002/anie.201406886.
- S. A. Moggach, S. Parsons and P. A. Wood, Crystallogr. Rev., 2008, 14, 143–184, DOI:10.1080/08893110802037945.
- A. V. Churakov, P. V. Prikhodchenko, J. A. K. Howard and O. Lev, Chem. Commun., 2009, 4224–4226, 10.1039/b906801e.
- P. V. Prikhodchenko, A. G. Medvedev, T. A. Tripol'skaya, A. V. Churakov, Y. Wolanov, J. A. K. Howard and O. Lev, CrystEngComm, 2011, 13, 2399–2407, 10.1039/c0ce00481b.
-
E. Farkas and I. Sóvágó, in Amino Acids, Peptides and Proteins, Royal Society of Chemistry, 2016, vol. 41, pp. 100–151 Search PubMed.
- W. H. Zhang, G. Otting and C. J. Jackson, Curr. Opin. Struct. Biol., 2013, 23, 581–587, DOI:10.1016/j.sbi.2013.06.009.
- P. Neumann-Staubitz and H. Neumann, Curr. Opin. Struct. Biol., 2016, 38, 119–128, DOI:10.1016/j.sbi.2016.06.006.
- W. D. G. Brittain and S. L. Cobb, Org. Biomol. Chem., 2018, 16, 10–20, 10.1039/C7OB02682J.
- I. Rilatt, L. Caggiano and R. F. W. Jackson, Synlett, 2005, 2701–2719, DOI:10.1055/s-2005-918950.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76, DOI:10.1002/1521-3773(20020104)41:1%3C48::AID-ANIE48%3E3.0.CO;2-U.
- G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565–573, DOI:10.1021/ar010054t.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127, DOI:10.1002/anie.200800128.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695, DOI:10.1021/ar400103r.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195, DOI:10.1021/cr500674c.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601, DOI:10.1021/acs.chemrev.5b00484.
- M. R. Scholfield, C. M. Vander Zanden, M. Carter and P. S. Ho, Protein Sci., 2013, 22, 139–152, DOI:10.1002/pro.2201.
- M. Erdélyi, Biochemistry, 2017, 56, 2759–2761, DOI:10.1021/acs.biochem.7b00371.
- M. R. Scholfield, M. C. Ford, A. C. C. Carlsson, H. Butta, R. A. Mehl and P. S. Ho, Biochemistry, 2017, 56, 2794–2802, DOI:10.1021/acs.biochem.7b00022.
- A. Sharma, S. Goswami, R. Rajagopalan and A. D. Konar, Supramol. Chem., 2015, 27, 669–678, DOI:10.1080/10610278.2015.1075021.
- A. S. Gangele, S. Goswami, A. K. Bar, P. Tiwari, S. Konar and A. D. Konar, Cryst. Growth Des., 2016, 16, 2130–2139, DOI:10.1021/acs.cgd.5b01803.
- E. Danelius, H. Andersson, P. Jarvoll, K. Lood, J. Gräfenstein and M. Erdélyi, Biochemistry, 2017, 56, 3265–3272, DOI:10.1021/acs.biochem.7b00429.
- A. Pizzi, N. Demitri, G. Terraneo and P. Metrangolo, CrystEngComm, 2018, 20, 5321–5326, 10.1039/C8CE01205A.
- P. Teng, G. M. Gray, M. Zheng, S. Singh, X. Li, L. Wojtas, A. van der Vaart and J. Cai, Angew. Chem., Int. Ed., 2019, 58, 7778–7782, DOI:10.1002/anie.201903259.
- A. Pizzi, L. Catalano, N. Demitri, V. Dichiarante, G. Terraneo and P. Metrangolo, Pept. Sci., 2020, 112, e24127, DOI:10.1002/pep2.24127.
- M. Mardirossian, M. Rubini, M. F. A. Adamo, M. Scocchi, M. Saviano, A. Tossi, R. Gennaro and A. Caporale, Molecules, 2021, 26, 7401, DOI:10.3390/molecules26237401.
- G. R. Desiraju, Chem. Commun., 2005, 2995–3001, 10.1039/b504372g.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524, DOI:10.1021/ar5001555.
- V. R. Pedireddi, D. S. Reddy, B. S. Goud, D. C. Craig, A. D. Rae and G. R. Desiraju, J. Chem. Soc., Perkin Trans. 2, 1994, 2353–2360, 10.1039/p29940002353.
- P. Metrangolo and G. Resnati, IUCrJ, 2014, 1, 5–7, DOI:10.1107/S205225251303491X.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19, 10.1039/p298700000s1.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13, DOI:10.1107/S0021889802022112.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. Van De Streek, J. Appl. Crystallogr., 2006, 39, 453–457, DOI:10.1107/S002188980600731X.
- M. J. Turner, S. P. Thomas, M. W. Shi, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2015, 51, 3735–3738, 10.1039/C4CC09074H.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011, DOI:10.1107/S1600576721002910.
-
D. Voet and J. G. Voet, Biochemistry, Wiley, New York, 4th edn, 2010 Search PubMed.
- W. Clegg and L. Horsburgh, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, o1925–o1926, DOI:10.1107/S1600536803025467.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587, DOI:10.1107/S205225251700848X.
- Y. V. Torubaev and I. V. Skabitsky, CrystEngComm, 2019, 21, 7057–7068, 10.1039/C9CE01174A.
- K. M. Bairagi, K. S. Ingle, R. Bhowal, S. A. Mohurle, A. Hasija, O. I. Alwassil, K. N. Venugopala, D. Chopra and S. K. Nayak, ChemPlusChem, 2021, 86, 1167–1176, DOI:10.1002/cplu.202100259.
- Y. V. Torubaev, I. V. Skabitsky, A. A. Anisimov and I. V. Ananyev, CrystEngComm, 2022, 24, 1442–1452, 10.1039/D1CE01487K.
- Y. V. Torubaev, D. Howe, G. Leitus and S. V. Rosokha, CrystEngComm, 2023, 25, 3380–3390, 10.1039/D3CE00316G.
- S. Ghosh and V. R. Pedireddi, Cryst. Growth Des., 2023, 23, 4591–4606, DOI:10.1021/acs.cgd.3c00350.
- Y. V. Torubaev and I. V. Skabitsky, Crystals, 2023, 13, 1555, DOI:10.3390/cryst13111555.
- A. Gavezzotti, J. Phys. Chem. B, 2002, 106, 4145–4154, DOI:10.1021/jp0144202.
- A. Gavezzotti, J. Phys. Chem. B, 2003, 107, 2344–2353, DOI:10.1021/jp022288f.
- A. Gavezzotti, New J. Chem., 2011, 35, 1360–1368, 10.1039/c0nj00982b.
- C. M. L. Vande Velde, M. Zeller and V. A. Azov, Acta Crystallogr., Sect. C: Struct. Chem., 2018, 74, 1692–1702, DOI:10.1107/S2053229618015309.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816, 10.1039/b704980c.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32, 10.1039/B818330A.
- M. Erdelyi and P. Metrangolo, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2017, 73, 135, DOI:10.1107/S205252061700484X.
- G. Resnati and W. T. Pennington, New J. Chem., 2018, 42, 10461–10462, 10.1039/C8NJ90054J.
- M. Hariharan, Cryst. Growth Des., 2022, 22, 2046–2049, DOI:10.1021/acs.cgd.2c00157.
- M. N. Kenworthy, J. P. Kilburn and R. J. K. Taylor, Org. Lett., 2004, 6, 19–22, DOI:10.1021/ol0360039.
- H. J. C. Deboves, C. A. G. N. Montalbetti and R. F. W. Jackson, J. Chem. Soc., Perkin Trans. 1, 2001, 1876–1884, 10.1039/b103832j.
- P. Danner, M. Morkunas and M. E. Maier, Org. Lett., 2013, 15, 2474–2477, DOI:10.1021/ol4009409.
- Y. Murakami, H. Yanuma and T. Usuki, Tetrahedron: Asymmetry, 2012, 23, 1557–1563, DOI:10.1016/j.tetasy.2012.10.009.
- Y. Koseki, H. Yamada and T. Usuki, Tetrahedron: Asymmetry, 2011, 22, 580–586, DOI:10.1016/j.tetasy.2011.03.002.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8, DOI:10.1107/S2053229614024218.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838, DOI:10.1107/S0021889899006020.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341, DOI:10.1107/S0021889808042726.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Frederick J. F.
Jacobs‡
,
Frederick J. F.
Jacobs‡