
Crystal-to-crystal polymorphic phase transition in a cocrystal accompanied by expansion and surface wettability change†
Plabon
Saikia
a,
Poonam
Gupta
 a,
Tridib R.
Nath
b and
Naba K.
Nath
*a
a,
Tridib R.
Nath
b and
Naba K.
Nath
*a
aDepartment of Chemical and Biological Sciences, National Institute of Technology, Meghalaya, India. E-mail: nabakamal.nath@gmail.com; nabakamal.nath@nitm.ac.in
bSophisticated Analytical Instrumentation Centre, Tezpur University, Tezpur 784001, Assam, India
First published on 16th July 2024
Abstract
In this study, cocrystallization of n-propylparaben and a ditopic bipyridine-based azine derivative in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio was carried out. The resulting single crystals of the cocrystal displayed crystal-to-crystal polymorphic phase transformation on heating. The phase transformation is associated with naked-eye visible expansion and cracking of the single crystals. The two polymorphs are distinct in their surface wettability.
:1 stoichiometric ratio was carried out. The resulting single crystals of the cocrystal displayed crystal-to-crystal polymorphic phase transformation on heating. The phase transformation is associated with naked-eye visible expansion and cracking of the single crystals. The two polymorphs are distinct in their surface wettability.
Polymorphism is a unique phenomenon exhibited by crystalline materials, where the molecules of the materials are packed in more than one crystalline arrangement, consequently leading to their distinct physical and chemical properties.1,2 Polymorphism is pertinent in industries such as drugs,3 agrochemicals,4 food,5 dyes and pigments,6etc. Cocrystals7,8 are multicomponent crystal forms containing more than one molecule in a stoichiometric ratio, bound by non-covalent interactions, and the properties of materials can be tuned9–11 by suitable selection of coformers. A cocrystal can also exhibit polymorphism,12 and both crystal forms are patentable.13,14 A polymorphic material can undergo crystal-to-crystal phase transformation from one form to another by applying heat15–22 and occasionally by mechanical stress23–25 or light.26,27
Heat-induced crystal-to-crystal polymorphic phase transition coupled with the thermosalient effect28–34 or crystal deformation such as bending,35,36 twisting,37 change in shape and size,38–40etc. has recently attracted much attention. This particular class of single crystals mainly undergoes two types of polymorphic phase transitions on heating: martensitic and reconstructive phase transformation.16,41–43 The martensitic phase transition involves cooperative and displacive molecular motions, most likely in a layer-to-layer fashion. In contrast, reconstructive transformation involves the formation of nuclei, and the transition transpires by slow molecular diffusion across the transition interface in a molecule-by-molecule fashion. A crystal undergoing nucleation and growth phase transition often loses its structural integrity after the transformation, whereas in the martensitic phase transition, the initial and transformed phases have structural similarity, and single crystals retain their integrity even after the transformation.
Heat-induced mechanical effects displayed by single crystals are rare, and only a handful of examples of cocrystals exhibiting such effects are reported.29,37,44 A recent study by Rawat et al. showed that the thermosalient nature of single component systems could be carried forward to multicomponent crystals by using suitable coformers.29 Liu et al. reported heat-responsive and self-healing single crystals of a cocrystal of coronene and TCNB.44 Gupta et al. cocrystallized probenecid and 4,4′-azopyridine in a 2:1 stoichiometric ratio; the resulting single crystals displayed mechanical responses to heat, light and mechanical force by displaying twisting, bending, and self-healing.37 Research on stimuli-responsive crystalline materials is evolving due to its importance in elucidating the fundamental understanding of the role of intermolecular interactions and molecular packing in material properties. This understanding further leads to the development of new materials that have vast potential in various domains, such as sensors, actuators, data storage, memory devices, etc.
Our previous work on the cocrystallization of a symmetrical ditopic hydrogen bond acceptor and coformer containing n-alkyl chains resulted in stimuli-responsive single crystals.37,45 In continuation, we have cocrystallized a bipyridine-based hydrogen bond acceptor and a coformer with n-alkyl substitution and serendipitously discovered a heat-responsive single crystal. In this study, we report a 1:0.5 stoichiometric cocrystal of n-propylparaben (PP) with (1E,2E)-1,2-bis(pyridin-4-ylmethylene)hydrazine (BPA) (Fig. 1) single crystals, which when heated displayed irreversible expansion and cracking, while undergoing crystal-to-crystal polymorphic phase transformation and wettability reversal.
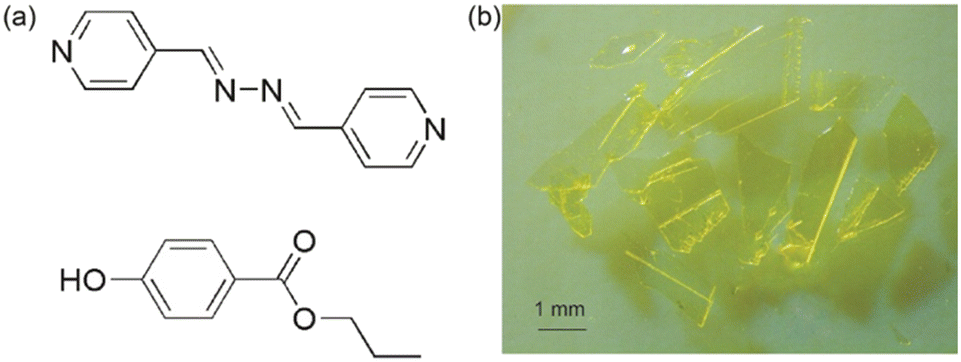 | ||
| Fig. 1 (a) Components of the cocrystal: BPA and PP. (b) Single crystals of the cocrystal. | ||
The coformers BPA and PP were synthesized (see the ESI† for details) and characterised by FT-IR (Fig. S1†) and 1H NMR (Fig. S2 and S3†). The cocrystal was obtained by slow evaporation (see the ESI† for details), and FT-IR (Fig. S1†) and X-ray diffraction (Fig. S4, Table S1†) were used to characterise the resulting single crystals.
The cocrystal crystallized in a triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with one molecule of PP and half a molecule of BPA in its asymmetric unit (Fig. S4, Table S1†). In the crystal structure, O–H groups of the two PP molecules form hydrogen bonds with the two pyridine nitrogen of BPA by the O–H⋯N hydrogen bond (d = 1.80 Å, D = 2.754(3) Å, θ = 171°) to create the basic structural unit (Fig. 2a). These units are connected via the C–H⋯O hydrogen bond (d = 2.75 Å, D = 3.485(4) Å, θ = 133°) between the ester C
space group with one molecule of PP and half a molecule of BPA in its asymmetric unit (Fig. S4, Table S1†). In the crystal structure, O–H groups of the two PP molecules form hydrogen bonds with the two pyridine nitrogen of BPA by the O–H⋯N hydrogen bond (d = 1.80 Å, D = 2.754(3) Å, θ = 171°) to create the basic structural unit (Fig. 2a). These units are connected via the C–H⋯O hydrogen bond (d = 2.75 Å, D = 3.485(4) Å, θ = 133°) between the ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group and the C–H of the n-propyl chain and offset π⋯π interactions (3.437(4) Å) between the pyridine rings. The stacks of the hydrogen-bonded units of PP and BPA connect to the neighbouring units via C–H⋯O (d = 2.80 Å, D = 3.366(4) Å, θ = 120°) and C–H⋯ π (d = 2.96 Å, D = 3.686(5) Å, θ = 132°) interactions (Fig. 2b) to complete the three-dimensional packing (Fig. 2c).
O group and the C–H of the n-propyl chain and offset π⋯π interactions (3.437(4) Å) between the pyridine rings. The stacks of the hydrogen-bonded units of PP and BPA connect to the neighbouring units via C–H⋯O (d = 2.80 Å, D = 3.366(4) Å, θ = 120°) and C–H⋯ π (d = 2.96 Å, D = 3.686(5) Å, θ = 132°) interactions (Fig. 2b) to complete the three-dimensional packing (Fig. 2c).
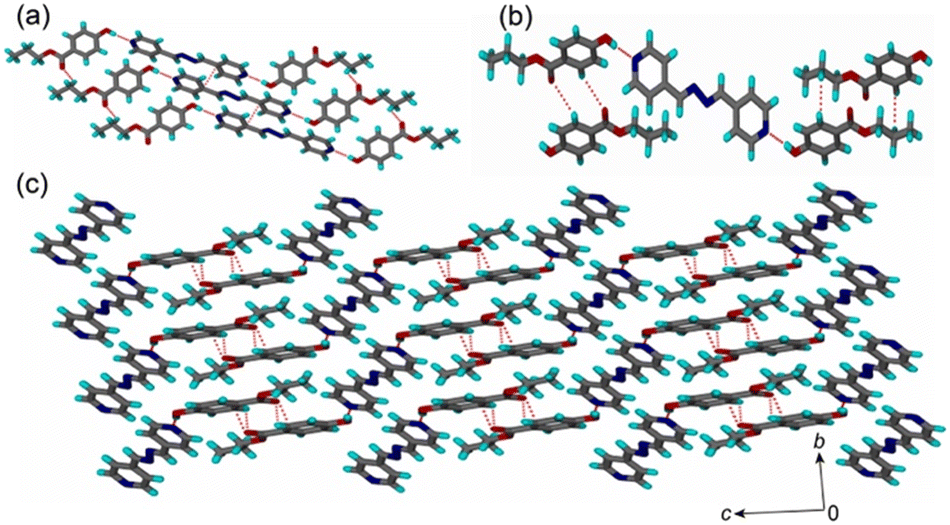 | ||
| Fig. 2 (a) Basic structural unit composed of O–H⋯N hydrogen-bonded BPA and PP stacks by π⋯π interactions. (b) The basic structural units connected to the nearest neighbours by various interactions. (c) Complete 3D packing of the BPA–PP cocrystal. | ||
When the cocrystal was characterised by performing differential scanning calorimetry on a crystalline sample, an endothermic peak appeared in the temperature range of 70–80 °C with a peak position at 72 °C, followed by a melting endotherm at 103 °C (Fig. 3). Thermogravimetric analysis revealed no weight loss in the temperature range of 40–105 °C (Fig. S5†), indicating the transition to be a polymorphic phase transformation.
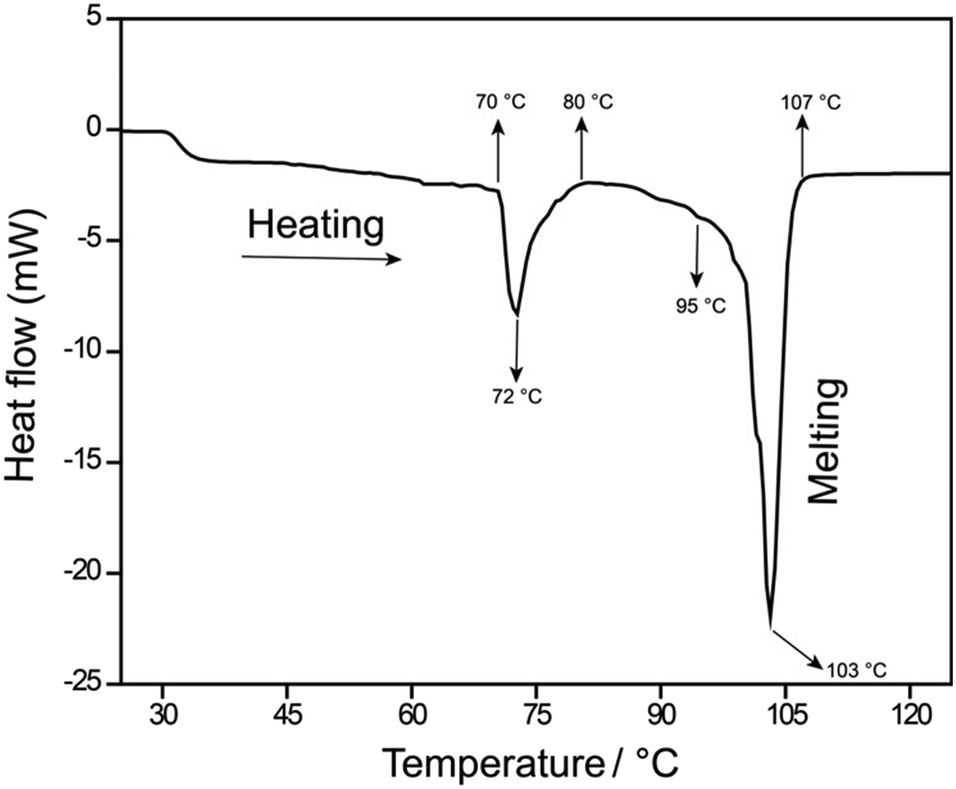 | ||
| Fig. 3 Differential scanning calorimetry showing two endotherms: one for the polymorphic phase transition at 72 °C and another for melting at 103 °C. | ||
To further confirm the polymorphic phase transformation, variable temperature powder X-ray diffraction was carried out on a sample of the bulk crystalline material at 35 °C, then at 90 °C by heating the sample in situ, followed by cooling it back to 35 °C (Fig. 4). The powder X-ray diffraction (PXRD) pattern of the bulk material at 35 °C matches with the calculated PXRD pattern obtained from the crystal structure (Fig. S6†). The PXRD pattern at 90 °C showed clear differences in the peak positions from the starting material, indicating a heat-induced polymorphic phase transition in the bulk crystalline material. The sample was cooled to room temperature, and again, the PXRD pattern was collected at room temperature, showing that the phase transformation is irreversible as the PXRD pattern did not change to that of the original form after cooling.
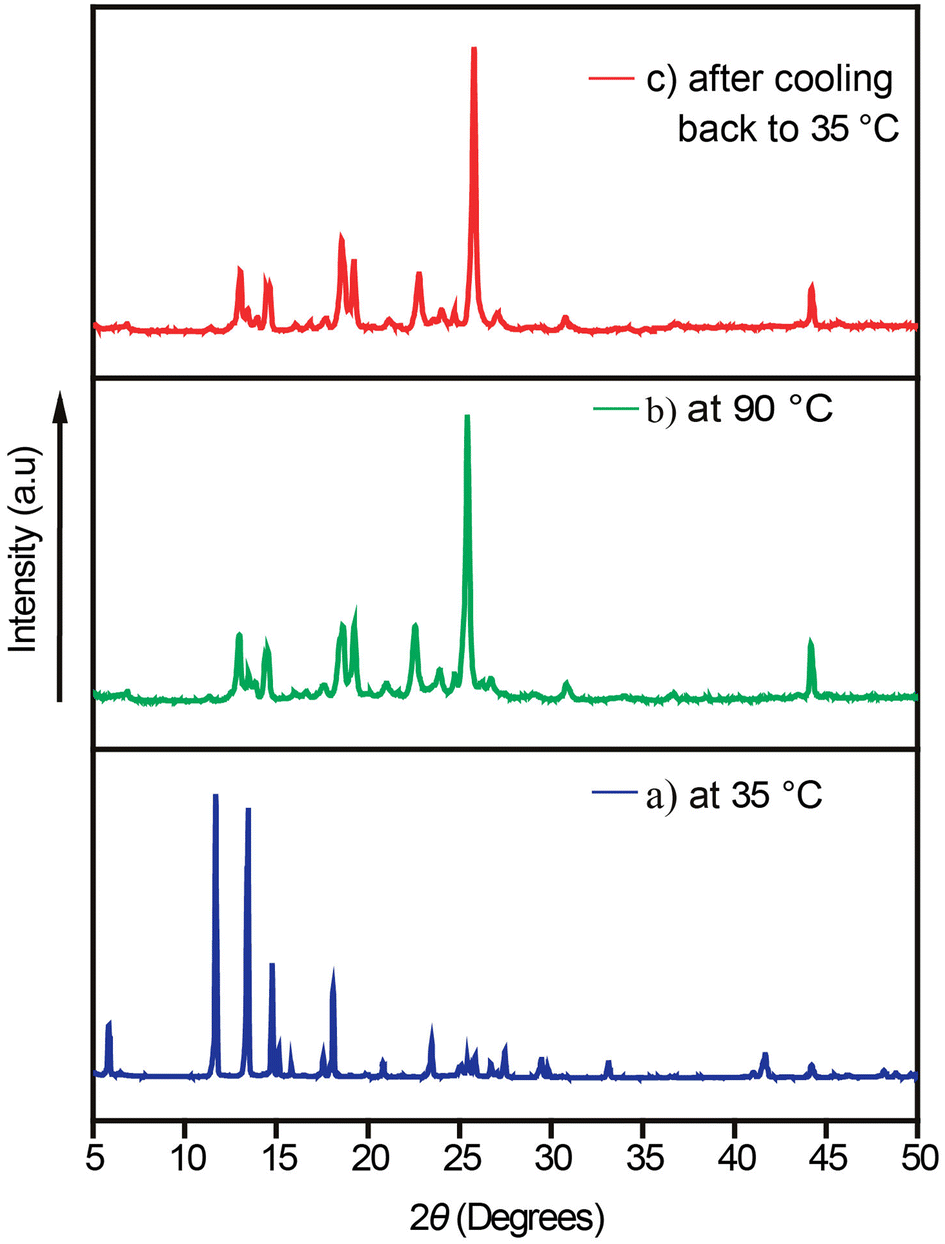 | ||
| Fig. 4 Variable temperature powder X-ray diffraction patterns of crystalline samples of the cocrystal. PXRD patterns (a) at 35 °C, (b) at 90 °C and (c) at 35 °C after cooling. | ||
The two polymorphs, polymorph 1, obtained by crystallization, and polymorph 2, obtained by heating polymorph 1, were further characterised by FT-IR spectroscopy, revealing clear differences in the CO and O–H stretching frequencies (Fig. S7†). To assess the stability, the crystals of the two polymorphs were first isolated and individual PXRD patterns were collected. The polymorphs were then subjected to solid-state grinding and solvent drop grinding using a few drops of acetonitrile solvent for 1 hour. The PXRD patterns of the ground materials of both polymorphs after solid-state and solvent drop grinding were then recorded and analysed. For polymorph 1, no significant changes were observed in the peak positions in the PXRD pattern (Fig. S8†). In contrast, the PXRD pattern of polymorph 2 changed, and the peak positions matched with those of polymorph 1 (Fig. S9†), indicating the higher thermodynamic stability of polymorph 1.
The polymorphic phase transition of the bulk material intrigued us to investigate the effect of heat on the single crystals. When a single crystal (polymorph 1) of the cocrystal is heated on a hot plate, it undergoes cracking and expansion during the phase transition at 70–75 °C along with the appearance of the naked-eye detectable growth phase (Fig. 5, Video S1†). The formation of nuclei first takes place, and then the growth phase of the crystal occurs, which leads to the transition of polymorph 1 to polymorph 2. The phase transformation generally arises from the corners of the crystals, cracked sites, twinned boundaries or defects in the single crystal. The speed of the growth phase is approximately 2.70 × 10−2 mm s−1, which is much slower than those observed in the martensitic phase transitions. During the phase transition, the crystals expand and crack at several sites (Fig. 5 and S10†), losing their integrity. The crystal structure of polymorph 2 could not be determined by single crystal X-ray diffraction due to the poor diffraction quality of the crystals after phase transition. Optical microscopy and SEM images of the crystal surfaces clearly showed the cracks that appeared after the phase transition (Fig. 5 and S10†). Among the 15 crystals studied under a thermal microscope, the maximum and minimum expansions (in%) exhibited by the crystals are 6.63 (l) and 13.54 (w), and 0.87 (l) and 0.90 (w), respectively (Table S2†).
 | ||
| Fig. 5 (a) The single crystal of the cocrystal, when heated from 25 °C to 75 °C, showing expansion along with the appearance of cracking on the crystal surface. SEM images of the crystal surface showing cracks (b) before and after (c) phase transition. | ||
We are particularly interested in the wettability of single crystal faces46,47 of thermally transformable materials, which could lead to wettability reversal and have applications in self-cleaning surfaces. The wettability of a surface by a liquid depends on several factors such as chemical composition, surface roughness, particle size, calculation method, etc. Single crystals are normally unsuitable for wettability studies due to the small surface areas of the crystal faces. However, the single crystals of the cocrystal of PP and BPA could be grown to sufficient sizes with a wider face (Fig. S11†), which can accommodate a 2 μL sized liquid droplet. To determine the wettability of the wider faces of the crystals of the two polymorphs, we have chosen two liquids of opposite polarities, viz. water and diiodomethane, and the contact angles were measured on 10 crystals each (Fig. S12 and S13†). It was found that the contact angles of water and diiodomethane on the wider face of polymorph 1 (average value, for water = 80.28° and diiodomethane = 33.05) in all the 10 crystals are higher compared to those of polymorph 2 (average value, for water = 71.58° and diiodomethane = 11.4) (Fig. 6, S12 and S13†). The surface energy of the single crystal of both polymorphs was determined by using the Owens–Wendt and Kaelble–Uy methods48–50 by using water and diiodomethane as probe liquids (Tables S3 and S4†). It was found that for polymorph 2, the surface energy is relatively higher than that of the single crystal of polymorph 1, which leads to more wettability in polymorph 2. The mean surface energy values obtained for polymorph 1 are 43.5 mJ m−2 (Owens–Wendt equation) and 42.98 mJ m−2 (Kaelble–Uy equation), and for polymorph 2, the values are 51.14 mJ m−2 (Owens–Wendt equation) and 50.5 mJ m−2 (Kaelble–Uy equation). This difference in wettability may be attributed to two factors: the exposure of functional groups on the wider face of the crystal and surface texture. The wider face of polymorph 1 is (001/00) (Fig. S11†), where the functional group CO, pyridine and alkyl chains are exposed (Fig. 6), resulting in a balance between hydrophilicity and hydrophobicity. On the other hand, after the polymorphic phase transition, the crystal surface did not become rough; instead, only cracks appeared, as visualized in the SEM images (Fig. 5c), along with an increase in the surface free energy of the wider faces. Hence, it may be concluded that the lowering of the contact angle value in polymorph 2 may be attributed to the exposure of hydrophilic centres due to the cracking of the crystal surface after the phase transformation. Another reason may be that after the phase transition, in polymorph 2, the hydrophilic groups such as O–H, pyridine and CO gain more exposure to the surface than the hydrophobic alkyl chains.
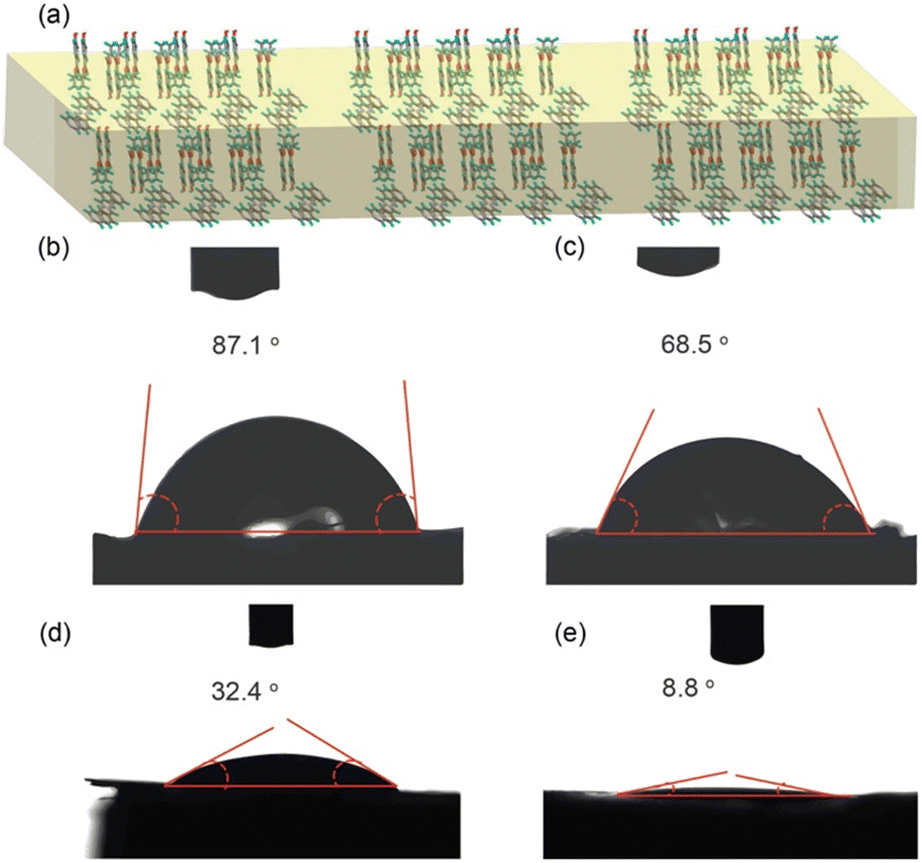 | ||
| Fig. 6 (a) The exposure of CO, pyridine, and alkyl chains on the wider face of the crystal. Contact angles of water for (b) polymorph 1 and (c) polymorph 2. The contact angles of diiodomethane for (d) polymorph 1 and (e) polymorph 2. | ||
Conclusions
Materials that exhibit thermomechanical properties are of particular interest, and hence, their characterization and optimization are important for future applications. The polymorphic crystal-to-crystal phase transition has been successfully demonstrated in a cocrystal composed of BPA and PP. The inherent irreversibility of the phase transformation facilitates the examination of the wider surface of the cocrystal before and after the phase transformation. Furthermore, during the phase transformation, naked-eye observable expansion and cracking of the crystal were observed. The changes in the wettability of the surface due to the polymorphic phase change also provide us with insight into their physical properties with temperature change. The precise mechanism of the phase transformation could not be determined due to the unavailability of structural data after the phase transformation. This study aims to enhance our comprehension of the intricate relationship between crystal polymorphism and material properties.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
The article is written with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PS and PG acknowledge NIT Meghalaya for the fellowship. NKN acknowledges SERB-CRG (file no: CRG/2019/004607) for funding. The Central Instrument Facility, NIT Meghalaya, the Department of Chemical and Biological Sciences of NIT Meghalaya and the Sophisticated Analytical Instrumentation Centre at Tezpur University provided the instrument facilities for this research.Notes and references
- A. J. Cruz-Cabeza and J. Bernstein, Chem. Rev., 2014, 114, 2170–2191 CrossRef CAS PubMed.
- A. Nangia, Acc. Chem. Res., 2008, 41, 595–604 CrossRef CAS PubMed.
- A. J. Cruz-Cabeza, S. M. Reutzel-Edens and J. Bernstein, Chem. Soc. Rev., 2015, 44, 8619–8635 RSC.
- D. Chopra, T. P. Mohan, K. S. Rao and T. N. G. Row, CrystEngComm, 2005, 7, 374–379 RSC.
- L. Bayés-García, T. Calvet, M. À. Cuevas-Diarte, E. Rovira, S. Ueno and K. Sato, Cryst. Growth Des., 2015, 15(8), 4045–4054 CrossRef.
- J. Bernstein and E. Goldstein, Mol. Cryst. Liq. Cryst., 1988, 164, 213–229 CrossRef CAS.
- G. Bolla and A. Nangia, Chem. Commun., 2016, 52, 8342–8360 RSC.
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha, M. L. Cheney, A. R. Choudhury, G. R. Desiraju, A. G. Dikundwar, R. Dubey, N. Duggirala, P. P. Ghogale, S. Ghosh, P. K. Goswami, N. R. Goud, R. R. K. R. Jetti, P. Karpinski, P. Kaushik, D. Kumar, V. Kumar, B. Moulton, A. Mukherjee, G. Mukherjee, A. S. Myerson, V. Puri, A. Ramanan, T. Rajamannar, C. Malla Reddy, N. Rodriguez-Hornedo, R. D. Rogers, T. N. G. Row, P. Sanphui, N. Shan, G. Shete, A. Singh, C. C. Sun, J. A. Swift, R. Thaimattam, T. S. Thakur, R. K. Thaper, S. P. Thomas, S. Tothadi, V. R. Vangala, N. Variankaval, P. Vishweshwar, D. R. Weyna and M. J. Zaworotko, Cryst. Growth Des., 2012, 12, 2147–2152 CrossRef CAS.
- D. Yan, A. Delori, G. O. Lloyd, T. Friščić, G. M. Day, W. Jones, J. Lu, M. Wei, D. G. Evans and X. Duan, Angew. Chem., Int. Ed., 2011, 50, 12483–12486 CrossRef CAS PubMed.
- Y. Ye, L. Gao, H. Hao, Q. Yin and C. Xie, CrystEngComm, 2020, 22, 8045–8053 RSC.
- N. J. Babu, P. Sanphui and A. Nangia, Chem. – Asian J., 2012, 7, 2274–2285 CrossRef CAS PubMed.
- S. Aitipamula, P. S. Chow and R. B. H. Tan, CrystEngComm, 2014, 16, 3451–3465 RSC.
- G. R. Desiraju and A. Nangia, Cryst. Growth Des., 2016, 16, 5585–5587 CrossRef CAS.
- A. V. Trask, Mol. Pharmaceutics, 2007, 4, 301–309 CrossRef CAS PubMed.
- Y. Abe, S. Karasawa and N. Koga, Chem. – Eur. J., 2012, 47, 15038–15048 CrossRef PubMed.
- H. Chung, S. Chen, N. Sengar, D. W. Davies, G. Garbay, Y. H. Geerts, P. Clancy and Y. Diao, Chem. Mater., 2019, 31, 9115–9126 CrossRef CAS.
- Q. Jiang, A. G. Shtukenberg, M. D. Ward and C. Hu, Cryst. Growth Des., 2015, 15, 2568–2573 CrossRef CAS.
- B. P. Krishnan and K. M. Sureshan, J. Am. Chem. Soc., 2015, 137, 1692–1696 CrossRef CAS PubMed.
- C. Ge, J. Liu, X. Ye, Q. Han, L. Zhang, S. Cui, Q. Guo, G. Liu, Y. Liu and X. Tao, J. Phys. Chem. C, 2018, 122, 15744–15752 CrossRef CAS.
- D. W. Davies, B. Seo, S. K. Park, S. B. Shiring, H. Chung, P. Kafle, D. Yuan, J. W. Strzalka, R. Weber, X. Zhu, B. M. Savoie and Y. Diao, Nat. Commun., 2023, 14, 1304 CrossRef CAS PubMed.
- D. Das, E. Engel and L. J. Barbour, Chem. Commun., 2010, 46, 1676–1678 RSC.
- K. Wang, C. Wang, M. K. Mishra, V. G. Young and C. C. Sun, CrystEngComm, 2021, 23, 2648–2653 RSC.
- H. Ito, M. Muromoto, S. Kurenuma, S. Ishizaka, N. Kitamura, H. Sato and T. Seki, Nat. Commun., 2013, 4, 2009 CrossRef PubMed.
- G. Liu, J. Liu, Y. Liu and X. Tao, J. Am. Chem. Soc., 2014, 136, 590–593 CrossRef CAS PubMed.
- D. P. Karothu, J. Weston, I. T. Desta and P. Naumov, J. Am. Chem. Soc., 2016, 138, 13298–13306 CrossRef CAS PubMed.
- T. Taniguchi, H. Sato, Y. Hagiwara, T. Asahi and H. Koshima, Commun. Chem., 2019, 2, 19 CrossRef.
- T. Seki, K. Sakurada, M. Muromoto and H. Ito, Chem. Sci., 2015, 6, 1491–1497 RSC.
- K. Omoto, T. Nakae, M. Nishio, Y. Yamanoi, H. Kasai, E. Nishibori, T. Mashimo, T. Seki, H. Ito, K. Nakamura, N. Kobayashi, N. Nakayama, H. Goto and H. Nishihara, J. Am. Chem. Soc., 2020, 142, 12651–12657 CrossRef CAS PubMed.
- H. Rawat, R. Samanta, B. Bhattacharya, S. Deolka, A. Dutta, S. Dey, K. B. Raju and C. M. Reddy, Cryst. Growth Des., 2018, 18, 2918–2923 CrossRef CAS.
- M. K. Panda, T. Runčevski, S. C. Sahoo, A. A. Belik, N. K. Nath, R. E. Dinnebier and P. Naumov, Nat. Commun., 2014, 5, 4811 CrossRef CAS PubMed.
- A. Khalil, E. Ahmed and P. Naumov, Chem. Commun., 2017, 53, 8470–8473 RSC.
- S. Mittapalli, D. S. Perumalla, J. B. Nanuboluc and A. Nangia, IUCrJ, 2017, 4, 812–823 CrossRef CAS PubMed.
- Y. Chen, J. Zhang, J. Zhang and X. Wan, J. Am. Chem. Soc., 2024, 146, 9679–9687 CrossRef CAS PubMed.
- S. C. Sahoo, M. K. Panda, N. K. Nath and P. Naumov, J. Am. Chem. Soc., 2013, 135, 12241–12251 CrossRef CAS PubMed.
- K. Takazawa, J.-I. Inoue and Y. Matsushita, Small, 2022, 18, 2204500 CrossRef CAS PubMed.
- M. Dharmarwardana, R. P. Welch, S. Kwon, V. K. Nguyen, G. T. McCandless, M. A. Omary and J. J. Gassensmith, Chem. Commun., 2017, 53, 9890–9893 RSC.
- P. Gupta, D. P. Karothu, E. Ahmed, P. Naumov and N. K. Nath, Angew. Chem., Int. Ed., 2018, 57, 8498–8502 CrossRef CAS PubMed.
- T. Minami, H. Sato and S. Matsumoto, CrystEngComm, 2018, 20, 2644–2647 RSC.
- D. P. Karothu, R. Ferreira, G. Dushaq, E. Ahmed, L. Catalano, J. M. Halabi, Z. Alhaddad, I. Tahir, L. Li, S. Mohamed, M. Rasras and P. Naumov, Nat. Commun., 2022, 13, 2823 CrossRef CAS PubMed.
- M. Dharmarwardana, S. Pakhira, R. P. Welch, C. Caicedo-Narvaez, M. A. Luzuriaga, B. S. Arimilli, G. T. McCandless, B. Fahimi, J. L. Mendoza-Cortes and J. J. Gassensmith, J. Am. Chem. Soc., 2021, 143, 5951–5957 CrossRef CAS PubMed.
- J. D. Dunitz, Pure Appl. Chem., 1991, 63, 177–185 CrossRef CAS.
- Y. V. Mnyukh, Mol. Cryst. Liq. Cryst., 1979, 52, 163–199 CrossRef.
- S. K. Park and Y. Diao, Chem. Soc. Rev., 2020, 49, 8287–8314 RSC.
- G. Liu, J. Liu, X. Ye, L. Nie, P. Gu, X. Tao and Q. Zhang, Angew. Chem., Int. Ed., 2016, 56, 198–202 CrossRef PubMed.
- N. K. Nath, M. Hazarika, P. Gupta, N. R. Ray, A. K. Paul and E. Nauha, J. Mol. Struct., 2018, 1160, 20 CrossRef CAS.
- S. S. Borah, M. Khan, P. Gogoi, N. Kalita, R. Thakuria and N. K. Nath, Chem. - Asian J., 2024, 19, e202301090 CrossRef CAS PubMed.
- D. Kitagawa and S. Kobatake, Chem. Sci., 2012, 3, 1445 RSC.
- M. Annamalai, K. Gopinadhan, S. A. Han, S. Saha, H. J. Park, E. B. Cho, B. Kumar, A. Patra, S.-W. Kim and T. Venkatesan, Nanoscale, 2016, 8, 5764 RSC.
- D. K. Owens and R. C. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741 CrossRef CAS.
- D. H. Kaelble, J. Adhes., 1970, 2, 66 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data for the cocrystal BPA + PP have been deposited at the CCDC under deposition number 2345382. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00407h |
| This journal is © The Royal Society of Chemistry 2024 |