
Covalent organic framework crystallization using a continuous flow packed-bed reactor†
Dayanni D.
Bhagwandin
 ab,
John H.
Dunlap
ab,
Ly D.
Tran
ab,
Alexander
Reidell
ab,
Drake
Austin
ab,
Amelia A.
Putnam-Neeb
ac,
Morgan
Loveday
ab,
Rahul
Rao
a,
Luke A.
Baldwin
*a and
Nicholas R.
Glavin
*a
ab,
John H.
Dunlap
ab,
Ly D.
Tran
ab,
Alexander
Reidell
ab,
Drake
Austin
ab,
Amelia A.
Putnam-Neeb
ac,
Morgan
Loveday
ab,
Rahul
Rao
a,
Luke A.
Baldwin
*a and
Nicholas R.
Glavin
*a
aMaterials and Manufacturing Directorate, Air Force Research Laboratory, Wright-Patterson AFB, Ohio 45433, USA. E-mail: luke.baldwin.1@afrl.af.mil; nicholas.glavin.1@afrl.af.mil
bUES, Inc., Beavercreek, Ohio 45432, USA
cNational Research Council Research Associate, Air Force Research Laboratory, Wright Patterson AFB, OH 45433, USA
First published on 5th December 2023
Abstract
Flow systems enable in-line synthesis and processing of organic materials in a continuous reaction pathway, which is advantageous for high-throughput and scale-up. In this work, a highly crystalline TAPB-OHPDA covalent organic framework (COF) was directly crystallized under continuous flow conditions in as little as 30 minutes. Brunauer–Emmett–Teller (BET) surface analysis reveals high surface areas greater than 1700 m2 g−1 can be afforded in 2 hours, resulting in a 36× faster processing time compared to a majority of other reported solvothermal methods. Additionally, the crystalline COF material was also washed with solvent in flow to reduce the required post-processing burden typically performed iteratively during purification and activation. The results presented herein provide foundational knowledge for COF syntheses under packed-bed flow conditions and reveal an opportunity to accelerate the formation and processing of highly crystalline COF materials.
Covalent organic frameworks (COFs) are a class of exceptionally versatile organic materials that have demonstrated their utility as responsive photodetectors,1,2 low-k dielectrics,3,4 ultrasensitive optical and electronic sensors,5,6 selective gas adsorption materials,7–9 high storage electrodes for batteries and supercapacitors,10,11 and in other emergent applications.12,13 Many of these applications take advantage of the properties arising from long range molecular order in the formation of nanoporous two-dimensional (2D) or three-dimensional (3D) structures. In the case of 2D COFs, the organic network is fabricated by selecting linker and vertex monomers with a planar structure that react to form thermodynamically reversible covalent bonds in-plane, and relatively weaker π–π interactions out-of-plane. The resultant structure is highly tuneable and can form pores with various shapes, sizes, framework chemistries, and decorated functional groups.14,15 This versatility has become especially valuable in optical applications, where the stacking mode between layers, the molecular chemical makeup, and pore-decorated functional groups have been exploited for bioimaging, photoemission, two-photon absorption, and photoresponsivity.16–18
While the applications and opportunities for crystalline COFs are expanding, the syntheses of high quality powders tend to be time-consuming, with subsequent processing steps often extending over several days as a result of the highly reversible nature of the covalent bonds linking the network together.19 As such, developing new processes that can enable rapid exploration of COF chemistries and crystallization techniques are of utmost importance to the field.
The implementation of flow chemistry rather than a traditional batch process, has been leveraged only a few times to prepare COF powders and thin films from monomer solutions. Rodríguez-San-Miguel et al.20 first demonstrated the utility of flow processing for COFs by synthesizing imine-based MT-COF-1 under continuous microfluidic conditions with higher crystallinity than the batch synthesized COF.21 Peng et al. demonstrated the successful synthesis of COFs in flow by injecting monomer solutions into a flow reactor to produce COF-LZU1, with the synthesized COFs exhibiting surface areas comparable to batch-synthesized analogues.22 Bisbey et al. demonstrated that maintaining the same concentration of oligomers with long induction periods in a flow cell could afford boronate-ester-linked COF films with good crystallinity,23 while Yang et al. revealed that 3D SBFdiyne-COF films could be synthesized using a similar experiment.24 While there are a couple of promising examples of continuous flow synthesis of COFs, thus far they have only focused on COF formation occurring from the interaction of monomers in the mobile phase in flow. As a result it can be challenging to find the appropriate conditions that do not clog the reactor as a result of precipitation from COF formation in the line. COF syntheses would therefore benefit from systems that allow for solid-state materials to be reacted in the stationary phase.
This work herein describes a unique continuous flow approach to rapidly produce high quality TAPB-OHPDA COF in a packed-bed flow setup (shown in Fig. 1a), through the crystallization of an amorphous polymer precursor in the solid phase. Scheme 1 summarizes the steps used in this work (pink) to afford crystalline COFs through an initial synthesis of the amorphous polymer followed by subsequent crystallization of the solid material using flow. Compared to the traditional monomer derived single-step solvothermal synthesis (Scheme 1, blue), this two-step flow chemistry process offers unique advantages. In this case, the TAPB-OHPDA COF crystallized in flow, reveals exceptionally high surface areas with values exceeding 1700 m2 g−1 in as little as 2 hours, representing an exciting opportunity to significantly decrease the synthesis and crystallization time of COF structures while retaining exceptional quality. As imine COFs such as TAPB-OHPDA COF have demonstrated dramatic color changes (orange to dark brown) in the presence of hydrogen-bond-donor solvents and vapors,25 as well as enhanced photocatalytic H2O2 evolution activity under visible light irradiation, the rapid synthesis described herein will be vital to accelerated material development and process refinement of exciting optical materials.
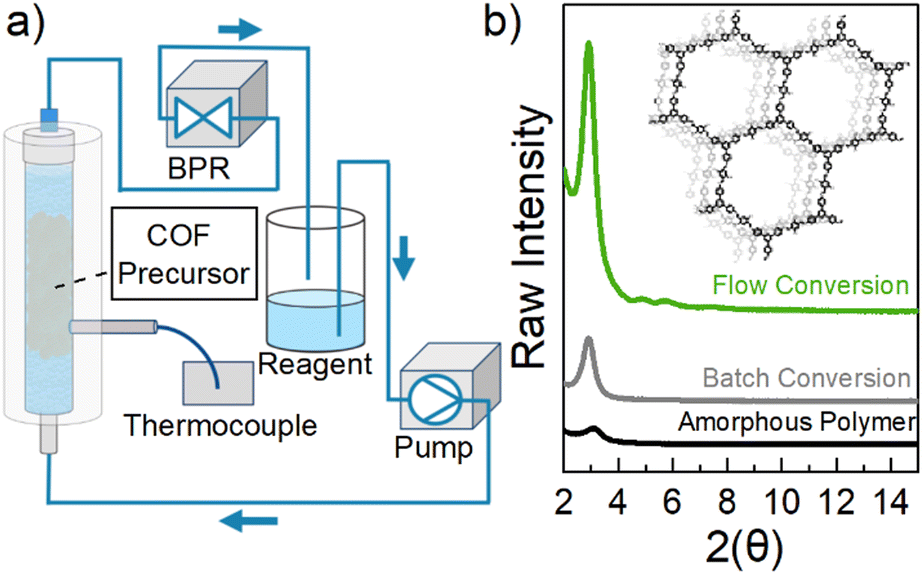 | ||
| Fig. 1 COF crystallization in flow. (a) Schematic of flow setup; (b) raw intensity powder X-ray diffraction (PXRD) of starting material/COF precursor (black), batch conversion at 70 °C for 17 hours (gray), flow conversion at 70 °C for 17 hours (green). | ||
 | ||
| Scheme 1 Synthesis of TAPB-OHPDA COF. Standard solvothermal synthesis (blue): OHPDA and TAPB monomers react to form crystalline COF; rapid amorphous polymer synthesis and subsequent packed-bed flow conversion in solid-state (pink): monomers are reacted to form amorphous polymer in 15 minutes, then solid material is converted to crystalline COF under flow conditions. | ||
The amorphous polymer precursor to TAPB-OHPDA COF was synthesized using a modified approach from previous work (see ESI† for synthetic details).26 Precursors 1,3,5-tris(4-aminophenyl)benzene (TAPB) and 2,5-dihydroxyterephthalaldehyde (OHPDA) were dissolved in a mixture of mesitylene, 1,4-dioxane, and water followed by the addition of acetic acid. After 15 minutes, the amorphous polymer precipitate was collected and washed. This initial rapid synthesis allowed enough time for the dissolved monomers to react and form an orange precipitate, but did not provide sufficient time for the supramolecular assembly to crystallize into a porous framework to form a COF.
Structural and chemical characterization of the amorphous polymer network was carried out using Fourier transform-infrared spectroscopy (FT-IR), Raman spectroscopy, and powder X-ray diffraction (PXRD). Both FT-IR and Raman spectra (further details shown in Fig. S6 and S7†) of the material revealed no remaining starting material and confirm the presence of imine bond formation.25,28 Furthermore, PXRD analysis shows a very weak signal at 3.1 2θ (Fig. 1b, black), indicating that only a very small amount of crystalline material is present. These combined techniques reveal that the synthesized material is in fact a polymer network connected by imine bonds and is mostly amorphous in nature.
To examine the potential of using continuous flow packed-bed processes to convert this amorphous precursor to crystalline COF, a Vapourtec R-Series Flow Chemistry System was used. In brief, the solid amorphous polymer (80 mg) was loaded into a fixed bed reactor and packed with glass beads to hold the material in place (Fig. 1a). Interestingly, we found that solvent recycling was critical for COF conversion and that if the solvent was used in a single pass construct, the amorphous solid slowly degraded into soluble species over time. Therefore, the reagent inlet and outlet lines were placed in the same vessel to recycle the solvent mixture which was comprised of dioxane, mesitylene, and 10 M acetic acid (6.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.7:1, v/v, 8 mL). Solvent flow was conducted using a peristaltic pump at 2.0 mL min−1 and a thermocouple was used to monitor reactor temperature. A back-pressure regulator (BPR) was placed after the fixed-bed reactor to maintain a system pressure of 5 bar. The temperature was increased by 20 °C intervals every 10 minutes until the bed reactor reached 70 °C. This ensured that the setup was heated evenly throughout and no dissolved polymer pieces precipitated within the reagent lines upon cooling. The reaction was heated at this temperature for various times (Fig. 2a, green). At the end of the reaction, the reagent was drained from the fixed bed reactor and the solid material was collected into a vial. Following similar procedures that have yielded high surface area COFs, the material was washed with methanol (20 mL) followed by acetone (20 mL) to remove any portion of the polymer that had dissolved into solution as small molecules.27 Then, methylene chloride (8 mL) was added and left at room temperature for 30 min. The solvent was decanted and this step was repeated two more times. Immediately after washing, n-hexane (8 mL) was added and left at room temperature overnight. Finally, this solvent was decanted and the material was placed under N2 flow and slowly heated (see further details in ESI†).
:1.7:1, v/v, 8 mL). Solvent flow was conducted using a peristaltic pump at 2.0 mL min−1 and a thermocouple was used to monitor reactor temperature. A back-pressure regulator (BPR) was placed after the fixed-bed reactor to maintain a system pressure of 5 bar. The temperature was increased by 20 °C intervals every 10 minutes until the bed reactor reached 70 °C. This ensured that the setup was heated evenly throughout and no dissolved polymer pieces precipitated within the reagent lines upon cooling. The reaction was heated at this temperature for various times (Fig. 2a, green). At the end of the reaction, the reagent was drained from the fixed bed reactor and the solid material was collected into a vial. Following similar procedures that have yielded high surface area COFs, the material was washed with methanol (20 mL) followed by acetone (20 mL) to remove any portion of the polymer that had dissolved into solution as small molecules.27 Then, methylene chloride (8 mL) was added and left at room temperature for 30 min. The solvent was decanted and this step was repeated two more times. Immediately after washing, n-hexane (8 mL) was added and left at room temperature overnight. Finally, this solvent was decanted and the material was placed under N2 flow and slowly heated (see further details in ESI†).
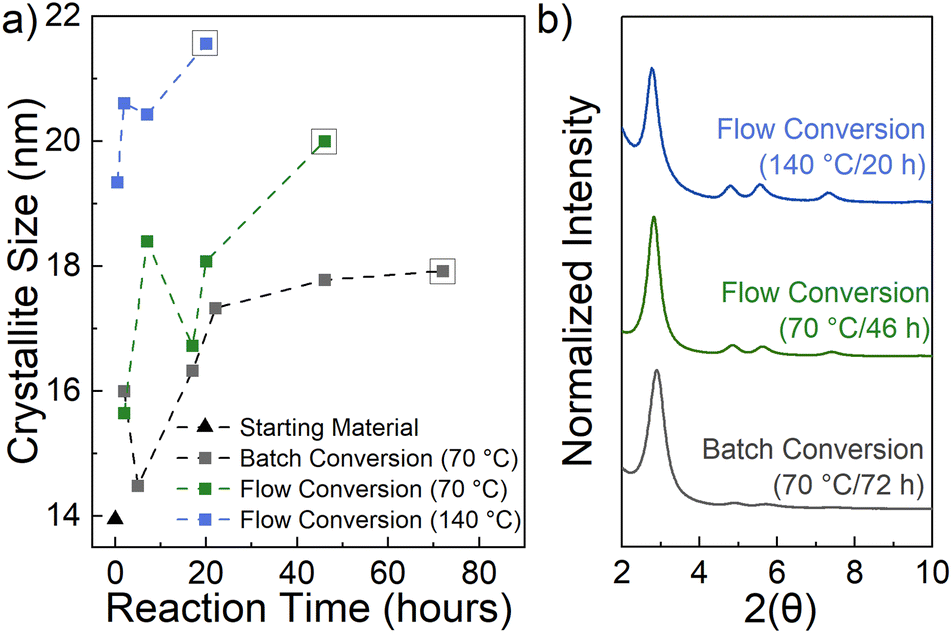 | ||
| Fig. 2 Time and temperature dependency of COF crystallinity in flow conversion. (a) Calculated crystallite size for flow and batch conversion; (b) PXRD of the best values for each conversion condition. | ||
FT-IR and Raman spectroscopy were used to confirm the absence of any remaining starting monomers and the presence of TAPB-OHPDA COF from the flow reaction (further information in Fig. S6 and S7†). PXRD analysis of this material crystallized in a packed-bed reactor reveals a peak at approximately 2.8 2θ that corresponds to the 〈100〉 plane of TAPB-OHPDA COF (Fig. 1b, green) and matches the reported COF PXRD pattern (2.8 2θ) synthesized under solvothermal conditions, indicating that both of the materials have the same crystal structure.28 As a comparison, a batch conversion was also tested by heating amorphous material in a scintillation vial to 70 °C (Fig. 2, gray).
The crystallite size was calculated using the Scherrer equation and is depicted in Fig. 2a. A Lorentzian peak fitting program was used to extrapolate the peak position and FWHM of the 〈100〉 peak from all collected data points (see further details in Table S1, Fig. S2–S5†). Results show that a shortened reaction time of 2 hours at 70 °C in flow (Fig. 2a, green) produces similar sized crystallites to that using the batch conversion method (Fig. 2a, gray). Running the reaction for 7 hours in flow increases the crystallite size beyond what the batch conversion can achieve after 72 hours. Furthermore, crystallite sizes approaching 20 nm can be formed using flow chemistry and a packed-bed reactor at 70 °C after 46 hours.
The flow system allows access to temperatures higher than the solvent boiling point, therefore 110 °C (see further details in Table S1, Fig. S4†) and 140 °C (Fig. 2, blue) reaction temperatures were also tested. Improvement in crystallite size in as little as 30 minutes occurs when converting at 140 °C. Fig. 2b shows the normalized PXRD corresponding to the largest crystallite size of each reaction temperature, with the flow conversion at 140 °C clearly showing improvement of weaker diffraction peaks at 4.8, 5.5, and 7.3 2θ, corresponding to the 〈110〉, 〈200〉, and 〈120〉 planes, respectively.28 This same highly crystalline TAPB-OHPDA COF also exhibits expected strong ultraviolet wavelength absorption and visible-range photoluminescence with a maximum around 420 nm (see details in Fig. S8†).
A time-consuming part of COF fabrication is the washing step, often employed after synthesis to increase the surface area. Typically during the washing step, the COF material is transferred to a new set up and washed with various solvents to remove any starting materials or impurities. In order to facilitate more automated processes without having to remove the solid material, these washing steps were performed in-line directly following the conversion reaction (see details in Fig. S1†). To accomplish this, after COF crystallization was complete, the reagent outlet was switched to a waste stream and the COF was washed with methanol, acetone, and methylene chloride. Then, the inlet and outlet reagent lines were added to the same vial and n-hexane was recycled overnight. After washing, the solvent was removed and the material was collected for the drying and heating step. Analysis of the 〈100〉 peak of this flow washed material indicates a crystallite size of 19.5 nm (Fig. 3a, purple), which is in good agreement to the COF crystallized in flow and washed via the traditional batch procedure. The flow system's ability to quickly and efficiently exchange solvents allows for sequential COF crystallization and washing, potentially leading to rapid COF development and scale up. In an attempt to capture sample to sample variability of this packed-bed reactor approach, the synthesis was performed in duplicate and the variability of the calculated crystallite size was reasonable (Fig. 3a, pink).
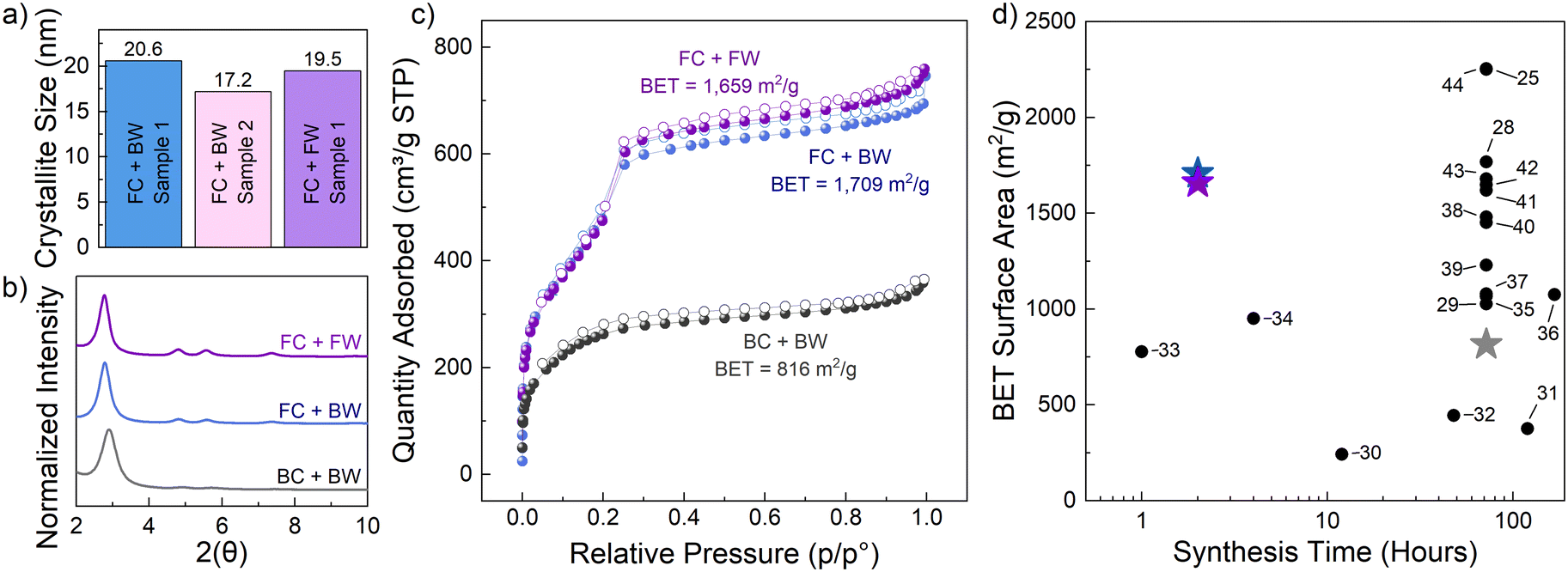 | ||
| Fig. 3 PXRD and surface area comparison of batch washing and in flow washing. a) Crystallite size of flow conversions where FC + BW indicates flow conversion + batch wash (140 °C/2 hours); FC + FW indicates flow conversion + flow wash (140 °C/2 hours); (b) normalized PXRD comparison of different combinations of flow and batch methods where BC + BW indicates batch conversion and batch wash (70 °C/72 hours); purple and blue PXRD correspond to the samples above; (c) respective BET surface area analysis (d) surface area comparison with existing literature (data labels indicate citation number listed in the references). | ||
To further confirm the efficacy of the packed-bed flow method for COF crystallization, BET analysis on the standard batch-washed and flow-washed samples was conducted. For the flow conversion/batch washed sample, results indicate a surface area of 1709 m2 g−1 (Fig. 3c, blue). This is well above the batch conversion/batch washed method at 70 °C for 72 hours which exhibited a surface area of only 816 m2 g−1 (Fig. 3c, gray). Using this new in-line wash-in-flow method a surface area of 1659 m2 g−1 was achieved. This surface area is nearly identical to the flow conversion/batch washing approach but offers the advantage of being able to be implemented in a continuous manufacturing approach, instead of relying on traditional human performed decanting steps. With respect to other synthetic procedures for TAPB-OHPDA COF powders (Fig. 3d),25,28–44 the results from this work clearly show that the implementation of flow chemistry allows for rapid conversion to crystalline material, reducing the standard 72-hour synthesis time by more than 36× for the same quality, or better. To further demonstrate generalizability, this method was extended to the synthesis of TAPB-PDA COF, which is derived from 1,3,5-tris(4-aminophenyl)benzene and terephthalaldehyde (PDA). After synthesis of the amorphous polymer, PXRD results confirm that our packed-bed flow chemistry approach can also produce highly crystalline TAPB-PDA COF powders (further details in Fig. S9†). Overall, COF powders rapidly crystallized in packed-bed flow reactors present an opportunity to significantly speed up growth and reduce processing times required for high quality materials. The flow processing component also allows for this method to be applied on a larger scale.45,46 The amount of COF crystallized in this setup could be increased by simply using a fixed-bed reactor with a larger volume. Furthermore, the flow set-up used in this study could be altered to implement an analogous film synthesis by placing an in-line flow cell with a substrate to perform the assembly of COF particles directly onto a surface.23,24 The mixing of reagents under continuous flow conditions allows for higher surface area-to-volume ratios22 and fine control of the crystallization and resulting nanostructure,20,34,47 which can aid in fine-tuning the required synthetic steps for future unexplored COF systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support of the Air Force Office of Scientific Research grant number 23RXCOR011. L. A. B. also acknowledges financial support provided by the Laboratory-University Collaboration Initiative (LUCI) Fellowship program from the U.S. Department of Defense Basic Research Office.Notes and references
- L. Cao, B. Guo, Y. Yu, X. Zhou, J. R. Gong and S. Lei, ACS Omega, 2019, 4, 18780–18786 CrossRef CAS.
- S. Bag, H. S. Sasmal, S. P. Chaudhary, K. Dey, D. Blätte, R. Guntermann, Y. Zhang, M. Položij, A. Kuc, A. Shelke, R. K. Vijayaraghavan, T. G. Ajithkumar, S. Bhattacharyya, T. Heine, T. Bein and R. Banerjee, J. Am. Chem. Soc., 2023, 145, 1649–1659 CrossRef CAS.
- A. M. Evans, A. Giri, V. K. Sangwan, S. Xun, M. Bartnof, C. G. Torres-Castanedo, H. B. Balch, M. S. Rahn, N. P. Bradshaw, E. Vitaku, D. W. Burke, H. Li, M. J. Bedzyk, F. Wang, J.-L. Brédas, J. A. Malen, A. J. H. McGaughey, M. C. Hersam, W. R. Dichtel and P. E. Hopkins, Nat. Mater., 2021, 20, 1142–1148 CrossRef CAS PubMed.
- P. Shao, J. Li, F. Chen, L. Ma, Q. Li, M. Zhang, J. Zhou, A. Yin, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2018, 57, 16501–16505 CrossRef CAS.
- H. Yuan, N. Li, J. Linghu, J. Dong, Y. Wang, A. Karmakar, J. Yuan, M. Li, P. J. S. Buenconsejo, G. Liu, H. Cai, S. J. Pennycook, N. Singh and D. Zhao, ACS Sens., 2020, 5, 1474–1481 CrossRef CAS PubMed.
- T. Skorjanc, D. Shetty and M. Valant, ACS Sens., 2021, 6, 1461–1481 CrossRef CAS PubMed.
- S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard, J. Am. Chem. Soc., 2008, 130, 11580–11581 CrossRef CAS PubMed.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS.
- S. Yuan, X. Li, J. Zhu, G. Zhang, P. Van Puyvelde and B. Van Der Bruggen, Chem. Soc. Rev., 2019, 48, 2665–2681 RSC.
- Y. Wu, Y. Wang, F. Xu, K. Qu, L. Dai, H. Cao, Y. Xia, L. Lei, K. Huang and Z. Xu, J. Membr. Sci., 2022, 659, 120799 CrossRef CAS.
- H. Wang, Z. Zeng, P. Xu, L. Li, G. Zeng, R. Xiao, Z. Tang, D. Huang, L. Tang, C. Lai, D. Jiang, Y. Liu, H. Yi, L. Qin, S. Ye, X. Ren and W. Tang, Chem. Soc. Rev., 2019, 48, 488–516 RSC.
- Q. Fang, C. Sui, C. Wang, T. Zhai, J. Zhang, J. Liang, H. Guo, E. Sandoz-Rosado and J. Lou, Matter, 2021, 4, 1017–1028 CrossRef CAS.
- Q. Fang, Z. Pang, Q. Ai, Y. Liu, T. Zhai, D. Steinbach, G. Gao, Y. Zhu, T. Li and J. Lou, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2208676120 CrossRef CAS.
- X. Liu, D. Huang, C. Lai, G. Zeng, L. Qin, H. Wang, H. Yi, B. Li, S. Liu, M. Zhang, R. Deng, Y. Fu, L. Li, W. Xue and S. Chen, Chem. Soc. Rev., 2019, 48, 5266–5302 RSC.
- D. D. Medina, T. Sick and T. Bein, Adv. Energy Mater., 2017, 7, 1700387 CrossRef.
- L. Zhang, L. Yi, Z. Sun and H. Deng, Aggregate, 2021, 2, e24 CrossRef CAS.
- L. K. Beagle, Q. Fang, L. D. Tran, L. A. Baldwin, C. Muratore, J. Lou and N. R. Glavin, Mater. Today, 2021, 51, 427–448 CrossRef CAS.
- L. D. Tran, K. F. Presley, J. K. Streit, J. Carpena-Núñez, L. K. Beagle, T. A. Grusenmeyer, M. J. Dalton, R. A. Vaia, L. F. Drummy, N. R. Glavin and L. A. Baldwin, Chem. Mater., 2022, 34, 529–536 CrossRef CAS.
- Y. Zeng, R. Zou, Z. Luo, H. Zhang, X. Yao, X. Ma, R. Zou and Y. Zhao, J. Am. Chem. Soc., 2015, 137, 1020–1023 CrossRef CAS PubMed.
- P. Martinez-Bulit, A. Sorrenti, D. Rodriguez San Miguel, M. Mattera, Y. Belce, Y. Xia, S. Ma, M.-H. Huang, S. Pané and J. Puigmartí-Luis, Chem. Eng. J., 2022, 435, 135117 CrossRef CAS.
- D. Rodríguez-San-Miguel, A. Abrishamkar, J. A. R. Navarro, R. Rodriguez-Trujillo, D. B. Amabilino, R. Mas-Ballesté, F. Zamora and J. Puigmartí-Luis, Chem. Commun., 2016, 52, 9212–9215 RSC.
- Y. Peng, W. K. Wong, Z. Hu, Y. Cheng, D. Yuan, S. A. Khan and D. Zhao, Chem. Mater., 2016, 28, 5095–5101 CrossRef CAS.
- R. P. Bisbey, C. R. DeBlase, B. J. Smith and W. R. Dichtel, J. Am. Chem. Soc., 2016, 138, 11433–11436 CrossRef CAS.
- Y. Yang, C. Schäfer and K. Börjesson, Chem, 2022, 8, 2217–2227 CAS.
- S. Jhulki, A. M. Evans, X.-L. Hao, M. W. Cooper, C. H. Feriante, J. Leisen, H. Li, D. Lam, M. C. Hersam, S. Barlow, J.-L. Brédas, W. R. Dichtel and S. R. Marder, J. Am. Chem. Soc., 2020, 142, 783–791 CrossRef CAS.
- B. J. Smith, A. C. Overholts, N. Hwang and W. R. Dichtel, Chem. Commun., 2016, 52, 3690–3693 RSC.
- L. D. Tran, B. J. Ree, A. Ruditskiy, L. K. Beagle, R. C. Selhorst, D. D. Bhagwandin, P. Miesle, L. F. Drummy, M. F. Durstock, R. Rao, H. Koerner, N. R. Glavin and L. A. Baldwin, Adv. Mater. Interfaces, 2023, 10, 2300042 CrossRef CAS.
- S. Kandambeth, V. Venkatesh, D. B. Shinde, S. Kumari, A. Halder, S. Verma and R. Banerjee, Nat. Commun., 2015, 6, 6786 CrossRef CAS.
- X. Xu, R. Xiong, Z. Zhang, X. Zhang, C. Gu, Z. Xu and S. Qiao, Chem. Eng. J., 2022, 447, 137447 CrossRef CAS.
- L. Yao, Z. Zhou, S. Wang, Q. Zou, H.-X. Wang, L.-X. Ma, S. Wang and X. Zhang, Chem. Sci., 2022, 13, 5902–5912 RSC.
- S. Zhang, N. Fang, X. Ji, Y. Gu, Z. Xu, S. Jin and Y. Zhao, JACS Au, 2022, 2, 1638–1650 CrossRef CAS PubMed.
- M. Gao, D. Wang, L. Deng, S. Liu, K. Zhang, T. Quan, L. Yang, X. Kang, Z. Xia and D. Gao, Chem. Mater., 2021, 33, 8036–8051 CrossRef CAS.
- W. Zhao, P. Yan, B. Li, M. Bahri, L. Liu, X. Zhou, R. Clowes, N. D. Browning, Y. Wu, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2022, 144, 9902–9909 CrossRef CAS PubMed.
- C. H. Feriante, S. Jhulki, A. M. Evans, R. R. Dasari, K. Slicker, W. R. Dichtel and S. R. Marder, Adv. Mater., 2020, 32, 1905776 CrossRef CAS PubMed.
- Y. Yang, J. Kang, Y. Li, J. Liang, J. Liang, L. Jiang, D. Chen, J. He, Y. Chen and J. Wang, New J. Chem., 2022, 46, 21605–21614 RSC.
- F. Tan, Y. Zheng, Z. Zhou, H. Wang, X. Dong, J. Yang, Z. Ou, H. Qi, W. Liu, Z. Zheng and X. Chen, CCS Chem., 2022, 4, 3751–3761 CrossRef CAS.
- J. Guo, W. Li, Y. Xu, Y. Mao, Z. Mei, H. Li, Y. He, X. San, K. Xu and X. Liang, Small Methods, 2023, 7, 2201371 CrossRef CAS.
- D. Zhu, J.-J. Zhang, X. Wu, Q. Yan, F. Liu, Y. Zhu, X. Gao, M. M. Rahman, B. I. Yakobson, P. M. Ajayan and R. Verduzco, Chem. Sci., 2022, 13, 9655–9667 RSC.
- Z. Li, Z. Liu, Z. Li, T. Wang, F. Zhao, X. Ding, W. Feng and B. Han, Adv. Funct. Mater., 2020, 30, 1909267 CrossRef CAS.
- R. Banerjee, et al., Chemically Stable Hollow Spherical COF And Synthesis Thereof, US10266634B2, 2019 April 23.
- Q. Sun, C.-W. Fu, B. Aguila, J. Perman, S. Wang, H.-Y. Huang, F.-S. Xiao and S. Ma, J. Am. Chem. Soc., 2018, 140, 984–992 CrossRef CAS.
- D. Zhu and R. Verduzco, ACS Appl. Mater. Interfaces, 2020, 12, 33121–33127 CrossRef CAS PubMed.
- Z.-J. Mu, X. Ding, Z.-Y. Chen and B.-H. Han, ACS Appl. Mater. Interfaces, 2018, 10, 41350–41358 CrossRef CAS.
- Q. Zhang, S. Dong, P. Shao, Y. Zhu, Z. Mu, D. Sheng, T. Zhang, X. Jiang, R. Shao, Z. Ren, J. Xie, X. Feng and B. Wang, Science, 2022, 378, 181–186 CrossRef CAS PubMed.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- G. Gambacorta, J. S. Sharley and I. R. Baxendale, Beilstein J. Org. Chem., 2021, 17, 1181–1312 CrossRef CAS.
- N. Contreras-Pereda, D. Rodríguez-San-Miguel, C. Franco, S. Sevim, J. P. Vale, E. Solano, W.-K. Fong, A. Del Giudice, L. Galantini, R. Pfattner, S. Pané, T. S. Mayor, D. Ruiz-Molina and J. Puigmartí-Luis, Adv. Mater., 2021, 33, 2101777 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ce01030a |
| This journal is © The Royal Society of Chemistry 2024 |