
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Adsorption of 5-fluorouracil, an anticancer drug, in faujasite-type zeolites: understanding storage and release with density functional theory calculations†‡
Michael
Fischer
 ab
ab
aCrystallography & Geomaterials Research, Faculty of Geosciences, University of Bremen, Klagenfurter Straße 2-4, 28359 Bremen, Germany. E-mail: michael.fischer@uni-bremen.de
bBremen Center for Computational Materials Science and MAPEX Center for Materials and Processes, University of Bremen, 28359 Bremen, Germany
First published on 10th June 2024
Abstract
Zeolites have been proposed as carrier materials for the encapsulation and controlled release of the anticancer drug 5-fluorouracil (5-FU). Besides, they could also find use in the adsorption-based removal of 5-FU from water, for example in the treatment of hospital effluents. In the present work, dispersion-corrected density functional theory (DFT) calculations and DFT-based ab initio molecular dynamics (AIMD) simulations are employed to study the interaction of 5-FU with faujasite-type zeolites having different Si/Al ratios. Comparing distinct local arrangements of Al atoms and charge-balancing protons, it is evaluated to what extent “multi-site” interactions, i.e., interactions of 5-FU with more than one proton, affect the adsorption energy. While the most pronounced increase in interaction strength occurs when moving from an all-silica zeolite to a protonic zeolite having a single proton in one twelve-membered ring, a significant additional stabilisation arises if a second framework proton is present in the same ring. Typically, several hydrogen bonds are formed between 5-FU and protonic zeolite frameworks, with 5-FU simultaneously acting as donor and acceptor. AIMD simulations confirm the stability of these hydrogen bonds at room temperature in the absence of water. Additionally, infrared spectra are predicted for selected low-energy configurations in order to facilitate an experimental identification of different bonding environments. AIMD simulations probing the competitive adsorption of 5-FU and water show that the high affinity of water to the framework protons causes a breaking of hydrogen bonds, framework deprotonation, and a displacement of 5-FU from its initial position. Exposure of dehydrated 5-FU@FAU composites to water might thus be a useful approach to trigger 5-FU release in drug delivery applications.
Introduction
5-Fluorouracil (Fig. 1, C4H3FN2O2, IUPAC name 5-fluoro-1H-pyrimidine-2,4-dione, CAS ID 51-21-8, PubChem CID 3385 (ref. 1)) is an antimetabolite drug that is widely used in the treatment of various cancers, including colorectal cancer and breast cancer. Several modulation strategies have been developed to improve the therapeutic effectiveness and cytotoxicity of 5-FU treatment.2,3 Although 5-FU is considered to be one of the safest chemotherapeutic agents, its toxicity can cause severe side effects (including fever, fatigue, and nausea) and toxic effects (including leukopenia and anaemia). Moreover, 5-FU treatment frequently results in the development of chemoresistant tumour cells.3 Use of carrier materials could ideally allow for a targeted release in the human body, increasing cytotoxicity while at the same reducing the amount of drug required for effective treatment, thereby limiting side effects and resistance development.4 The variety of materials that have been investigated as potential 5-FU delivery vehicles is the subject of dedicated review articles.4–6 Organic materials that have been proposed for this purpose include polymer microspheres7 and functionalised block copolymers that release 5-FU under UV irradiation,8 among many other systems. Examples from the field of inorganic (nano)materials include functionalised silica nanoparticles,9,10 layered hydroxyapatite,11 as well as purified natural clay minerals (e.g., montmorillonite12) and clays modified with organic surfactants13 or other agents.14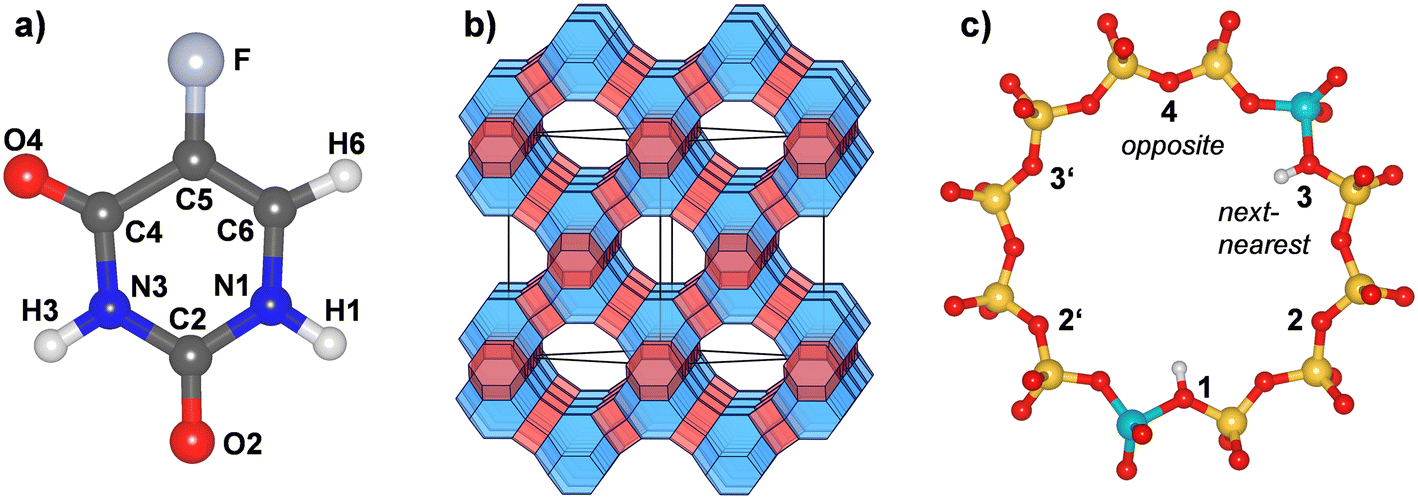 | ||
| Fig. 1 a) Molecular structure and atom labelling scheme of 5-fluorouracil. The labelling follows earlier work.15 b) Schematic representation of the FAU framework. Sodalite cages are shown in blue, double six-membered rings in red. “Supercages” connected by twelve-membered rings (12MRs) are transparent. c) Labelling scheme of proton positions within a single 12MR. The shown example corresponds to a _1_3 arrangement. Yellow = Si, turquoise = Al, red = O, white = H. | ||
Zeolites are crystalline inorganic materials whose structures are built up by a framework of corner-sharing TO4 tetrahedra.16 Most natural zeolites and the majority of synthetic zeolites have an aluminosilicate composition (T = Si, Al), resulting in a negative framework charge that is balanced by extra-framework cations or framework protons. However, neutral-framework all-silica zeolites and zeolite-like materials having other elements on the T sites have also been reported.17,18 The presence of accessible cavities or channels is a key feature of zeolite crystal structures, and the arising intrinsic porosity is pivotal for their large-scale use in catalysis, ion exchange, and separation processes.17 Besides these established applications, zeolites have also received considerable interest as potential drug delivery materials, along with other possible applications in medicine.19 With regard to the specific task of 5-FU delivery, Datt et al. studied the storage and release of 5-FU from zeolite Y samples (FAU framework type16) having Si/Al ratios of 2.5, 15, and 30.20 They observed similar loadings for all three samples, on the order of 0.1 g 5-FU per gram of zeolite, but rather different release characteristics, with the most Al-rich sample (Si/Al = 2.5) releasing only a small fraction of the adsorbed 5-FU under physiological conditions. In contrast, the more Si-rich samples released more than 50% within a few minutes. 27Al-NMR spectroscopy provided evidence for a binding of 5-FU to extra-framework aluminium (EFAL) sites, which were most abundant in the most Al-rich sample.
Subsequently, a number of other authors studied the 5-FU delivery properties of various zeolites.21–27 Neves, Baltazar and co-workers investigated sodium-exchanged zeolite Y (NaY) and compared it to other systems including Linde type L and Linde type A zeolites (LTL and LTA framework, respectively) and ZSM-5 (MFI framework).21,24 They also demonstrated the cytotoxicity of the 5-FU@zeolite composites to different cancer cell lines in cell viability studies, whereas the pristine zeolites were shown to be non-cytotoxic. Spanakis et al. compared the 5-FU release behaviour of zeolite NaX (FAU framework) and zeolite beta (BEA framework) and employed molecular dynamics (MD) simulations using a classical force field (FF) to explain the observed differences.22 Apart from commercially available synthetic zeolites, which can be relatively expensive, zeolites synthesised from inexpensive and widespread starting materials (metakaolin)25 and natural zeolites27 were also investigated as 5-FU carrier materials. Another avenue of research is constituted by the preparation and characterisation of functionalised zeolites and zeolite-containing nanocomposites. For example, it was shown that surface functionalisation of synthetic zeolites can enhance the bioavailability of encapsulated 5-FU.28 Magnetic nanocomposites of zeolites and Fe3O4 nanoparticles are of interest as they could be directed to the location of the tumour through application of an external magnetic field.29,30 Furthermore, the use of silver-exchanged zeolites as 5-FU carriers could help to prevent microbial infections in the tumour microenvironment due to the antimicrobial activity of Ag+ cations.31 Looking beyond zeolites to other crystalline porous materials, metal–organic frameworks (MOFs) and MOF-containing nanocomposites have also been investigated as materials permitting controlled 5-FU delivery.32–34
Apart from their potential use as 5-FU carrier materials, zeolites could also be employed as adsorbents for the selective removal of 5-FU from wastewaters. While little focus has so far been placed on 5-FU in this context, a number of experimental investigations have dealt with the removal of other pharmaceutically active ingredients from wastewaters using hydrophobic high-silica zeolites.35–44 Although 5-FU is not commonly ranked among the pharmaceuticals of most concern with regard to their environmental impact,45,46 it has been identified as a priority cytostatic drug for environmental monitoring studies due to its reported persistence and ecotoxicity.47 In addition to occasional reports of low levels of 5-FU (< 10 ng L−1) in wastewater treatment plant influents,48 5-FU has been detected primarily in hospital effluents,49 in at least one case in high concentrations exceeding 100 μg L−1.50 The toxicity (including genotoxicity) of 5-FU, alone and in mixtures, has been investigated for various aquatic organisms, and estimations of the arising environmental hazard potential generally agree that its potential environmental impact is non-negligible.51–55 This highlights the need to develop efficient removal strategies from contaminated waters, especially hospital effluents. Among other technological options, adsorption-based processes using activated carbons have been proposed for 5-FU removal.56,57 While they are more expensive to produce than activated carbons, hydrophobic zeolites might constitute an interesting alternative due to the reduced pore blockage by co-adsorbed natural organic matter38 and good adsorbent regeneration properties.41 Due to their narrow pore size distribution, they are especially well suited for the highly efficient removal of molecules that fit well into the pores.
In addition to its potential relevance for different applications, highlighted above, 5-FU adsorption in zeolites is also interesting from a more fundamental point of view. Due to the presence of two negatively polarised carbonyl oxygen atoms and two amine groups (Fig. 1), 5-FU can act as hydrogen bond donor and acceptor, and a stabilisation in the zeolite pores through various hydrogen bonds can be envisaged, especially for protonic zeolites having (at least) two framework protons in relatively close proximity. Experimentally, the nature and extent of such “multi-site” interactions is rather difficult to probe, not least due to the important influence of defects on the adsorption properties, as highlighted above when discussing the work of Datt et al.20 In contrast, atomistic modelling methods are ideally suited to investigate such interactions in a systematic fashion, giving access to adsorption energies, low-energy structures, and derived quantities like vibrational spectra. Molecular dynamics simulations allow to study the dynamic behaviour of the host-guest system. It is clear that the use of idealised models of perfect zeolite crystals constitutes a certain limitation, as not all features that may play a role in real materials (defects, intrinsic mesoporosity, …) are incorporated in the modelling. So far, atomistic modelling methods have been employed to study the adsorption of pharmaceuticals in zeolites, both in the context of “screening” studies addressing a range of compounds in different zeolites with computationally efficient force field (FF) methods58,59 and in studies of individual compounds using computationally more expensive density functional theory (DFT) calculations.37,60–63 The specific topic of 5-FU adsorption in zeolites has so far received limited attention from the computational perspective, the previously mentioned study of 5-FU diffusion by Spanakis et al. being an exception.22 However, it is worth mentioning that dispersion-corrected DFT has been used to investigate the adsorption of 5-FU at a hydroxylated cristobalite surface.64 That work highlighted the important role of hydrogen bonds in stabilising the surface-adsorbed 5-FU molecule. Moreover, a DFT-based composite electronic structure method was employed to shed light on the local structure and vibrational properties of 5-FU adsorbed in HKUST-1, a copper-containing MOF.34
In the present work, dispersion-corrected DFT calculations are employed to investigate the adsorption of 5-FU in FAU-type zeolites, considering aluminosilicate models having different amounts of Al atoms and charge-balancing framework protons as well as a reference all-silica FAU model. Focus is placed on FAU for various reasons: first, FAU-type zeolites have been addressed in several previous drug delivery investigations, as highlighted above. Second, they are available across the whole range of Si/Al ratios (FAU type systems with Si/Al < 1.5 are commonly designated “zeolite X”, those with Si/Al 1.5 are dubbed “zeolite Y”), whereas many other framework types are synthetically accessible only within a certain range. Finally, the relatively simple structure, with only one non-equivalent T site, renders the generation of models having different proton arrangements relatively straightforward. The scope of the present study remains restricted to protonic, rather than cationic zeolites because the propensity of metal cations to associate with 6MRs of the FAU structure65 renders interactions of one 5-FU molecule with several cations unlikely, except, potentially, at high cation concentrations (approaching a Si/Al ratio of 1). The aims can be summarised as follows:
• The possible extent of “multi-site” interactions is probed through calculations for FAU models with different proton arrangements. In addition to comparing adsorption energies, an analysis of the low-energy configurations gives insights into the role of hydrogen bonds.
• A calculation of infrared (IR) spectra of adsorbed 5-FU is used to identify features that would allow for the experimental detection of different bonding situations using IR spectroscopy.
• Static calculations are complemented by DFT-based AIMD calculations, which allow to assess the lability/stability of the hydrogen bonds at room temperature. Further AIMD simulations are employed to investigate the competitive adsorption of 5-FU and water.
It is clear that the model systems employed in the present work are somewhat simplified, imposing specific arrangements of Al atoms and protons in the zeolite structure, rather than accounting for the coexistence of various local arrangements in real samples. However, the detailed insights obtained through a systematic comparison of different local environments, which bring about different bonding situations, do not only enhance the fundamental understanding, but they might also be relevant for the development of new zeolite applications.
Computational details
Zeolite models and preliminary force field calculations
The crystal structure of all-silica zeolite Y (SiO2-FAU) was taken from experimental data.66 Aluminosilicate models containing different amounts of framework Al atoms and charge-balancing protons, labelled H-FAU_NH (with N = 1, 2, 3, 16, 32) were generated from this initial structure. On the one hand, highly siliceous models containing 1, 2, or 3 Al atoms and protons located within a single twelve-membered (12MR) of the FAU unit cell were considered, including two possible arrangements for H-FAU_2H (_1_3 = next-nearest and _1_4 = opposite, as shown in Fig. 1c). On the other hand, more Al-rich models having one/two Al atoms and protons in every 12MR were prepared. It was always assumed that the framework protons are bonded to O1 oxygen atoms, as this constitutes the energetically most favourable proton site in the accessible pore space of FAU.63 Details of all models are compiled in Table 1. Each structure model was optimised using GULP,67 relaxing all atomic coordinates and cell parameters, but fixing the cell dimensions to a cubic metric. These optimisations used the SLC (Sanders–Leslie–Catlow) potential parameters.68,69 In the subsequent DFT and AIMD calculations, the cell parameters were fixed at the FF-optimised values listed in Table 1.| Label | Unit cell composition | Si/Al ratio | Proton arrangement | a/Å |
|---|---|---|---|---|
| SiO2-FAU | Si192O384 | Infinity | — | 24.227 |
| H-FAU_1H | HAlSi191O384 | 191 | 1 H in one 12MR | 24.237 |
| H-FAU_2H_1_3 | H2Al2Si190O384 | 95 | 2 H in one 12MR, located at next-nearest O1 atoms | 24.248 |
| H-FAU_2H_1_4 | H2Al2Si190O384 | 95 | 2 H in one 12MR, located at opposite O1 atoms | 24.248 |
| H-FAU_3H | H3Al3Si189O384 | 63 | 3 H in one 12MR, located at next-nearest O1 atoms | 24.258 |
| H-FAU_16H | H16Al16Si176O384 | 11 | 1 H in each 12MR | 24.398 |
| H-FAU_32H_1_3 | H32Al32Si160O384 | 5 | 2 H in each 12MR, located at next-nearest O1 atoms | 24.579 |
| H-FAU_32H_1_4 | H32Al32Si160O384 | 5 | 2 H in each 12MR, located at opposite O1 atoms | 24.566 |
The structure of 5-FU was taken from the PubChem database.1 While a number of tautomers exist, the diketo form visualised in Fig. 1a is the most stable one, which accounts for >99.9% of the tautomer population at 375 K (evaporation temperature).70 The structure of 5-FU was initially optimised using PCFF parameters.71,72 Different adsorption configurations of 5-FU in the pores of the FAU models were generated using FF-based Monte Carlo (MC) simulations, making use of the Sorption module integrated in the Materials Studio suite (DS BIOVIA).73 Details of the MC calculations are provided in the ESI,† section S1.1. For each adsorbent model, between 3 and 10 distinct, pre-optimised 5-FU@FAU configurations were used as starting points for DFT optimisations, which are described in the following subsection. For AIMD simulations including co-adsorbed water molecules, which were performed only for a subset of FAU models, a hydration shell of 16 H2O molecules was generated using a FF-based simulated annealing procedure as implemented in the Adsorption Locator module of Materials Studio. Details of these simulations are presented in the ESI,† section S1.2.
All FF-based simulations used PCFF parameters to describe the guest molecules (5-FU, water),71,72 and partial charges and Lennard-Jones parameters reported by Emami et al. to represent framework Si and O atoms.74 Since that work does not include parameters for framework protons, Al atoms, and O atoms of the AlO4 tetrahedra, partial charges for these atoms were derived from the DFT electrostatic potential of H-FAU_1H. The REPEAT method was used to compute charges,75 which were then scaled and slightly adjusted. The whole procedure is described in more detail in the ESI,† section S1.3. Lennard-Jones parameters of H, Al, and O(AlO4) atoms were estimated using parameters of similar atom types included in the work of Emami et al. All partial charges and Lennard-Jones parameters are compiled in Table S3 of the ESI.†
Density functional theory calculations and DFT-based molecular dynamics
All DFT calculations and DFT-based AIMD simulations were carried out using the Gaussian and plane wave (GPW) method implemented in the Quickstep code, which is part of the CP2K package (version 9.1).76,77 The dispersion-corrected rev-vdW-DF2 functional proposed by Hamada78 was employed due to its good performance demonstrated in an earlier comparative study.79 Due to the size of the FAU unit cell, the sampling of the first Brillouin zone was restricted to the Γ point, only. “Molecularly optimised” Gaussian basis sets developed by VandeVondele and Hutter80 were used in conjunction with Gaussian-type Goedecker–Teter–Hutter pseudopotentials developed by Krack.81 Double-zeta short-range (DZVP-MOLOPT-SR, shortened to DZVP throughout this article) basis sets were used in DFT optimisations, calculations of vibrational spectra, and AIMD simulations, in which the plane wave energy cutoff was set to 600 Ry. Adsorption energies were obtained from single-point calculations on previously optimised structures, using triple-zeta (TZVP-MOLOPT, shortened to TZVP) basis sets and an energy cutoff of 900 Ry. As previous work on acetaminophen adsorption indicated that the use of TZVP-MOLOPT basis sets results in a relatively small basis set superposition error (BSSE) on the order of 4 to 5% of the total adsorption energy,79 no BSSE correction was included. Adsorption energies were calculated as:| ΔEads = EDFT,5FU@FAU − EDFT,FAU − EDFT,5FU |
All atomic positions were relaxed in structure optimisations, which used a BFGS optimiser, imposing a maximal gradient of 5 × 10−6 Ha a0−1 and a maximal displacement between optimisation steps of 2.5 × 10−5a0 (a0 = 0.529177 Å) as convergence criteria. Calculations of the vibrational modes and IR intensities employed the finite displacement method, using a partial Hessian approach in which only displacements of the atoms of 5-FU and (where relevant) of framework protons and framework oxygen atoms participating in hydrogen bonds were considered. Structure visualisations were prepared using VESTA.82 The analysis of vibrational properties made use of MOLDEN, using a broadening with a full width at half maximum of 10 cm−1 for plots of IR spectra.83
All AIMD simulations were carried out for a temperature of 298 K in the NVT ensemble, using a Nosé–Hoover thermostat84,85 with a timestep of 0.5 fs and a time constant of 50 fs. Three types of tasks were tackled with AIMD simulations:
(1) AIMD simulations starting from optimised 5-FU@FAU configurations were performed in order to determine internal energies of adsorption ΔUads and to investigate the stability/lability of hydrogen bonds. For this purpose, simulations were run for the 5-FU@FAU configuration, the guest-free zeolite, and for 5-FU in a box, computing three independent trajectories of 12.5 ps (25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps) for each system (four trajectories were computed for 5-FAU@SiO2-FAU, see Results). After discarding the first 2.5 ps as equilibration phase, internal energies of adsorption as well as estimated upper and lower boundary values were computed from the production part of the trajectories. The procedure, which follows earlier work,62,63 is described in more detail in section S1.4 of the ESI.† For atom combinations of interest, radial distribution functions (RDFs) were calculated for individual trajectories using VMD,86 and then averaged over all 3 (or 4) trajectories.
000 steps) for each system (four trajectories were computed for 5-FAU@SiO2-FAU, see Results). After discarding the first 2.5 ps as equilibration phase, internal energies of adsorption as well as estimated upper and lower boundary values were computed from the production part of the trajectories. The procedure, which follows earlier work,62,63 is described in more detail in section S1.4 of the ESI.† For atom combinations of interest, radial distribution functions (RDFs) were calculated for individual trajectories using VMD,86 and then averaged over all 3 (or 4) trajectories.
(2) To investigate the behaviour of adsorbed 5-FU in the presence of water, AIMD simulations were run for models in which 16 H2O molecules were added in the vicinity of the 5-FU molecule. While the specific amount of H2O molecules included in these calculations is, to a degree, arbitrary, the simulations of 5-FU in liquid water, described below, showed that there are typically about 13 to 14 H2O molecules within a distance of 5 Å from the ring atoms of 5-FU. On this basis, 16 H2O molecules appear as a plausible choice that should correspond to an essentially complete first hydration shell of the adsorbed 5-FU molecule. The simulations of co-adsorbed 5-FU and water considered only H-FAU_1H, H-FAU_2H_1_3, and H-FAU_2H_1_4. Since these simulations were aimed primarily at qualitative insights, only a single trajectory was computed for each system, but for an extended time period of 25 ps.
(3) To analyse the behaviour of 5-FU in liquid water, three independent trajectories were computed for a single 5-FU molecule in a box of 256 H2O molecules, with the size of the box adjusted to match the experimental density of liquid water at ambient conditions (0.997 g cm−3). Starting configurations were generated using the Amorphous Cell module of Materials Studio, using PCFF parameters.
Results and discussion
1) Adsorption energies
Fig. 2 shows adsorption energies from DFT optimisations, reporting Boltzmann-weighted energy values ΔĒads,Boltz obtained with TZVP (blue) and DZVP (red) basis sets. The adsorption energies of individual configurations are compiled in the ESI,† Table S4. The Boltzmann-weighted values always fall within 2 kJ mol−1 of the adsorption energies of the lowest-energy configurations ΔĒads,lowest, which are not plotted separately in Fig. 2. The Boltzmann average is dominated by a single configuration for many FAU models, but there are exceptions where two or three configurations are very close in energy, sometimes showing different ordering depending on the choice of basis sets. While the discussion in the following sections will place most emphasis on the TZVP lowest-energy configurations, other low-energy configurations will also be considered in those cases where they exhibit pronounced differences from the lowest-energy ones. Generally, the TZVP adsorption energies are between 7% and 13% less negative than the corresponding DZVP values. Larger differences on the order of 17% to 35% were found in a previous rev-vdW-DF2 study considering the adsorption of larger organics having molar masses between 150 and 300 g mol−1 (compared to 130.08 g mol−1 for 5-FU).79 Altogether, the present work corroborates the previous conclusion that (at least) triple-zeta basis sets are necessary to compute reasonably accurate adsorption energies for sizeable organic molecules in zeolites, which is why the following paragraph will focus on the TZVP results.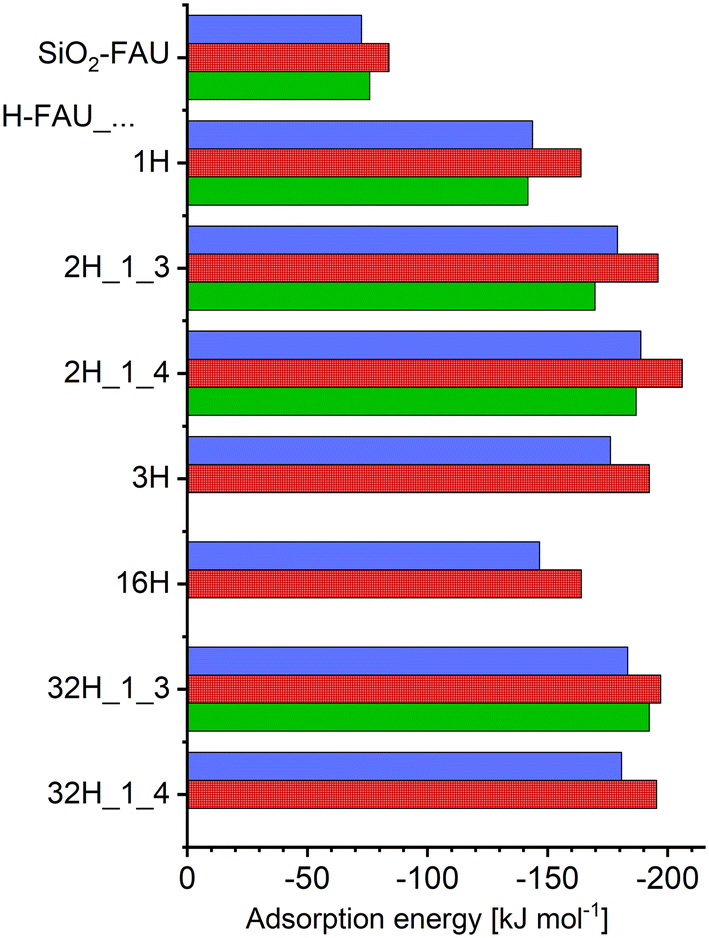 | ||
| Fig. 2 Boltzmann-weighted adsorption energies obtained from DFT calculations using TZVP basis sets (blue bars, top) and DZVP basis sets (red bars, centre) and internal energies of adsorption obtained from AIMD simulations using DZVP basis sets (green bars, bottom). | ||
Looking at the numerical values, an adsorption energy of −73 kJ mol−1 is predicted for purely siliceous FAU. The presence of a single framework proton in H-FAU_1H results in a near-doubling of the interaction strength, with a ΔĒads,Boltz value of −144 kJ mol−1. Inclusion of a second proton in the 12MR results in a further increase by about 25%/31% for the “1_3” and “1_4” proton arrangements of H-FAU_2H. If a third proton is present in the same 12MR, however, the adsorption energy is somewhat less negative than for H-FAU_2H, an evolution that can be understood when looking at the local environment of the adsorbed 5-FU molecule (discussed in the following section). For the more Al-rich models containing 16 and 32 protons, the computed adsorption energies are within ±5% of the values obtained for the corresponding low-Al models having the same number and arrangement of protons in a single 12MR (e.g., H-FAU_2H_1_3: −179 kJ mol−1, H-FAU_32H_1_3: −183 kJ mol−1). This indicates that the adsorption energy is dominantly influenced by the local environment, and that the overall Si/Al ratio and the more distant environment have only a minuscule effect (as long as framework topology [and, consequently, framework density] remain [essentially] constant).
For those FAU models where AIMD simulations were run, internal energies of adsorption are also included in Fig. 2 (green bars). Numerical values of ΔUads, including the estimated upper and lower boundary values, are given in the ESI† (Table S5). Typical error bars associated with the AIMD results are on the order of ±10 kJ mol−1, with the larger error of about ±20 kJ mol−1 for H-FAU_2H_1_4 being an exception. For 5-FU@SiO2-FAU, where two configurations are very close in energy (Config1 and Config2, see Fig. 3), a total of four AIMD simulations were run, two starting from each configuration. The resulting internal energy of adsorption is about 9% less negative than the ΔĒads,Boltz value computed with DZVP basis sets. Relative differences obtained for the protonic FAU models show significant scatter, falling between 2% and 13%. Given the error bars of the AIMD-derived ΔUads values, it is clear that these numbers should not be overinterpreted. Nevertheless, it is worth noting that the reduction in interaction strength caused by the inclusion of thermal motion in the simulations is considerably smaller than observed in previous, methodologically analogous investigations of carbamazepine and triclosan adsorption, where differences were on the order of 20% to 30% for all-silica FAU, and amounted to 16% for triclosan in H-FAU_1H.62,63
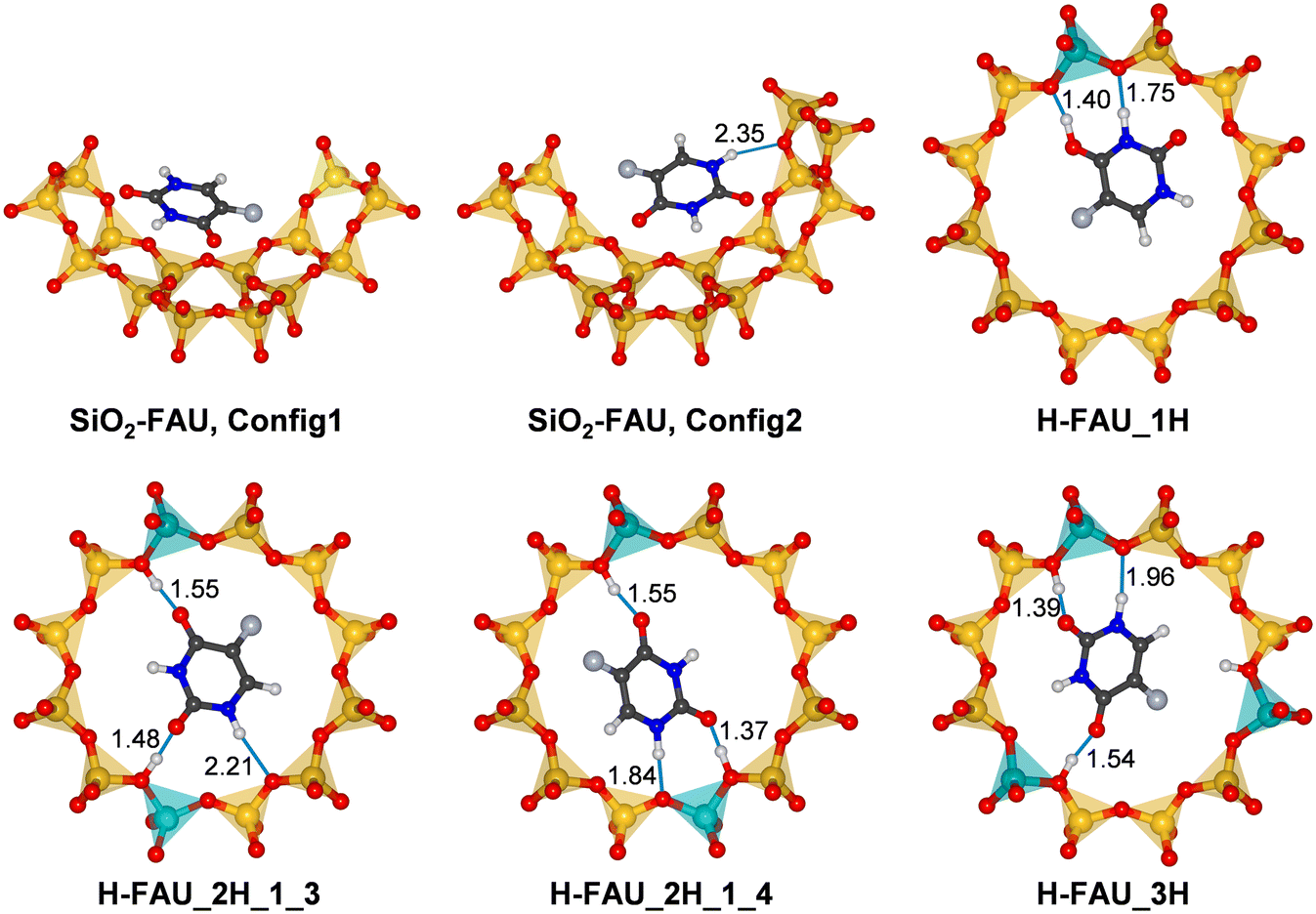 | ||
| Fig. 3 Lowest-energy configurations of 5-FU in FAU. For SiO2-FAU, the surrounding portion of the framework is shown, whereas only the 12MR containing the Al atom(s) is shown for protonic FAU models. Hydrogen bonds are displayed as blue lines, with distances given in Å. | ||
2) Lowest-energy configurations and hydrogen bonds
In order to analyse the host-guest interactions in more detail, lowest-energy configurations of adsorbed 5-FU were visualised, together with the surrounding part of the framework. Fig. 3 shows such visualisations for SiO2-FAU and H-FAU models with 1, 2, or 3 protons per unit cell, with further figures for more Al-rich models being included in Fig. S1 (ESI†). For those cases where AIMD simulations were run, radial distribution functions of relevant combinations of atoms are compiled in Fig. S2–S6.†For 5-FU@SiO2-FAU, two configurations that are very close in energy are shown in Fig. 3. In the first configuration, which is energetically slightly favoured according to DZVP results, the 5-FU molecule lies more or less flat above one 6MR and one 4MR forming the wall of the FAU supercage. As no hydrogen bonds are present, it can be concluded that the molecule interacts with the zeolite predominantly through dispersion interactions. In the second configuration, more favourable according to the calculation using TZVP basis sets, the molecule assumes a more tilted orientation with respect to the curvature of the pore wall, forming a relatively long hydrogen bond via the H1 atom to a framework oxygen atom (Ofw). AIMD simulations starting from both configurations (two from Config1, two from Config2) resulted in fairly similar RDFs. In particular, the H1⋯Ofw RDF (Fig. S2†), averaged over all four trajectories, exhibits only a shoulder in the distance range between 2 and 2.5 Å, not a pronounced maximum. This indicates that hydrogen bonds to oxygen atoms of the all-silica framework are relatively weak and transient, as observed previously for triclosan in all-silica FAU and MOR.63
In the lowest-energy configuration obtained for 5-FU@H-FAU_1H, the framework is deprotonated by the adsorbed molecule, with the proton now being bonded to the O4 atom of 5-FU. As is visible in Fig. 3, the protonated 5-FU species is stabilised by two short hydrogen bonds from the H@O4 and H3 atoms to two Ofw atoms neighbouring the framework Al site. Protonation of O2 is observed in two other configurations (Config1 and Config5 of Table S4†), which are, however, about 10 kJ mol−1 less stable. This agrees with the previous finding by Wielińska et al. that protonation of O4 is energetically favoured.15 Configurations in which a neutral 5-FU molecule is hydrogen-bonded to the framework are even less stable. The Ofw⋯Hfw and Hfw⋯O4 RDFs visualised in Fig. S3† confirm that the proton remains closer to O4 than to Ofw for the majority of the simulation time. However, an overlap of the maxima in the distance range of 1.0 to 1.3 Å points to a certain degree of dynamic exchange.
In contrast to the scenario observed for H-FAU_1H, the 5-FU molecule is only hydrogen-bonded to framework protons in the lowest-energy configurations for both models having two protons within one 12MR (Fig. 3). Among the DFT-optimised structures, only a single configuration with a protonated 5-FU molecule was found (for H-FAU_2H_1_3), which is about 25 kJ mol−1 higher in energy than the most stable configuration. In both configurations shown in Fig. 3, the hydrogen bond involving the O2 atom is somewhat shorter than that involving the O4 atom. Repulsive interactions between the F atom and the framework are likely to prevent the formation of a very short hydrogen bond to O4. Moreover, different partial charge calculation schemes consistently deliver a more negative polarisation of the O2 atom compared to O4 (Table S6†). Additional hydrogen bonds occur between the H1 atom of 5-FU and framework oxygen atoms. In H-FAU_2H_1_4, this hydrogen bond is fairly short, involving an O atom that is part of the AlO4 tetrahedron, whereas a longer bond to an O atom of a Si–O–Si linkage is present in H-FAU_2H_1_3. It is plausible to infer that the presence of a shorter hydrogen bond to a more negatively polarised Ofw atom contributes to the higher stability of the former configuration. The RDFs (Fig. S4 and S5†) indicate that the hydrogen bonds highlighted in Fig. 3 are stable on the timescale of the AIMD simulations. Although there appears to be some fluctuation between the hydrogen-bonded state and short-lived framework deprotonation in the vicinity of the O2 atom, the cumulative RDFs indicate that the proton is located within 1.2 Å of O2 for less than 10% of the total simulation time.
The lowest-energy configuration for H-FAU_3H is qualitatively similar to the one obtained for H-FAU_2H_1_3. Although the H1⋯Ofw hydrogen bond is shorter, the adsorption energy is slightly less negative. As the third proton of the 12MR lies at a distance of 3.25 Å from the H6 atom of 5-FU, repulsive electrostatic interactions between them are likely to be responsible for the somewhat reduced stability. For the more Al-rich models (Fig. S1†), the lowest-energy configurations are generally very similar to those observed for the corresponding H-FAU_1H/2H models, with framework deprotonation occurring in the system with one proton per 12MR (H-FAU_16H) and solely hydrogen-bonded 5-FU in models with two protons per 12MR (H-FAU_32H_1_3/_1_4). Among the whole set of optimised configurations, there are two cases where 5-FU is hydrogen-bonded to framework protons pointing into different 12MRs (one for H-FAU_16H, the other for H-FAU_32H_1_3, they are labelled with asterisks in Table S4†). However, these configurations are energetically less favourable than those where all significant interactions occur within a single 12MR. This indicates that the inclusion of different proton arrangements within one ring may be sufficient to cover the most relevant low-energy bonding situations for a relatively simply and rigid molecule like 5-FU.
3) IR spectra and molecular structure of adsorbed 5-FU molecules
Diffraction methods are often unable to resolve the locations of guest molecules in zeolite pores due to their disorder over different possible positions and orientations. In such cases, spectroscopic methods can be very useful to gain insights into host–guest interactions and adsorption-induced changes in the molecular structure. As an IR-spectroscopic characterisation of FAU samples loaded with 5-FU has been reported,20 DFT-computed IR spectra were generated for free 5-FU and three representative models visualised in Fig. 3, namely 5-FU@SiO2-FAU, Config2 (weak H1⋯Ofw hydrogen bond), 5-FU@H-FAU_1H (deprotonated 5-FU), and 5-FU@H-FAU_2H_1_3 (5-FU stabilised by three hydrogen bonds). Fig. 4 shows the calculated IR spectra for the wavenumber range from 1200 to 1800 cm−1, emphasising the four most intense bands of free 5-FU (two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching modes, C5C6 and C5–F stretching modes) as well as the corresponding bond distances. Intramolecular bond distances and, where assignable, frequencies of associated stretching modes are compiled in the ESI,† Table S7. Relevant bending modes are also included in this table. Since some modes involve significant displacements of more than two or three atoms, it has to be stressed that this assignment is somewhat simplified and primarily intended to facilitate the following discussion.
O stretching modes, C5C6 and C5–F stretching modes) as well as the corresponding bond distances. Intramolecular bond distances and, where assignable, frequencies of associated stretching modes are compiled in the ESI,† Table S7. Relevant bending modes are also included in this table. Since some modes involve significant displacements of more than two or three atoms, it has to be stressed that this assignment is somewhat simplified and primarily intended to facilitate the following discussion.
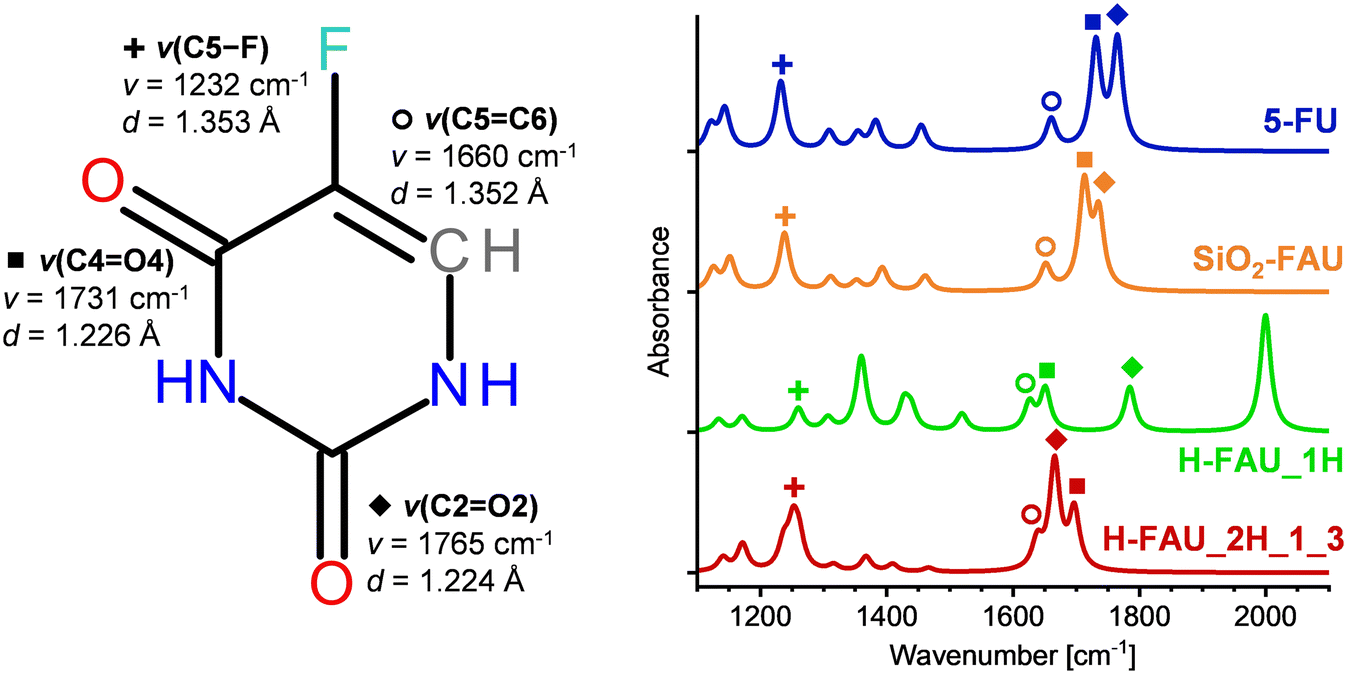 | ||
| Fig. 4 Left: 5-FU molecule including bond distances and stretching frequencies of the four most prominent modes in the wavenumber range from 1200 to 1800 cm−1. Right: DFT-calculated IR spectra of free 5-FU (top curve) and adsorbed 5-FU. Spectra are normalised to have the same maximal intensity in the shown wavenumber range. | ||
Tables S8† compares the calculated frequencies in the range of 1200 to 1800 cm−1 to experimental results obtained for 5-FU in an Ar matrix and to DFT-B3LYP calculations, both reported by Ivanov et al.,70 and to experimental results obtained for solid 5-FU by Datt et al. (available only for the four main modes shown in Fig. 4).20 The root mean square error of the DFT frequencies from the present work with respect to experimental results for 5-FU in an Ar matrix amounts to 17 cm−1 (including only the modes falling between 1200 and 1800 cm−1 listed in Table S8†). This agreement appears satisfactory, especially when considering that the DFT frequencies were directly taken from the calculations, without the application of any scaling factor. Scaling of vibrational frequencies computed with electronic structure methods by a method-dependent scaling factor is often done to minimise systematic deviations from experimental frequencies.87
Upon adsorption in SiO2-FAU, the most significant change in the IR spectrum is a slight red-shift of the two ν(CO) modes, correlated with a modest elongation of the CO bonds. In the higher-frequency region (not included in the figure), the ν(N1–H1) mode is red-shifted due to the presence of the H1⋯Ofw hydrogen bond (Fig. 3). Much more pronounced changes are visible in the IR spectrum of 5-FU@H-FAU_1H: the deprotonation of the framework by 5-FU results in the emergence of a new ν(O4–H) stretching mode at 2000 cm−1, which is very prominently visible. The splitting of the ν(CO) modes increases drastically, with the ν(C4O4) mode being red-shifted due to the bond elongation upon protonation of O4, whereas the ν(C2O2) mode is (slightly) blue-shifted. The ν(C5C6) mode is red-shifted, and the O4–H bending mode appears as a fairly intense signal at 1360 cm−1. A closer look at Table S7† shows that several of the bond distances in the 5-FU ring change rather significantly upon protonation. These changes can explain the shift of a mode associated with ring deformations from 1383 cm−1 in free 5-FU to 1519 cm−1. Altogether, the changes in the IR spectra are so pronounced that it should be straightforward to identify protonated 5-FU species in zeolite pores, even if only a fraction of the adsorbed molecules are protonated. For the 5-FU@H-FAU_2H_1_3 case, where 5-FU is solely hydrogen-bonded, the changes are less distinctive, but still clearly visible: here, both ν(CO) modes are red-shifted, with the larger shift occurring for the ν(C2O2) mode that participates in the stronger hydrogen bond. The ν(CC) mode is also red-shifted, but not as strongly, resulting in partial overlap of the signals arising from these three modes. The Ofw–Hfw bending modes appear at 1237 and 1261 cm−1, respectively, thus overlapping with the ν(C5–F) mode.
For the four modes highlighted in Fig. 4, experimental frequencies of 5-FU adsorbed in H-FAU samples having Si/Al ratios of 2.5, 15, and 30 were reported by Datt et al.20 Since the results for all three samples were essentially identical, it is sufficient to discuss those obtained for the most Si-rich sample: taking free 5-FU in Ar matrix as reference, these authors observed a slight red-shift of the ν(C2O2) mode by −5 cm−1 and a more prominent red-shift of the ν(C4O4) mode by −25 cm−1. These changes are much smaller than those predicted by the DFT calculations for hydrogen-bonded 5-FU adsorbed in H-FAU_2H_1_3, as well as being qualitatively different from those computed for the protonated 5-FU case. Experimentally, a slight blue-shift of the ν(CC) mode by +5 cm−1 was observed, whereas the ν(C5–F) mode was completely unaffected. Neither of these results agree with the computational predictions summarised above. These apparent discrepancies can be explained when taking into account that Datt et al. concluded that 5-FU adsorbed in their samples primarily interacted with Lewis acidic EFAL sites, not with framework protons. While a direct corroboration would require further calculations, the present results support the findings by Datt et al. that adsorption of 5-FU at Brønsted acidic framework protons was not the dominant process occurring in their samples.
4) Behaviour of adsorbed 5-FU in the presence of water
Up to this point, all calculations assumed an adsorption of isolated 5-FU in the zeolite pores, neglecting the possible co-adsorption of water. While this is probably a reasonable approximation for highly hydrophobic all-silica zeolites, the framework charge of aluminosilicate zeolites results in an increased hydrophilicity, and co-adsorption of water will be inevitable in many real-world scenarios. In a previous computational study using an analogous computational setup, an adsorption energy of −86 kJ mol−1 was obtained for the adsorption of a single H2O molecule at the framework proton of H-FAU_1H.63 While this is considerably less negative than the adsorption energy obtained for 5-FU, one needs to consider that the adsorption of water in protonic zeolites usually involves the interaction of a cluster of several H2O molecules with the framework.88–91 Hence, a more meaningful comparison can be made by considering the adsorption energy of a (H2O)n cluster having roughly the same size as a single 5-FU molecule. Based on the crystal structure of the most stable polymorph, the molecular volume of 5-FU can be approximated as 121 Å3,92 compared to 32.5 Å3 for H2O.93 Thus, a cluster of four H2O molecules should occupy a similar volume as one 5-FU molecule. According to previously reported calculations, the total adsorption energy for such a cluster is on the order of −274 kJ mol−1 (−68.5 kJ mol−1 per H2O),63 being considerably more negative than the ΔĒads,Boltz value of −144 kJ mol−1 computed for 5-FU@H-FAU_1H.These considerations already indicate that the competitive adsorption of water should result in a displacement of 5-FU away from the framework protons. AIMD simulations were employed to investigate the co-adsorption of both species in more detail. First, preliminary FF-based simulations were used to add 16 H2O molecules to the lowest-energy configurations computed for 5-FU in H-FAU_1H, H-FAU_2H_1_3, and H-FAU_2H_1_4. AIMD simulations with a total duration of 25 ps were then run for each of these (5-FU + 16H2O)@FAU systems. In the following, key observations for each trajectory are presented, focussing on representative snapshots from different stages of the simulation and selected RDFs (including time-resolved RDFs for different time intervals where relevant).
Fig. 5 shows the environment of the adsorbed 5-FU molecule in (5-FU + 16H2O)@H-FAU_1H after 0.5 ps and 2.5 ps and at the end of the AIMD simulation (after 25 ps). At the beginning of the simulation, the protonated 5-FU molecule is hydrogen-bonded to the framework, as in the lowest-energy configuration of 5-FU@H-FAU_1H in the absence of water (Fig. 3). After approximately 1.5 ps, the proton moves from the 5-FU molecule to one of the surrounding water molecules, and the RDFs computed over different time intervals, collected in Fig. S11,† show that no new O4–H bonds form at later stages of the simulation. The 5-FU molecule progressively moves away from its initial location near the framework Al atom to another part of the cage, whereas the vicinity of the Al atom is occupied by a positively charged cluster formed by water molecules and one hydronium ion. The 5-FU molecule that was initially bonded to the framework now forms numerous hydrogen bonds to surrounding water molecules. Additional RDFs compiled in Fig. S10† indicate that the O2 atom of 5-FU remains unprotonated, and that there is no formation of any new Ofw–Hfw bond; in other words, the “excess” proton remains part of the water cluster throughout the simulation.
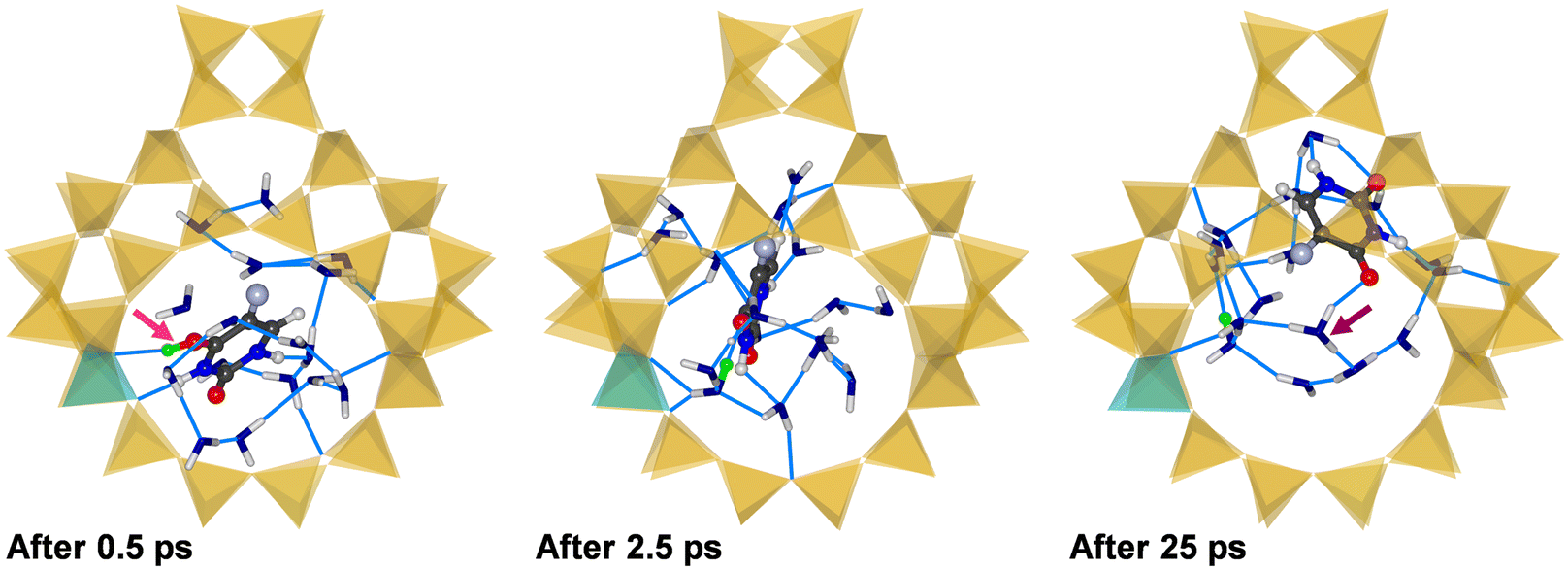 | ||
| Fig. 5 Snapshots from three different stages of the AIMD simulation of (5-FU + 16H2O)@H-FAU_1H. To enhance the clarity, only one FAU supercage is shown and H2O molecules are visualised in a stick representation, with OH2O atoms shown in blue. The proton that was initially attached to the framework is shown in green. The pink arrow highlights protonation of 5-FU, the purple arrow highlights formation of a hydronium ion. | ||
In the case of (5-FU + 16H2O)@H-FAU_2H_1_3, the following chain of events can be identified based on representative snapshots shown in Fig. 6 (after 0.5, 2.5, 25 ps): at the very beginning of the simulation, the framework proton that participated in a hydrogen bond to the O4 atom of 5-FU in the water-free structure (Fig. 3) is removed from the framework, protonating the O4 atom. While the other framework proton is still bonded to the framework in the first snapshot shown in Fig. 6, it is removed shortly afterwards, forming a covalent O2–Hfw bond. At approximately the same time, the O4 atom is deprotonated again, with the proton being transferred to a water molecule. This is the situation after 2.5 ps. After about 6 ps, the O2 atom is also deprotonated. At the end of the simulation, the vicinity of the Al atoms is occupied by the cluster consisting of H2O molecules and two hydronium ions, and the 5-FU molecule is displaced considerably from its initial location. The time-resolved RDFs shown in Fig. S13† further illustrate these findings.
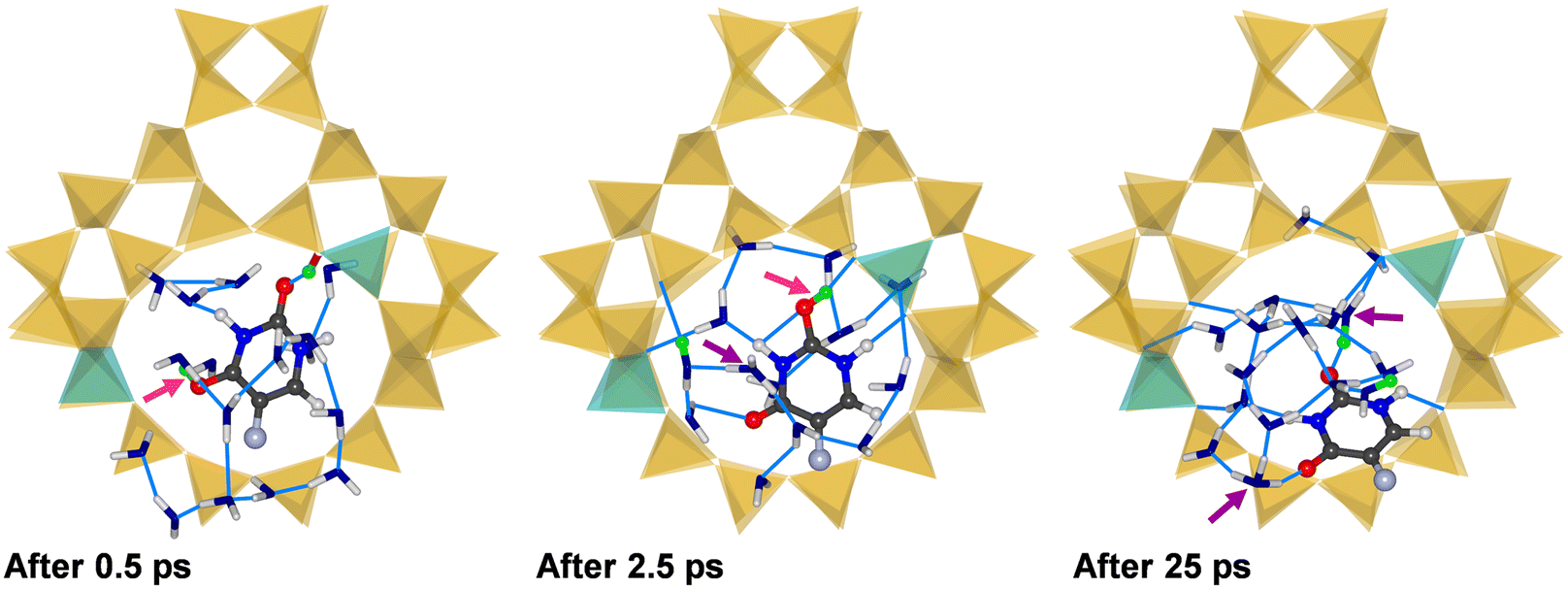 | ||
| Fig. 6 Snapshots from three different stages of the AIMD simulation of (5-FU + 16H2O)@H-FAU_2H_1_3. | ||
Three representative snapshots of (5-FU + 16H2O)@H-FAU_2H_1_4 are shown in Fig. 7, again visualising the system after 0.5, 2.5, and 25 ps. In this case, one of the framework protons moves to the O2 atom of 5-FU in the very initial stages of the simulation, while the O4 atom remains hydrogen-bonded to the other framework proton after 0.5 ps. After 2.5 ps, however, a rather unexpected situation is observed: at this point, both the O2 and O4 atom of 5-FU are protonated, and the N3 atom is deprotonated. Carrying a net charge of +1, 5-FU is now in a different tautomeric form than observed in all previous calculations involving protonated 5-FU. Although this tautomer was found to be less stable than the form having a single protonated O atom in a previous computational study,15 it does not seem implausible to surmise that it can be stabilised, at least temporarily, through localised interactions in a specific environment. At the end of the 25 ps trajectory, the proton that was previously attached to the O4 atom has been transferred to a water molecule, whereas the O2 and N3 atoms remain protonated and deprotonated, respectively. The absence of additional peaks in the RDFs in the time interval from 20 to 25 ps confirms the stability of this scenario on the picosecond timescale. Although the 5-FU molecule still forms a hydrogen bond to a Ofw atom neighbouring one of the two Al sites, Fig. 7 clearly shows the movement away from its initial location at the centre of the 12MR.
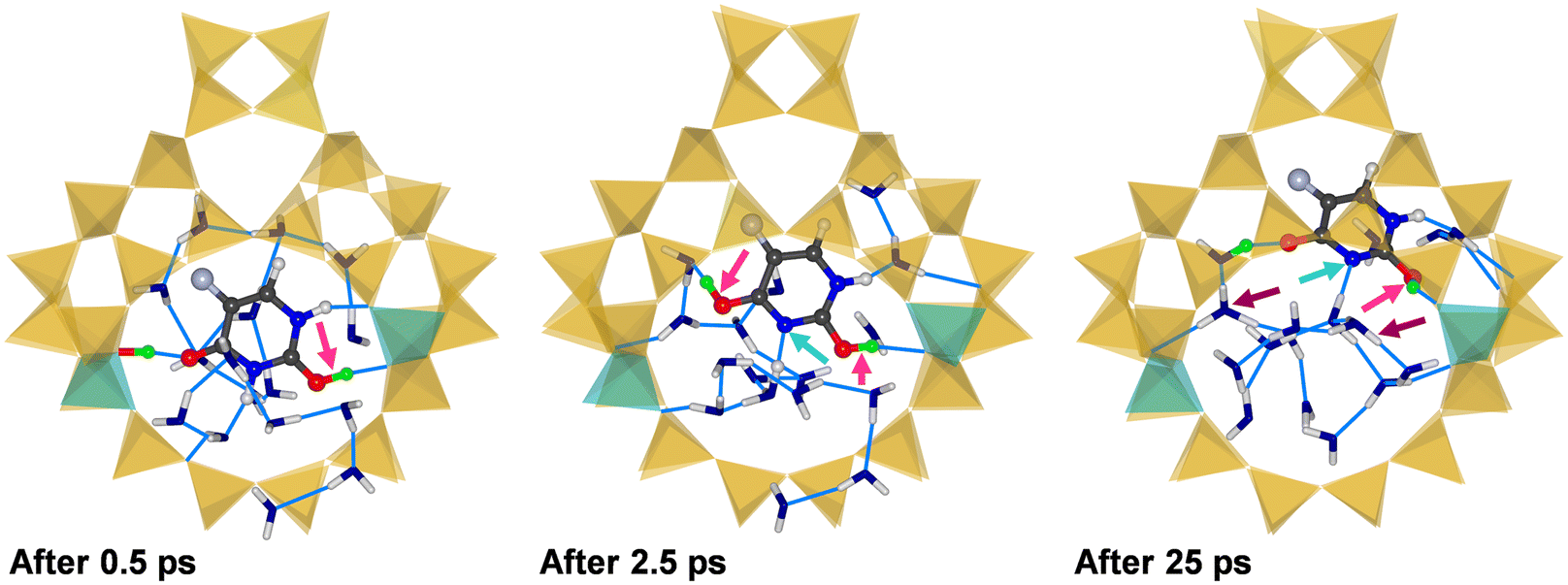 | ||
| Fig. 7 Snapshots from three different stages of the AIMD simulation of (5-FU + 16H2O)@H-FAU_2H_1_4. Light blue arrows highlight deprotonation of the ring N atom. | ||
Although there are non-negligible differences among the individual AIMD trajectories, some common features can be identified: in all three cases, the co-adsorption of water and 5-FU results in framework deprotonation, a finding that falls in line with previous observations made for water clusters89–91 and for co-adsorbed water and triclosan in protonic zeolites.63 Although protonated 5-FU occurs as an intermediate species during the simulations, the protons are transferred to water molecules over the course of the 25 ps simulations. In the final snapshots, 5-FU is always in the neutral form, interacting through hydrogen bonds with the positively charged water cluster and the deprotonated zeolite framework.
It can be inferred from these observations that the behaviour of co-adsorbed 5-FU and water in the zeolite pores is strongly impacted by the Brønsted acidity of the framework protons. It is therefore interesting to compare this behaviour to that of 5-FU in aqueous solution. For this purpose, additional AIMD simulations were run for a box containing one 5-FU and 256 H2O molecules, considering three different starting configurations. The RDFs computed from these trajectories, shown in Fig. S16,† provide no indications for protonation of 5-FU oxygen atoms or deprotonation of nitrogen atoms, in other words, the neutral tautomer shown in Fig. 1 is stable in aqueous solution. This corroborates that the proton(s) that the zeolite provides are responsible for the proton transfers between 5-FU and its environment observed in the AIMD simulations.
Conclusions
The DFT calculations presented in this work show that 5-FU adsorbed in all-silica FAU is primarily stabilised through dispersion interactions, with hydrogen bonds to framework oxygen atoms playing only a minor role. In protonic zeolites, a significant stabilisation arises from interactions of carbonyl O atoms with the framework protons. While framework deprotonation and concurrent formation of a positively charged 5-FU species was observed upon interaction with a single framework proton, the availability of two framework protons in proximity resulted in the formation of adsorption complexes stabilised by several hydrogen bonds, with neutral 5-FU simultaneously acting as acceptor and donor. AIMD simulations confirmed the stability of these hydrogen bonds at room temperature (in the absence of water). From a broader perspective, these results show that “multi-site” interactions, simultaneous interactions with more than one framework proton, can make a considerable contribution to the stabilisation of functional organic molecules in the pores of protonic zeolites. Such scenarios should thus be taken into account in computational studies and in the analysis of experimental data, primarily for species possessing two or more negatively polarised areas that could interact with the framework protons (or, analogously, with cations in cation-exchanged zeolites, as discussed recently for the example of the organic UV filter octyl methoxycinnamate in zeolite NaX94).Both in terms of adsorption energies and equilibrium structures, fairly similar results were obtained for zeolite models having vastly different Si/Al ratios, but identical proton arrangements in the 12MR where 5-FU is adsorbed. This indicates that models containing only a few framework protons can be useful to represent specific local environments. However, it should be kept in mind that the presence of 2 or 3 protons in the same 12MR at Si/Al ratios of 95 and 63, respectively, appears extremely improbable when assuming a more or less statistic distribution. Thus, these structures should be seen as representatives of possible local arrangements at lower Si/Al ratios, rather than being realistic models. An additional simplification made in the present study was the assumption that the framework protons are always bonded to O1 oxygen atoms, neglecting the possibility to occupy different sites at finite temperature.
The computational prediction of IR spectra clearly showed that different bonding environments of 5-FU could be easily distinguished by means of IR spectroscopy. Although significant discrepancies between the DFT results and previously reported experimental data were apparent, these are most probably attributable to the interaction of 5-FU with EFAL sites in the experimentally studied samples. Future computational work will look into a DFT-based modelling of the interaction between 5-FU and EFAL sites, taking EFAL models proposed in the recent literature as starting points.95,96 While it might appear counterintuitive that all three samples studied experimentally in prior work show identical shifts of IR-active modes, it has to be considered that even the most Si-rich sample contained, on average, 6 Al atoms per unit cell (Si/Al ratio of 30). Thus, it still seems possible that a significant fraction of the adsorbed 5-FU molecules can interact with EFAL sites. However, these aspects cannot be elucidated with calculations alone, and a more detailed understanding of the role of different aluminium environments would require further experimental investigations of 5-FU adsorption in zeolite samples having well-quantified amounts of framework and extra-framework Al atoms.
Finally, AIMD simulations including co-adsorbed water molecules showed that the water molecules have a higher affinity towards the framework protons, displacing 5-FU from their vicinity. Thus, it seems unlikely that interactions with the framework protons would contribute significantly to the stabilisation of 5-FU in zeolite pores in the presence of water. In the context of contaminant removal, it is therefore pivotal to maximise adsorbent hydrophobicity, as already discussed in earlier work, for example, for the case of triclosan adsorption.63,97 For drug delivery, on the other hand, there are some additional implications: if 5-FU was loaded in the absence of water (e.g., from non-aqueous solvents like acetone, as reported in some previous studies21,24,26) and the 5-FU@zeolite composite stored without exposure to humidity, the drug could be released upon contact with water, for example, in the human body. Potentially, the release kinetics could be tailored through a variation of the Si/Al ratio and/or the framework topology. In the prior experimental investigation by Datt et al., the sample having the lowest Si/Al ratio showed essentially no 5-FU release in simulated body fluid, pointing to a strong binding of 5-FU to EFAL sites.20 Based on the present work, it is suggested to focus on protonic zeolites containing few EFAL sites in future experimental investigations of zeolites as 5-FU carrier materials. An additional possibility to tailor the affinity towards 5-FU (or other drug molecules of interest) is the use of cation-exchanged zeolites containing different metal cations. A systematic exploration using an analogous computational protocol is planned for future work.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Deutsche Forschungsgemeinschaft (German Research Foundation, DFG) through a Heisenberg fellowship (project no. 455871835). The author gratefully acknowledges the computing time granted by the Resource Allocation Board and provided on the supercomputers Lise and Emmy at NHR@ZIB and NHR@Göttingen as part of the NHR infrastructure. The calculations for this research were conducted with computing resources under the project hbc00062.References
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, Nucleic Acids Res., 2023, 51, D1373–D1380 CrossRef PubMed.
- D. B. Longley, D. P. Harkin and P. G. Johnston, Nat. Rev. Cancer, 2003, 3, 330–338 CrossRef CAS PubMed.
- S. Vodenkova, T. Buchler, K. Cervena, V. Veskrnova, P. Vodicka and V. Vymetalkova, Pharmacol. Ther., 2020, 206, 107447 CrossRef CAS PubMed.
- A. A. Valencia-Lazcano, D. Hassan, M. Pourmadadi, A. Shamsabadipour, R. Behzadmehr, A. Rahdar, D. I. Medina and A. M. Díez-Pascual, Eur. J. Med. Chem., 2023, 246, 114995 CrossRef CAS PubMed.
- J. L. Arias, Molecules, 2008, 13, 2340–2369 CrossRef CAS PubMed.
- S. P. Chandran, S. B. Natarajan, S. Chandraseharan and M. S. B. Mohd Shahimi, J. Cancer Res. Pract., 2017, 4, 45–48 CrossRef.
- A. Lamprecht, H. Yamamoto, H. Takeuchi and Y. Kawashima, J. Controlled Release, 2003, 90, 313–322 CrossRef CAS PubMed.
- Q. Jin, F. Mitschang and S. Agarwal, Biomacromolecules, 2011, 12, 3684–3691 CrossRef CAS PubMed.
- K. Liu, Z. Q. Wang, S. J. Wang, P. Liu, Y. H. Qin, Y. Ma, X. C. Li and Z. J. Huo, Int. J. Nanomed., 2015, 10, 6445–6454 CAS.
- T. Moodley and M. Singh, Pharmaceutics, 2019, 11, 288 CrossRef CAS PubMed.
- H. Luo, D. Ji, C. Li, Y. Zhu, G. Xiong and Y. Wan, Int. J. Pharm., 2016, 513, 17–25 CrossRef CAS PubMed.
- F. H. Lin, Y. H. Lee, C. H. Jian, J. M. Wong, M. J. Shieh and C. Y. Wang, Biomaterials, 2002, 23, 1981–1987 CrossRef CAS PubMed.
- S. A. Gârea, A. I. Mihai, A. Ghebaur, C. Nistor and A. Sârbu, Int. J. Pharm., 2015, 491, 299–309 CrossRef PubMed.
- D. Tan, P. Yuan, F. Dong, H. He, S. Sun and Z. Liu, Appl. Clay Sci., 2018, 159, 102–106 CrossRef CAS.
- J. Wielińska, A. Nowacki and B. Liberek, Molecules, 2019, 24, 3683 CrossRef PubMed.
- C. Baerlocher, D. Brouwer, B. Marler and L. B. McCusker, Database of Zeolite Structures, 2024, https://www.iza-structure.org/databases/ Search PubMed.
- P. A. Wright, Microporous Framework Solids, Royal Society of Chemistry, Cambridge, 2007 Search PubMed.
- S. Kulprathipanja, Zeolites in Industrial Separation and Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010 Search PubMed.
- L. Bacakova, M. Vandrovcova, I. Kopova and I. Jirka, Biomater. Sci., 2018, 6, 974–989 RSC.
- A. Datt, E. A. Burns, N. A. Dhuna and S. C. Larsen, Microporous Mesoporous Mater., 2013, 167, 182–187 CrossRef CAS.
- N. Vilaça, R. Amorim, A. F. Machado, P. Parpot, M. F. R. Pereira, M. Sardo, J. Rocha, A. M. Fonseca, I. C. Neves and F. Baltazar, Colloids Surf., B, 2013, 112, 237–244 CrossRef PubMed.
- M. Spanakis, N. Bouropoulos, D. Theodoropoulos, L. Sygellou, S. Ewart, A. M. Moschovi, A. Siokou, I. Niopas, K. Kachrimanis, V. Nikolakis, P. A. Cox, I. S. Vizirianakis and D. G. Fatouros, Nanomedicine, 2014, 10, 197–205 CrossRef CAS PubMed.
- R. A. Al-Thawabeia and H. A. Hodali, J. Chem., 2015, 2015, 403597 Search PubMed.
- N. Vilaça, A. F. Machado, F. Morais-Santos, R. Amorim, A. Patrícia Neto, E. Logodin, M. F. R. Pereira, M. Sardo, J. Rocha, P. Parpot, A. M. Fonseca, F. Baltazar and I. C. Neves, RSC Adv., 2017, 7, 13104–13111 RSC.
- A. G. Abd-Elsatar, M. M. Farag, H. F. Youssef, S. A. Salih, M. M. Mounier and E. El-Meliegy, Prog. Biomater., 2019, 8, 101–113 CrossRef CAS PubMed.
- O. Y. Golubeva, Y. A. Alikina, E. Y. Brazovskaya and V. V. Ugolkov, Appl. Clay Sci., 2020, 184, 105401 CrossRef CAS.
- M. A. Sayed, H. M. El-Zeiny, J. S. Khim, J. S. Ajarem, A. A. Allam and M. R. Abukhadra, ACS Omega, 2022, 7, 6991–7001 CrossRef CAS PubMed.
- N. Vilaça, A. R. Bertão, E. A. Prasetyanto, S. Granja, M. Costa, R. Fernandes, F. Figueiredo, A. M. Fonseca, L. De Cola, F. Baltazar and I. C. Neves, Mater. Sci. Eng., C, 2021, 120, 111721 CrossRef PubMed.
- T. Sağir, M. Huysal, Z. Durmus, B. Z. Kurt, M. Senel and S. Isik, Biomed. Pharmacother., 2016, 77, 182–190 CrossRef PubMed.
- E. Y. Brazovskaya and O. Y. Golubeva, Pet. Chem., 2023, 63, 820–828 CrossRef CAS.
- A. R. Bertão, V. Ivasiv, C. Almeida-Aguiar, P. R. Correia, A. M. Fonseca, M. Bañobre-López, F. Baltazar and I. C. Neves, Microporous Mesoporous Mater., 2024, 364, 112871 CrossRef.
- H. P. Nguyen Thi, H. D. Ninh, C. Van Tran, B. T. Le, S. V. Bhosale and D. D. La, ChemistrySelect, 2019, 4, 2333–2338 CrossRef CAS.
- Z. Hu, C. Qiao, Z. Xia, F. Li, J. Han, Q. Wei, Q. Yang, G. Xie, S. Chen and S. Gao, ACS Appl. Mater. Interfaces, 2020, 12, 14914–14923 CrossRef CAS PubMed.
- B. E. Souza, L. Donà, K. Titov, P. Bruzzese, Z. Zeng, Y. Zhang, A. S. Babal, A. F. Möslein, M. D. Frogley, M. Wolna, G. Cinque, B. Civalleri and J. C. Tan, ACS Appl. Mater. Interfaces, 2020, 12, 5147–5156 CrossRef CAS PubMed.
- A. Rossner, S. A. Snyder and D. R. U. Knappe, Water Res., 2009, 43, 3787–3796 CrossRef CAS PubMed.
- I. Braschi, S. Blasioli, L. Gigli, C. E. Gessa, A. Alberti and A. Martucci, J. Hazard. Mater., 2010, 178, 218–225 CrossRef CAS PubMed.
- I. Braschi, G. Gatti, G. Paul, C. E. Gessa, M. Cossi and L. Marchese, Langmuir, 2010, 26, 9524–9532 CrossRef CAS PubMed.
- D. J. De Ridder, J. Q. J. C. Verberk, S. G. J. Heijman, G. L. Amy and J. C. Van Dijk, Sep. Purif. Technol., 2012, 89, 71–77 CrossRef.
- A. Martucci, L. Pasti, N. Marchetti, A. Cavazzini, F. Dondi and A. Alberti, Microporous Mesoporous Mater., 2012, 148, 174–183 CrossRef CAS.
- S. Fukahori, T. Fujiwara, N. Funamizu, K. Matsukawa and R. Ito, Water Sci. Technol., 2013, 67, 319–325 CrossRef CAS PubMed.
- I. Braschi, S. Blasioli, E. Buscaroli, D. Montecchio and A. Martucci, J. Environ. Sci., 2016, 43, 302–312 CrossRef CAS PubMed.
- N. Jiang, R. Shang, S. G. J. Heijman and L. C. Rietveld, Water Res., 2018, 144, 145–161 CrossRef CAS PubMed.
- M. Fu, J. Wang, B. Heijman and J. P. van der Hoek, J. Water Process Eng., 2021, 44, 102403 CrossRef.
- X. Zheng, N. Jiang, H. Zheng, Y. Wu and S. G. J. Heijman, Sep. Purif. Technol., 2022, 282, 120009 CrossRef CAS.
- P. de Voogt, M.-L. Janex-Habibi, F. Sacher, L. Puijker and M. Mons, Water Sci. Technol., 2009, 59, 39–46 CrossRef CAS PubMed.
- T. aus der Beek, F.-A. Weber, A. Bergmann, S. Hickmann, I. Ebert, A. Hein and A. Küster, Environ. Toxicol. Chem., 2016, 35, 823–835 CrossRef CAS PubMed.
- V. Booker, C. Halsall, N. Llewellyn, A. Johnson and R. Williams, Sci. Total Environ., 2014, 473–474, 159–170 CrossRef CAS PubMed.
- M. Isidori, M. Lavorgna, C. Russo, M. Kundi, B. Žegura, M. Novak, M. Filipič, M. Mišík, S. Knasmueller, M. L. de Alda, D. Barceló, B. Žonja, M. Česen, J. Ščančar, T. Kosjek and E. Heath, Environ. Pollut., 2016, 219, 275–287 CrossRef CAS PubMed.
- P. Verlicchi, A. Galletti, M. Petrovic and D. Barceló, J. Hydrol., 2010, 389, 416–428 CrossRef CAS.
- S. N. Mahnik, B. Rizovski, M. Fuerhacker and R. M. Mader, Anal. Bioanal. Chem., 2004, 380, 31–35 CrossRef CAS PubMed.
- A. Parrella, M. Lavorgna, E. Criscuolo, C. Russo, V. Fiumano and M. Isidori, Chemosphere, 2014, 115, 59–66 CrossRef CAS PubMed.
- M. Novak, B. Žegura, B. Modic, E. Heath and M. Filipič, Sci. Total Environ., 2017, 601–602, 293–300 CrossRef CAS PubMed.
- M. Mišík, M. Filipic, A. Nersesyan, M. Kundi, M. Isidori and S. Knasmueller, Water Res., 2019, 164, 114953 CrossRef PubMed.
- M. Jureczko and J. Kalka, Eur. J. Pharmacol., 2020, 866, 172816 CrossRef CAS PubMed.
- C. Venâncio, B. Monteiro, I. Lopes and A. C. A. Sousa, Environ. Sci. Pollut. Res., 2023, 30, 58841–58854 CrossRef PubMed.
- L. Kovalova, D. R. U. Knappe, K. Lehnberg, C. Kazner and J. Hollender, Environ. Sci. Pollut. Res., 2013, 20, 3607–3615 CrossRef CAS PubMed.
- E. Macedo, M. S. F. Santos, F. J. Maldonado-Hódar, A. Alves and L. M. Madeira, Ind. Eng. Chem. Res., 2018, 57, 3932–3940 CrossRef CAS.
- M. Fischer, Mater. Adv., 2020, 1, 86–98 RSC.
- J. Brauer and M. Fischer, ChemPhysChem, 2024 DOI:10.1002/cphc.202400347.
- X. Wei, Y. Wang, A. J. Hernández-Maldonado and Z. Chen, Green Energy Environ., 2017, 2, 363–369 CrossRef.
- A. Datt, D. Fields and S. C. Larsen, J. Phys. Chem. C, 2012, 116, 21382–21390 CrossRef CAS.
- M. Fischer, ChemPhysChem, 2023, 24, e202300022 CrossRef CAS PubMed.
- M. Fischer, Environ. Sci.: Adv., 2023, 2, 1082–1098 CAS.
- S. Simonetti, A. D. Compañy, E. Pronsato, A. Juan, G. Brizuela and A. Lam, Appl. Surf. Sci., 2015, 359, 474–479 CrossRef CAS.
- T. Frising and P. Leflaive, Microporous Mesoporous Mater., 2008, 114, 27–63 CrossRef CAS.
- J. A. Hriljac, M. M. Eddy, A. K. Cheetham, J. A. Donohue and G. J. Ray, J. Solid State Chem., 1993, 106, 66–72 CrossRef CAS.
- J. D. Gale and A. L. Rohl, Mol. Simul., 2003, 29, 291–341 CrossRef CAS.
- M. J. Sanders, M. Leslie and C. R. A. Catlow, J. Chem. Soc., Chem. Commun., 1984, 1271–1273 RSC.
- K.-P. Schröder, J. Sauer, M. Leslie, C. Richard, A. Catlow and J. M. Thomas, Chem. Phys. Lett., 1992, 188, 320–325 CrossRef.
- A. Y. Ivanov, V. S. Leontiev, L. F. Belous, Y. V. Rubin and V. A. Karachevtsev, Low Temp. Phys., 2017, 43, 400–408 CrossRef CAS.
- H. Sun, S. J. Mumby, J. R. Maple and A. T. Hagler, J. Am. Chem. Soc., 1994, 116, 2978–2987 CrossRef CAS.
- H. Heinz, T.-J. Lin, R. Kishore Mishra and F. S. Emami, Langmuir, 2013, 29, 1754–1765 CrossRef CAS PubMed.
- D. S. BIOVIA, BIOVIA Materials Studio 2021, DS Biovia, 2021 Search PubMed.
- F. S. Emami, V. Puddu, R. J. Berry, V. Varshney, S. V. Patwardhan, C. C. Perry and H. Heinz, Chem. Mater., 2014, 26, 2647–2658 CrossRef CAS.
- C. Campaná, B. Mussard and T. K. Woo, J. Chem. Theory Comput., 2009, 5, 2866–2878 CrossRef PubMed.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- T. D. Kühne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Glöβ, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed.
- I. Hamada, Phys. Rev. B, 2014, 89, 121103 CrossRef.
- M. Fischer and J. Brauer, ChemistryOpen, 2024 DOI:10.1002/open.202300273.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- M. Krack, Theor. Chem. Acc., 2005, 114, 145–152 Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- G. Schaftenaar and J. H. Noordik, J. Comput.-Aided Mol. Des., 2000, 14, 123–134 CrossRef CAS PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- M. K. Kesharwani, B. Brauer and J. M. L. Martin, J. Phys. Chem. A, 2015, 119, 1701–1714 CrossRef CAS PubMed.
- M. V. Vener, X. Rozanska and J. Sauer, Phys. Chem. Chem. Phys., 2009, 11, 1702–1712 RSC.
- A. Vjunov, M. Wang, N. Govind, T. Huthwelker, H. Shi, D. Mei, J. L. Fulton and J. A. Lercher, Chem. Mater., 2017, 29, 9030–9042 CrossRef CAS.
- S. Eckstein, P. H. Hintermeier, R. Zhao, E. Baráth, H. Shi, Y. Liu and J. A. Lercher, Angew. Chem., Int. Ed., 2019, 58, 3450–3455 CrossRef CAS PubMed.
- E. Grifoni, G. Piccini, J. A. Lercher, V.-A. Glezakou, R. Rousseau and M. Parrinello, Nat. Commun., 2021, 12, 2630 CrossRef CAS PubMed.
- A. T. Hulme, S. L. Price and D. A. Tocher, J. Am. Chem. Soc., 2005, 127, 1116–1117 CrossRef CAS PubMed.
- S. W. Peterson and H. A. Levy, Acta Crystallogr., 1957, 10, 70–76 CrossRef CAS.
- M. Fischer, R. Fantini, R. Arletti, J. Brauer and L. Mino, J. Phys. Chem. C, 2023, 117, 24242–24252 CrossRef.
- M. Jin, M. Ravi, C. Lei, C. J. Heard, F. Brivio, Z. Tošner, L. Grajciar, J. A. van Bokhoven and P. Nachtigall, Angew. Chem., Int. Ed., 2023, 62, e202306183 CrossRef PubMed.
- J. L. Mancuso and V. Van Speybroeck, J. Catal., 2024, 429, 115211 CrossRef CAS.
- N. Jiang, R. Shang, S. G. J. Heijman and L. C. Rietveld, Sep. Purif. Technol., 2020, 235, 116152 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The supplementary PDF contains details of force field simulations (S1.1 to S1.3) and of calculations of internal energies of adsorption (S1.4), tables of adsorption energies (S2.1) and internal energies of adsorption (S2.2), a figure showing additional low-energy configurations (S2.3), figures showing input structures used in AIMD simulations of (5 FU + 16H2O)@FAU (S2.7), RDF plots (S2.4, S2.8, S2.9) as well as tables reporting additional results (partial charges, S2.5; intramolecular bond distances and vibrational modes, S2.6). See DOI: https://doi.org/10.1039/d4ce00344f |
| ‡ Input files and datasets are available from https://doi.org/10.6084/m9.figshare.25541446. The deposited ZIP archives include sample CP2K input files (Sample_inputs.zip), low-energy configurations and output files from TZVP calculations (Configs_TZVP_results.zip), results of vibrational calculations (Vibrational_calculations.zip), and results of AIMD simulations (5-FU_FAU_AIMD_results.zip, 5-FU_H2O_FAU_AIMD_results.zip, 5-FU_H2O_AIMD_results.zip). |
| This journal is © The Royal Society of Chemistry 2024 |