
Crystal engineering of monometallic lanthanide(III) supramolecular systems within the N3-tridentate hydrazone Schiff-base ligand†
Dominika
Prętka
 *a,
Dawid
Marcinkowski
a,
Agnieszka
Siwiak
a,
Maciej
Kubicki
a,
Giuseppe
Consiglio
b,
Violetta
Patroniak
a and
Adam
Gorczyński
*a
*a,
Dawid
Marcinkowski
a,
Agnieszka
Siwiak
a,
Maciej
Kubicki
a,
Giuseppe
Consiglio
b,
Violetta
Patroniak
a and
Adam
Gorczyński
*a
aFaculty of Chemistry, Department of Functional Nanostructure Synthesis, Adam Mickiewicz University in Poznań, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland. E-mail: dominika.pretka@amu.edu.pl; adam.gorczynski@amu.edu.pl
bDipartimento di Scienze Chimiche, Università di Catania, I-95125 Catania, Italy
First published on 4th July 2024
Abstract
A comprehensive series of mononuclear Ln(III) complexes based on N3-tridentate Schiff-base ligand L have been synthesized using triflate or nitrate lanthanide(III) metal salts. X-ray structural characterization shows that the obtained complexes exhibit a variety of coordination modes around the Ln(III) centers tuned through appropriate counterions and reaction conditions. Examination of the above-mentioned factors reveals that, in the case of all lanthanide assemblies, we observe anion-driven structural diversity. Tuning of the crystal structure of the formed supramolecular systems is also highly influenced by the lanthanide contraction phenomenon, directly resulting in a series of isostructural groups, with general formulae of [ML2(OTf)nX3−n](OTf)3−n, if n = 2, X = MeCN (except for Yb), and if n = 1, X = MeOH in the triflate series, and [ML(NO3)3(X)](Y), where X and Y represent MeOH, MeCN or H2O, respectively, in the nitrate series. The present study gives insight into the lanthanide contraction phenomenon within the tridentate N-heterocyclic coordination architectures of Ln(III) systems and can serve for targeted manipulation of properties through molecular tectonics and crystal engineering aspects in the domains, where subtle changes of the Ln(III) coordination spheres are important like molecular magnetism or catalysis.
Introduction
Self-assembly and self-organization of individual molecules into highly ordered supramolecular systems with specific arrangements of separate molecular moieties forming given assemblies are essential processes in supramolecular chemistry.1–4 However, while these phenomena are well-recognized in many scientific fields and for numerous chemical compounds,5–12 it remains a challenge to control the characteristics and complexity of creating supramolecular systems. Consequently, various factors that could significantly impact self-sorting and self-assembly events such as external variables (reaction conditions, temperature, concentration, solvent, stoichiometry, pH, etc.) and so-called ‘molecular codes’ (size and shape, steric effect, hydrogen bond complementarity, charge transfer and coordination sphere)13 have been studied over many years. The aim is to gain better insights into possibilities of forming a wide range of structures for final supramolecular architectures, related to the presence of different intermolecular forces like hydrogen bonds, metal–ligand interactions, or other weak non-covalent interactions such electrostatic, van der Waals and π-interactions.14–18 Therefore, the above-mentioned dependencies are crucial when it comes to the rational design of supramolecular systems, especially concerning their related properties resulting from their rich structural diversity. Hence, it is necessary to continue exploring such influences to better understand the interactions arising at the molecular level. In the literature, numerous examples of different coordination compounds exist, whose properties depend entirely on their crystal structure motifs. The specific influence of the coordination sphere around the metal ions has been widely studied recently, notably in terms of magnetic behavior characteristics, where slow magnetic relaxation processes can be tuned by the coordination geometry determined by (i) the ligand field, (ii) the presence of additional coordinated solvent molecules, (iii) coordination anion effects, (iv) electrostatic effects, e.g. the symmetry of charge distribution around the metal ions, or by (v) guest molecules, e.g. the lattice solvents.19–25 Among various types of ligand molecules that display flexibility and structural tunability, our attention has focused on hydrazone-type ligands, which continuously remain of great interest due to facile synthesis methods and subsequent formation of metallosupramolecular architectures with multifunctional characteristics. Considering the design of metal complexes based on f-electron metal ions, the lanthanide contraction phenomenon has been taken into consideration for its influence on preferences for coordination motifs of individual metal ions in the presence of different coordinating ligand molecules. Additionally, lanthanide contraction can directly influence properties, which is another crucial aspect in the field of coordination chemistry, where structural diversity is a determining factor of possible applications of Ln(III) supramolecular systems.26,27 The complexes for the further study of luminescence/magnetism properties among different coordination modes/metal ions have been synthesized based on the following aspects. The chemistry of lanthanide-based assemblies is constantly being developed towards fascinating coordination properties of lanthanide ions with the possibility of creating various supramolecular architectures (mononuclear/polynuclear systems) and also their intriguing photophysical and magnetic properties, which determine their possibilities of multifunctional applications related to NIR emission, CPL studies, optical probes and sensing/imaging materials, host–guest chemistry featuring luminescence characteristics or storing and processing information at the molecular level featuring magnetic properties.28The N3-tridentate hydrazone Schiff-base ligand has been structurally designed in a way to minimize the possibility of the creation of more complicated systems such as hydrogen bonded supramolecular networks, which could have been made possible due to the introduction of methyl groups on nitrogen atoms into the ligand scaffold, which blocked such types of supramolecular interactions and formed monometallic, single molecule complexes. This is especially directed in the aspects of designed complexes with magnetic properties and possible applications as effective Single-Ion Magnets. Based on our experience and the literature knowledge, we suppose that designed monometallic lanthanide complexes based on hydrazone ligand L will demonstrate wide tuning of properties with slight structural changes. This supposition is supported by known examples of such complexes based on similar ligands with characteristics like magnetic behavior,29–31 biomimetic systems,32 biological activities,33,34 catalytic35–37 and luminescence properties.38,39 Previously, we demonstrated that monometallic complexes of Er(III) and Yb(III) with ligand L can exhibit field-induced Single-Ion Magnet behavior, resulting from the appropriate tuning of the ligand donor atoms around the prolate lanthanide(III) ions.40 Due to the importance of these aspects in observing slow magnetic relaxation and the temperature at which it occurs, we decided to investigate how the parent composition of these complexes changes within the same ligand scaffold. In our investigation, we will consider: (i) the lanthanide contraction phenomenon, (ii) the influence of counterions and (iii) synthetic conditions.
Here, we report the synthesis of monometallic lanthanide complexes with tridentate ligand L, using a number of triflate or nitrate lanthanide(III) metal salts, with the aim of structurally characterizing them and examining the influence of utilizing appropriate counterions/lanthanide ions on tuning the crystal structure of the formed supramolecular systems. All of the complexes were characterized via FT-IR and mass spectra analysis, particularly emphasizing single crystal X-ray diffraction studies so that even subtle changes in the supramolecular compound architectures can be discerned as a function of the Ln(III) salt as well as the reaction conditions. We observe multi-level control of the structure of simple Ln–L complexes, finding that the crystal structures of these assemblies clearly depend on several factors used during the synthesis (Fig. 1). Firstly, the counterion chosen during the synthesis directly influenced the stoichiometry of the ligand to metal ratio of the obtained complexes – 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in the triflate series or 1:1 in the nitrate series. Secondly, the choice of the lanthanide(III) ion affects the specific coordination sphere of the central ion, mainly coordinating a different number of triflate anions, different coordination modes – monodentate or bidentate of the nitrate counterions, or preferences of the Ln(III) center to be coordinated by the different donor atoms from various solvent molecules, as a consequence of lanthanide contraction. These detailed dependences are essential for designing supramolecular systems with the desired structure and for fine-tuning the properties of the formed lanthanide(III) complexes at the molecular level.
:1 in the triflate series or 1:1 in the nitrate series. Secondly, the choice of the lanthanide(III) ion affects the specific coordination sphere of the central ion, mainly coordinating a different number of triflate anions, different coordination modes – monodentate or bidentate of the nitrate counterions, or preferences of the Ln(III) center to be coordinated by the different donor atoms from various solvent molecules, as a consequence of lanthanide contraction. These detailed dependences are essential for designing supramolecular systems with the desired structure and for fine-tuning the properties of the formed lanthanide(III) complexes at the molecular level.
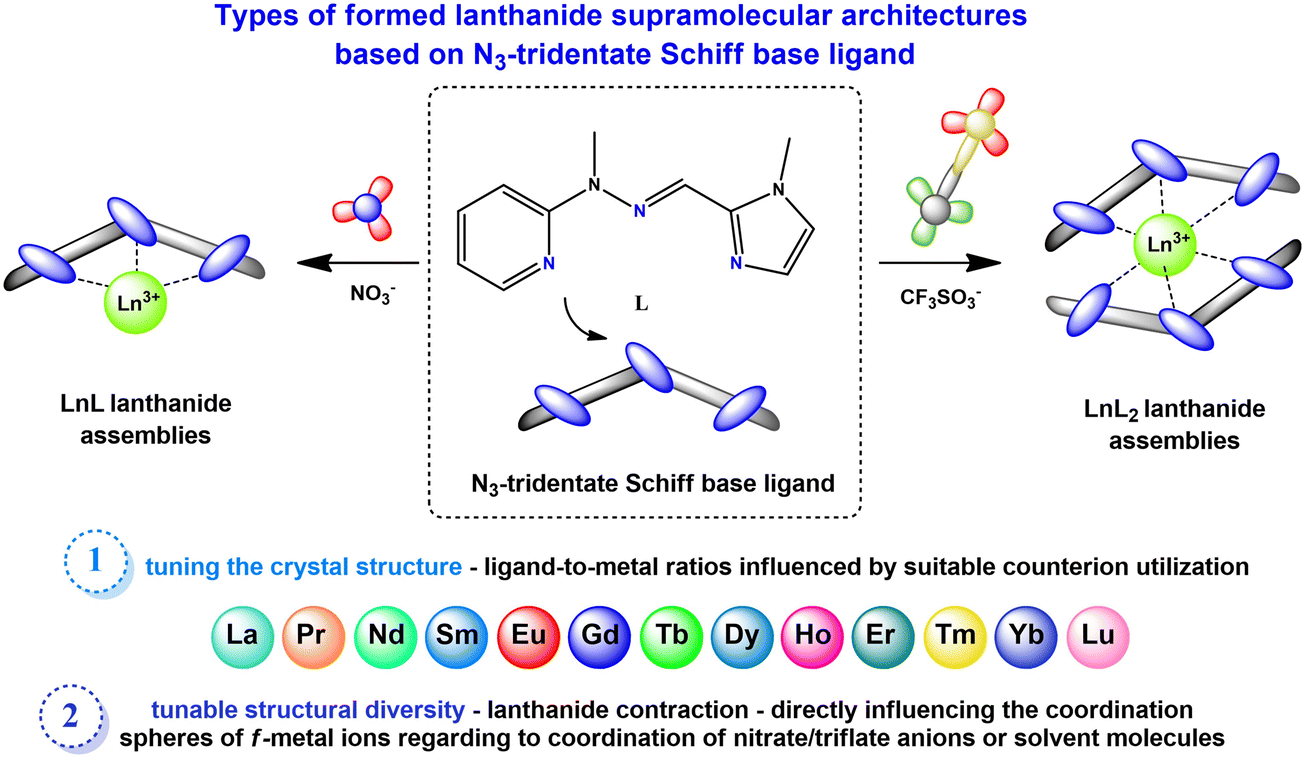 | ||
| Fig. 1 The coordination behavior of the N3-tridentate Schiff-base ligand L with various lanthanide(III) salts influences the formation of supramolecular architectures. Schematic representations illustrate the rich structural diversity resulting from the influence of variable factors. | ||
Experimental
Reagents and physical measurements
Organic compounds, metal salts and solvents were of analytical grade quality and were supplied by Aldrich. All manipulations were performed under aerobic conditions using solvents and chemicals as received unless otherwise stated. ESI mass spectra of methanolic solutions ∼10−4 M were determined using a Waters Micromass ZQ spectrometer. IR spectra were obtained with a Bruker FT-IR IFS 66/s spectrophotometer and peak positions are reported in cm−1. All NMR spectra were recorded on a Bruker-600 MHz-AvanceNEO spectrometer.Synthesis of the lanthanide complexes
Ligand L was prepared as reported by us previously.41 However, we obtained a single crystal suitable for X-ray analysis in another form of the ligand, upon transformation to its hydrobromide salt and using slow diffusion of iPr2O into the methanolic solution at 4 °C. Changing the ratio of L:Ln(III) salt has not resulted in compounds with different stoichiometry but only to a decrease in the overall yield. Therefore, the presented procedure allows for the most efficient synthesis of compounds 1–25. The molar ratio of the ligand to the corresponding metal salt for complexes 1–12 was 2:1. For complexes 13–25, the molar ratio of the ligand to the corresponding salt was 1:1. Complexes 10, 11, 22 and 24 were obtained by us previously40 and for the sake of completeness, they are presented here as well. Synthetic procedure details and characterization of the lanthanide complexes are depicted in the ESI.†
X-ray crystallography
Diffraction data were collected by the ω-scan technique on Agilent Technologies four-circle diffractometers: for 1, 7a, 19 and 23 at 295(2) K on a SuperNova with an Atlas CCD detector, equipped with a Nova microfocus CuKα radiation source (λ = 1.54178 Å), for 2, 5, 6, 11, 15 and 20 at room temperature, for 9, 13, 16, 17, 18, 22 and 24 at 120(1) K, for 2a, 3, 4, 7, 8, 10, 14 and 21 at 100(1) K on an Xcalibur with an Eos CCD detector and graphite-monochromated MoKα radiation (λ = 0.71069 Å), for Tb-perchlorate using graphite-monochromated MoKα radiation (λ = 0.71073 Å), at 100(1) on a Rigaku XCalibur four-circle diffractometer with an EOS CCD detector and for LH·Br− and Eu-hydrogen at 130(1) K, and for 25 at room temperature, with mirror-monochromated CuKα radiation (λ = 1.54178 Å), at 100(1) on a Rigaku SuperNova four-circle diffractometer with an Atlas CCD detector. The data were corrected for Lorentz-polarization as well as for absorption effects.42 Precise unit-cell parameters were determined by a least-squares fit of reflections of the highest intensity, chosen from the whole experiment. The structures were solved with SIR92 (ref. 43) and refined with the full-matrix least-squares procedure on F2 using SHELXL-2013.44 The scattering factors incorporated in SHELXL97 were used. All non-hydrogen atoms were refined anisotropically, and hydrogen atoms in the complexes as well as methyl and hydroxyl hydrogens in LH·Br− were placed in idealized positions and refined as ‘riding models’ with isotropic displacement parameters set at 1.2 (1.5 for methyl or hydroxyl groups) times Ueq of appropriate carrier atoms. In the structures of 7, 8, 10, and 11, voids filled with diffused electron density were found; as the modelling of solvent molecules was in these cases unsuccessful, the SQUEEZE procedure45 was applied. Crystals of 17, 22 and 24 were found to be two-component twins and this was taken into account both in data reduction and structure refinement. The crystallographic data and parameters are presented in Tables S1–S3.† Values of the Flack parameter46 for 2–5 (Pca21) do not allow the absolute structure to be determined. It should be noted, however, that due to the presence of mirror planes in the point-group symmetry, both the molecule and its mirror image are present in the crystal structures. Large values of the Flack parameter may be caused not only by experimental reasons but also by pseudo-centrosymmetry of the structures (80–90%). Crystallographic data (excluding structure factors) for the structural analysis have been deposited with the Cambridge Crystallographic Data Centre, no. CCDC-2338931 (LH·Br−), CCDC-1005948 (1), CCDC-1005949 (2), CCDC-1052303 (2a), CCDC-1052299 (3), CCDC-1005950 (4), CCDC-1005951 (5), CCDC-1005952 (6), CCDC-1052307 (7), CCDC-1052306 (7a), CCDC-1052308 (8), CCDC-1435578 (9), CCDC-1052304 (10), CCDC-1435579 (11), CCDC-1435580 (13), CCDC-1052302 (14) CCDC-1435581 (15), CCDC-1435582 (16), CCDC-1435583 (17), CCDC-1435584 (18), CCDC-1052300 (19), CCDC-1006326 (20), CCDC-1052301 (21), CCDC-1435585 (22), CCDC-1052305 (23), CCDC-1435586 (24), CCDC-2338933 (25), CCDC-2338932 (Tb-perchlorate) and CCDC-2338934 (Eu-hydrogen).Results and discussion
Synthesis of monometallic lanthanide complexes
The reaction between the N3-tridentate hydrazone Schiff-base ligand L and trivalent lanthanide salts (Ln(CF3SO3)3 or Ln(NO3)3) in a mixture of acetonitrile/methanol at room temperature leads to isolation of various monometallic lanthanide(III) complexes, which can be divided into several groups depending on the structure formed according to the following general formulae: [ML2(OTf)nX3−n](OTf)3−n, where n is 1, 2 or 3 respectively and X denotes the coordinating solvent molecules or [ML(NO3)3(X)](Y), where X and Y denote the coordinating solvent molecules. The triflate-based systems exhibit a 2:1 ligand-to-metal ion ratio, while the nitrate systems demonstrate a 1:1 ratio, noting that eventual variations do not change the composition of the complex, only the final yield of the isolated architectures. Specifically, the triflate group can be divided into 4 sub-groups: (i) [ML2(OTf)3] n = 3 (M – La, Pr, Nd, Sm, Eu, Gd – 1–6); (ii) [ML2(OTf)2(MeCN)](OTf) n = 2 (M – Tb – 7a; Ho – 9); (iii) [ML2(OTf)(MeOH)2](OTf)2n = 1 (Tb – 7; Dy – 8; Er – 10); (iv) [YbL2(OTf)2](OTf) – 11, where lanthanide contraction is the primary factor for variation of counterion/solvent molecules in the first/second coordination sphere of the Ln polyhedron. The nitrate group is less diversified, with 2 sub-groups: (i) [ML(NO3)3(X)](Y), where all nitrates remain as bidentate modes (M – La, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho – 13–21); (ii) [ML(NO3)3(X)](Y), where one of the nitrate anions is present in the monodentate mode (M – Er – 22, Yb – 24, Lu – 25), while X and Y denote solvent molecules: MeOH, MeCN, H2O or DMSO, respectively. All complexes have been fully characterized by spectroscopic (FT-IR) and spectrometric (ESI-MS) methods, followed by X-ray crystallography, clearly indicating the formation of specific assemblies. The infrared spectra (Fig. S1†) and mass spectra analysis (Fig. S2†) for one complex from both the nitrate and triflate series are presented as representative examples. Analysis of the IR spectra (Fig. S1†) of complexes from both the triflate (complex 8) and nitrate series (complex 20) confirms the presence of the Schiff-base ligand within the structure by the appearance of bands in the range from 1438–1322 cm−1 attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching mode. The bands at 3404 cm−1 (8) and 3374 cm−1 (20) are assigned to the O–H stretching mode of the methanol molecule present in the crystal structure of the complexes, coordinated or not coordinated to the Ln(III) center. In the IR spectrum of complex 8, characteristic bands indicating the presence of triflate anions are observed. Specifically, the ν(SO3) and ν(CF3) asymmetric stretching bands appeared at 1303 cm−1 and 1231–1222 cm−1, respectively, while the ν(SO3) and ν(CF3) symmetric stretching bands were observed at 1169 cm−1 and 1029 cm−1. For complex 20, the spectrum displays vibrations indicating the presence of coordinated nitrate groups. The asymmetric stretching band ν(NO2) appeared at 1497 cm−1, the symmetric stretching band ν(NO2) ranged from 1291–1238 cm−1 and the band at 1049 cm−1 was attributed to the ν(NO) stretching from nitrate anions. The mass spectra of terbium(III) complex 7 with triflate counterions (red frame) and complex 19 with nitrate counterions (blue frame) showed distinct mass fragmentation patterns with different ion peak intensities, where we can distinguish peaks originating from the protonated ligand molecule and lanthanide complexes with variable arrangements of ligand molecules, counterions or solvent molecules (Fig. S2†). Considering representative examples of mass spectrum analyses for Tb(III) complexes with different counterions and the synthesized lanthanide series, it is suggested that nitrate analogues with only one coordinating ligand L are more stable than triflate complexes. In some cases, the intensity of the signal from the protonated ligand molecule – 216 [LH]+ is less intense in nitrate complexes, but more notably, the intensity of signals from complex entities – 315 (100) [TbL(NO3)3(H2O)2(CH3OH) + 2H+]2+ and 498 (45) [TbL(NO3)2]+ is higher than in triflate analogues – 554 (20) [TbL(CF3SO3)(CH3OH)–H+]+ and 887 (15) [TbL2(CF3SO3)2]+. Characterization for the diamagnetic Lu(III) triflate (12) and Lu(III) nitrate (25) complexes as representative examples was further supported by 1H NMR spectroscopy. The investigated 1H NMR spectra of both complexes clearly show significant shifts in proton signals compared to the 1H NMR spectrum of ligand L recorded in acetonitrile-d3 (Fig. S3†). The notable shifts were observed for protons from the pyridine: H1–H4 (downfield), imine: H6 (downfield) and imidazole moieties: H8 (upfield) and H9 (downfield), which additionally confirms the coordination of the lanthanide(III) ions in both triflate and nitrate Lu(III) compounds through nitrogen atoms from the pyridine, imine and imidazole moieties from the ligand molecule. Comparing proton signals in the NMR spectra of complexes 12 and 25, we are able to observe slight differences in shifts, suggesting that in the case of these two complexes, different systems are formed. In the case of the nitrate complex (25), it is a 1:1 ligand-to-metal ratio system, which has been additionally confirmed by the obtained crystal structure, where the lutetium(III) ion is coordinated by one ligand molecule, three nitrate counterions and an additional DMSO molecule. In the case of the triflate complex (12), as the spectrum differs from the representative spectrum of the nitrate analogue of the 1:1 system, we can predict that in this complex, two ligand molecules take part in the coordination of the lanthanide(III) ion, as has been observed in other crystal structures of the triflate complexes through the whole lanthanide series synthesized here.
N stretching mode. The bands at 3404 cm−1 (8) and 3374 cm−1 (20) are assigned to the O–H stretching mode of the methanol molecule present in the crystal structure of the complexes, coordinated or not coordinated to the Ln(III) center. In the IR spectrum of complex 8, characteristic bands indicating the presence of triflate anions are observed. Specifically, the ν(SO3) and ν(CF3) asymmetric stretching bands appeared at 1303 cm−1 and 1231–1222 cm−1, respectively, while the ν(SO3) and ν(CF3) symmetric stretching bands were observed at 1169 cm−1 and 1029 cm−1. For complex 20, the spectrum displays vibrations indicating the presence of coordinated nitrate groups. The asymmetric stretching band ν(NO2) appeared at 1497 cm−1, the symmetric stretching band ν(NO2) ranged from 1291–1238 cm−1 and the band at 1049 cm−1 was attributed to the ν(NO) stretching from nitrate anions. The mass spectra of terbium(III) complex 7 with triflate counterions (red frame) and complex 19 with nitrate counterions (blue frame) showed distinct mass fragmentation patterns with different ion peak intensities, where we can distinguish peaks originating from the protonated ligand molecule and lanthanide complexes with variable arrangements of ligand molecules, counterions or solvent molecules (Fig. S2†). Considering representative examples of mass spectrum analyses for Tb(III) complexes with different counterions and the synthesized lanthanide series, it is suggested that nitrate analogues with only one coordinating ligand L are more stable than triflate complexes. In some cases, the intensity of the signal from the protonated ligand molecule – 216 [LH]+ is less intense in nitrate complexes, but more notably, the intensity of signals from complex entities – 315 (100) [TbL(NO3)3(H2O)2(CH3OH) + 2H+]2+ and 498 (45) [TbL(NO3)2]+ is higher than in triflate analogues – 554 (20) [TbL(CF3SO3)(CH3OH)–H+]+ and 887 (15) [TbL2(CF3SO3)2]+. Characterization for the diamagnetic Lu(III) triflate (12) and Lu(III) nitrate (25) complexes as representative examples was further supported by 1H NMR spectroscopy. The investigated 1H NMR spectra of both complexes clearly show significant shifts in proton signals compared to the 1H NMR spectrum of ligand L recorded in acetonitrile-d3 (Fig. S3†). The notable shifts were observed for protons from the pyridine: H1–H4 (downfield), imine: H6 (downfield) and imidazole moieties: H8 (upfield) and H9 (downfield), which additionally confirms the coordination of the lanthanide(III) ions in both triflate and nitrate Lu(III) compounds through nitrogen atoms from the pyridine, imine and imidazole moieties from the ligand molecule. Comparing proton signals in the NMR spectra of complexes 12 and 25, we are able to observe slight differences in shifts, suggesting that in the case of these two complexes, different systems are formed. In the case of the nitrate complex (25), it is a 1:1 ligand-to-metal ratio system, which has been additionally confirmed by the obtained crystal structure, where the lutetium(III) ion is coordinated by one ligand molecule, three nitrate counterions and an additional DMSO molecule. In the case of the triflate complex (12), as the spectrum differs from the representative spectrum of the nitrate analogue of the 1:1 system, we can predict that in this complex, two ligand molecules take part in the coordination of the lanthanide(III) ion, as has been observed in other crystal structures of the triflate complexes through the whole lanthanide series synthesized here.
Single crystal X-ray analysis
:1 ligand-to-metal ratio systems. This is due to the strong affinity of lanthanide ions to oxygen atoms and the strong preference of Ln(III) centers to adopt a coordination number of 9 or 10 in such systems. The coordinated nitrate ions (usually in bidentate mode) then push the second ligand molecule out of the coordination sphere of lanthanide(III) ions and form stable complexes. As a result, the coordination sites are filled with only one ligand molecule, three nitrate counterions and one solvent molecule, in contrast to the triflate series, where two ligand molecules to a metal ion are always coordinated.
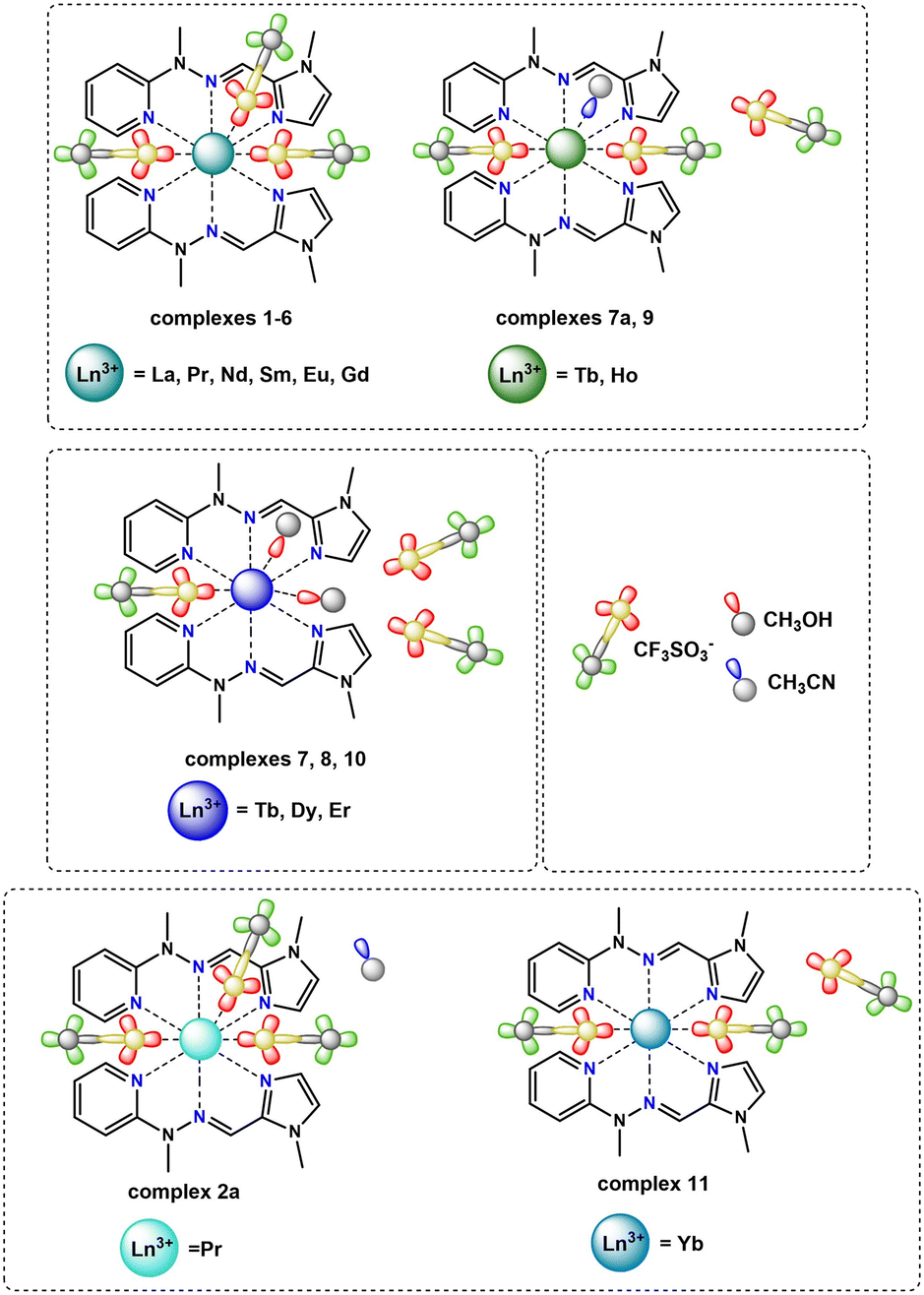 | ||
| Fig. 2 Coordination modes present in the crystal structures of monometallic complexes from the triflate series. | ||
The complexes forming specific groups (1–3) are isomorphous and crystallize in identical space groups: orthorhombic Pca21 (complexes 1–6), monoclinic P21/c (complexes 7a, 9) and monoclinic P21/n (complexes 7, 8, 10), respectively. The other two complexes crystallize in monoclinic P21 and triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space groups for complex 2a and 11. Molecular structures for representative isostructural and single complexes from the triflate series are provided in Fig. S4.† For all lanthanide(III) complexes from this series, some similarities and general differences are observable, which will be described upon in detail. When the triflate lanthanide(III) salts are used during the synthesis, the formed complexes always consist of the Ln(III) center coordinated by six nitrogen atoms originating from pyridine (atoms N1A/B), imine (atoms N8A/B) and imidazole (atoms N14A/B) groups – three from each of the two ligand molecules. Generally, the remaining coordination sites are filled by oxygen atoms from triflate anions or, in some cases, donor atoms of oxygen/nitrogen from solvent molecules (methanol, acetonitrile). As a result, the coordination number is mostly 9, except for the complex with the Yb(III) ion (11), where a lower coordination number of 8 is observed. The choice of different lanthanide(III) ions also influences the coordination modes present in monometallic complexes. Apart from the coordination of two ligand molecules to the metal ion, the coordination spheres are further filled by various additional molecules (counterions or solvent molecules), with their arrangement being a fundamental distinction in these supramolecular systems. In general, four groups of compounds can be identified, where metal ions are coordinated by the same donor atoms. This diversity in coordination modes for selected lanthanide ions is represented in Fig. 3a–d, shown as representative examples.
space groups for complex 2a and 11. Molecular structures for representative isostructural and single complexes from the triflate series are provided in Fig. S4.† For all lanthanide(III) complexes from this series, some similarities and general differences are observable, which will be described upon in detail. When the triflate lanthanide(III) salts are used during the synthesis, the formed complexes always consist of the Ln(III) center coordinated by six nitrogen atoms originating from pyridine (atoms N1A/B), imine (atoms N8A/B) and imidazole (atoms N14A/B) groups – three from each of the two ligand molecules. Generally, the remaining coordination sites are filled by oxygen atoms from triflate anions or, in some cases, donor atoms of oxygen/nitrogen from solvent molecules (methanol, acetonitrile). As a result, the coordination number is mostly 9, except for the complex with the Yb(III) ion (11), where a lower coordination number of 8 is observed. The choice of different lanthanide(III) ions also influences the coordination modes present in monometallic complexes. Apart from the coordination of two ligand molecules to the metal ion, the coordination spheres are further filled by various additional molecules (counterions or solvent molecules), with their arrangement being a fundamental distinction in these supramolecular systems. In general, four groups of compounds can be identified, where metal ions are coordinated by the same donor atoms. This diversity in coordination modes for selected lanthanide ions is represented in Fig. 3a–d, shown as representative examples.
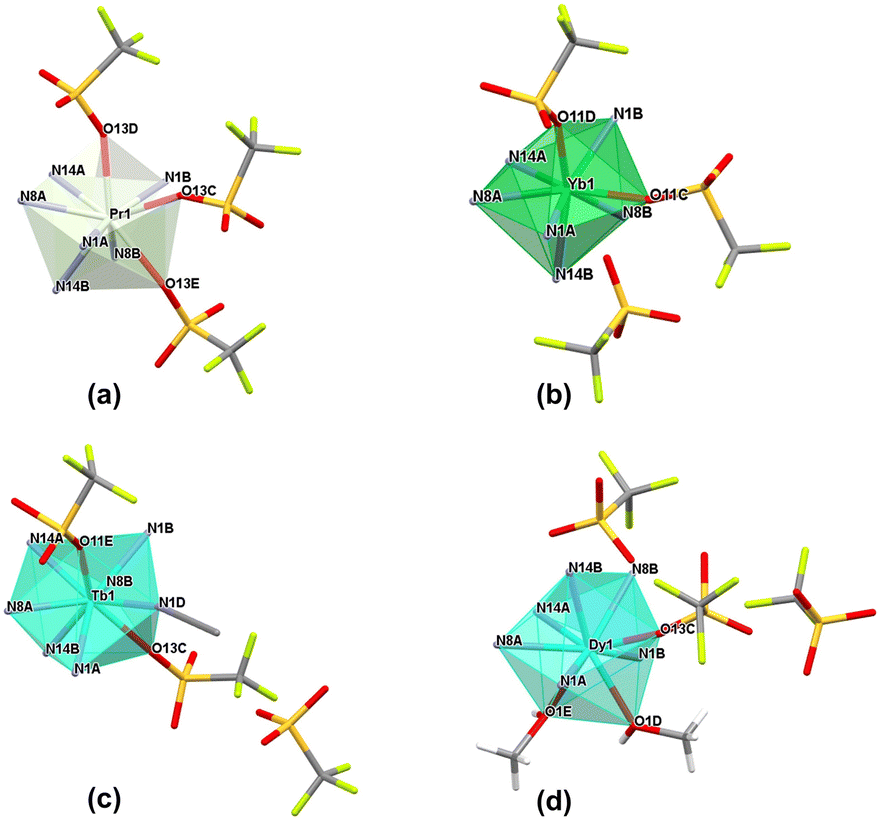 | ||
| Fig. 3 Coordination polyhedron representation for (a) Pr1 (2), (b) Yb1 (11), (c) Tb1 (7a) and (d) Dy1 (8) as representative examples of coordination modes present in monometallic lanthanide complexes in the triflate series, considering coordinating to the lanthanide ions oxygen atoms from methanol or triflate molecules and nitrogen atoms from acetonitrile and ligand molecules; for clarity, only coordinating nitrogen atoms (N1A/B, N8A/B and N14A/B) from the ligand molecule are shown, with the rest of the molecule omitted. | ||
When we get insight into the isostructural groups, we can observe common features among cationic complexes with terbium (7, 7a), holmium (9), dysprosium (8) and erbium (10) metal ions. These complexes exhibit crystal structures that also contain diffuse electron density regions, probably filled by the disordered water molecules, which is not observed in the case of neutral complexes with other lanthanide complexes from the triflate series. Based on the crystal structures of the obtained triflate complexes, generally there are compounds where besides nitrogen atoms from the ligand molecule, coordination sites are filled differently: 1) three oxygen atoms from monocoordinated CF3SO3− anions are present (complexes 1–6), 2) only two oxygen atoms from monodentate CF3SO3− anions are observed (complex 11), 3) two oxygen atoms from monodentate CF3SO3− anions and an additional nitrogen atom from the acetonitrile molecule are found (complexes 7a, 9) and 4) solely one oxygen atom from a monodentate CF3SO3− anion and two additional oxygen atoms from methanol molecules are identified (complexes 7, 8, 10). Therefore, for most systems in this series, we can observe the relationship that if three triflate anions present in the structure of the complex participate in the coordination lanthanide ion, the coordination sphere remains unoccupied by any other solvent molecules. The only exception to this observation occurs in complex 2a, where the crystal structure shows an additional acetonitrile molecule, not coordinated to the central ion. According to this, for the complex with the praseodymium(III) ion, two similar supramolecular systems were obtained. This difference directly results from the varied selection of crystallization conditions, which influence the crystal packing of the mononuclear assemblies (Fig. S5†). This phenomenon is quite interesting, because a minor change, such as an additional solvent molecule in the crystal structure of the supramolecular system, gives a non-centrosymmetric group to the 2a lanthanide(III) complex. Another dependency observed in these systems is that when the lanthanide ion is coordinated by only one triflate counterion, it is consistently coordinated also by two oxygen atoms from methanol molecules. On the other hand, when the lanthanide ion is coordinated by two triflates, it is typically also coordinated by a nitrogen atom from an acetonitrile molecule. In such cases, the crystal structure contains one or two additional triflate counterions, which are not coordinated to the central ion and balance the charge of monocationic or dicationic assemblies, respectively. This dependency is not observed in complex 11, where the crystal structures exhibit only two coordinated oxygen atoms originating from monodentate CF3SO3− counterions. Therefore, this complex forms a cationic system where the positive charge is balanced by a single triflate counterion. Unlike the other complexes that involve two coordinated triflate anions, there are no solvent molecules coordinated to the Ln(III) center in complex 11. In accordance with the lanthanide contraction, a noticeable trend is the displacement of triflate anions from the coordination sphere in favor of solvent molecules. This change significantly influences the final charge of the obtained complexes, a crucial factor in various scientific fields with regard to inducing desirable properties in designed supramolecular systems. We can observe these dependences for example in the field of molecular magnetism, where certain structural differences in the crystal structures, like the cationic or neutral character of the metal complex, can affect the magnetic anisotropy of these compounds. This in turn can determine whether they behave as a Single-Molecule Magnet or not, as demonstrated for different metal complexes with d- or f-electron systems.48,49 Moreover, the overall charge of metal complexes significantly influences catalysis, when the charge-controlled catalyst can influence greatly specific activation processes and types of reactions having a direct impact on the activity and selectivity of the catalyst, important factors in defining an effective catalyst.50,51
Complexes from the triflate series, despite crystallizing in different space groups belonging to distinct crystallographic classes, exhibit quite similar geometries. However, several differences are observed at the level of the crystal structure architecture. The crystal packing motifs of each isostructural group of complexes are presented in Fig. 4.
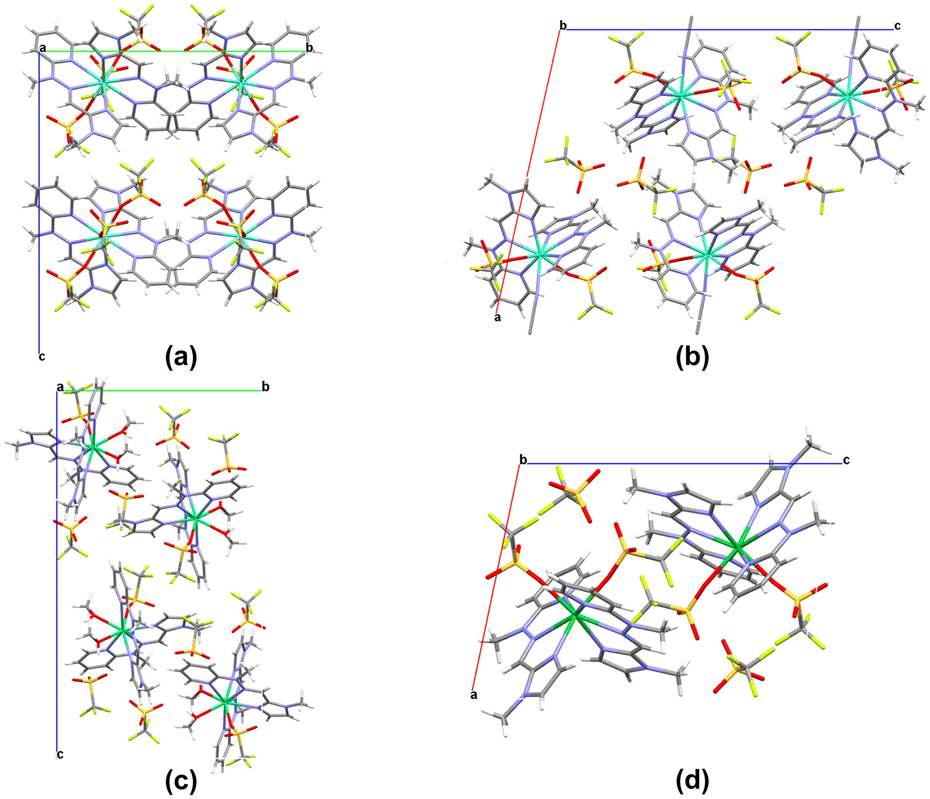 | ||
| Fig. 4 Crystal packing representation for each isostructural group of complexes from the triflate series: (a) 6, (b) 7a, (c) 10, and (d) 11 along the a axis for (a) and (c) and along the b axis for (b) and (d). | ||
The crystal packing for distinct isostructural groups differs mainly in the distribution of individual complex molecules within the crystal lattice. Notably, in the demonstrated representative complexes 6, 7a and 10, the unit cells contain four molecules as a repetitive entity. In contrast, complex 11 contains only two molecules of the individual lanthanide complex. The type of crystal structure observed in the lanthanide assemblies significantly influences the final supramolecular motifs in these systems. It is noteworthy that in the triflate series, hydrogen bonding is present only in the dicationic complexes (7, 8, 10). Within the crystal structure of these complexes, the O–H⋯O hydrogen bonds are formed between the triflate oxygen atom (not coordinated to the Ln(III) center) and the methanol oxygen atom involved in the coordination sphere (Fig. S6†). This observation highlights the impact of crystal packing effects in these supramolecular systems, particularly the presence of H-bond interactions in the triflate complexes. The presence of hydrogen bonds is crucial in supramolecular chemistry, as these non-covalent interactions provide the structural complexity and molecular diversity of the obtained supramolecular assemblies, offering opportunities to influence and control their specific properties.52–54
space group belonging to the triclinic crystal system. This contrasts with the triflate series, where distinct crystallographic groups were observed for specific metal ions. Besides the lower ligand-to-metal ratio and generally higher coordination numbers, a significant difference distinguishes these supramolecular systems from the complexes in the triflate series. The complexes in this series can mostly be categorized into very similar, but slightly different isostructural groups with the general formula: [LnL(NO3)3(X)](Y), where X and Y denote solvent molecules (such as methanol, acetonitrile, water or dimethyl sulfoxide) present in the crystal structures of monometallic lanthanide(III) assemblies (Fig. 5).
 | ||
| Fig. 5 Coordination modes present in the crystal structures of monometallic complexes from the nitrate series. | ||
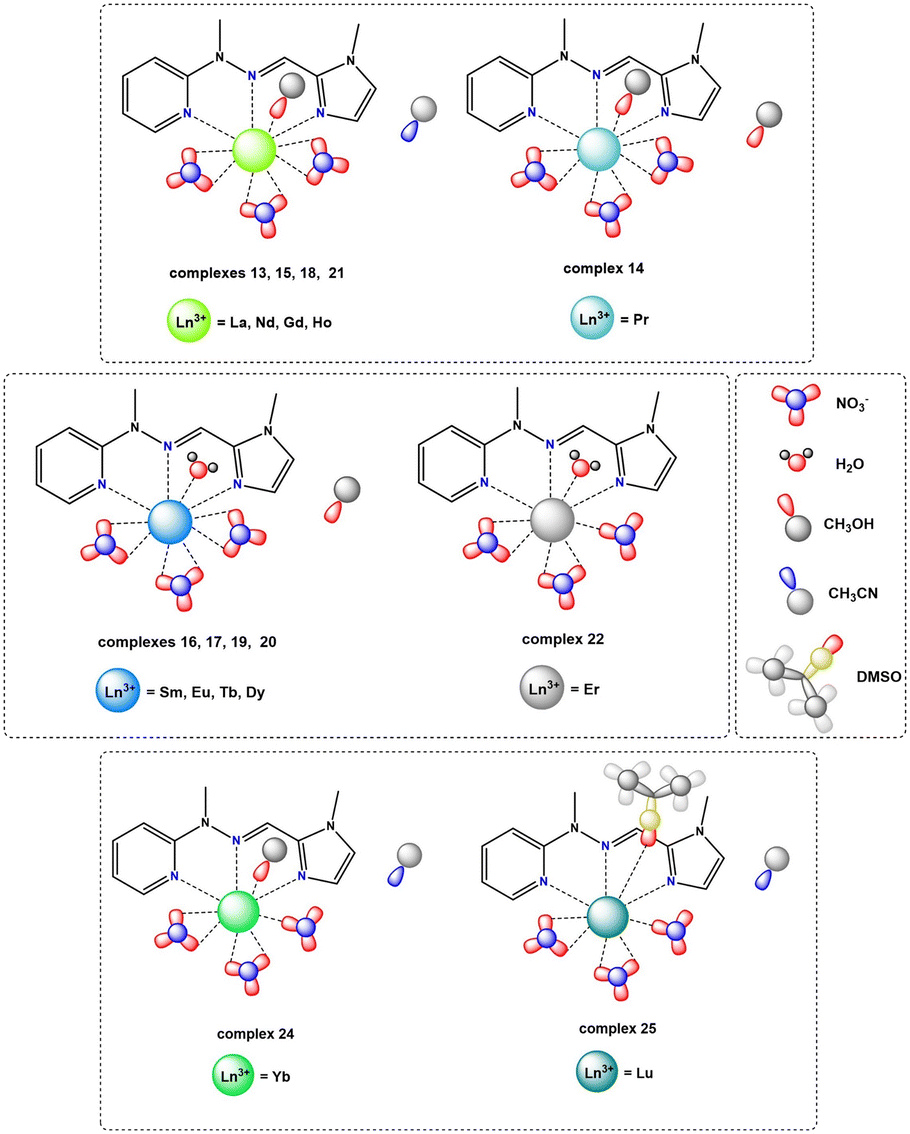
In the case of the nitrate complexes, the coordination of the Ln(III) center involves three nitrogen atoms from the ligand molecule: specifically, from pyridine (atom N1A), imine (atom N8A) and imidazole (atom N14A) moieties. Besides that, the main differences are variable solvent molecules, particularly those coordinating to the metal ions, as well as those present in the outer coordination sphere, not coordinated to the Ln(III) center. Distinct groups of compounds can be identified, where coordination sites are completed with solvent molecules of water or methanol. When the Ln(III) center is coordinated by one oxygen atom from a water molecule, the outer coordination sphere predominantly contains a methanol molecule. This observation is consistent for complexes 16 (Sm), 17 (Eu), 19 (Tb), and 20 (Dy) with the formula [LnL(NO3)3(H2O)]CH3OH. In contrast, when one oxygen atom from a methanol molecule participates in the coordination of a metal ion, the general observation is that the outer coordination sphere is occupied by acetonitrile molecule, which we can observe in complexes with the formula [LnL(NO3)3CH3OH)]CH3CN, specifically, complexes 13 (La), 15 (Nd), 18 (Gd), 21 (Ho) and 24 (Yb). However, there is one exception in complex 14 (Pr), where instead of an acetonitrile molecule, another methanol molecule is present with half occupancy, not coordinated to the central ion. Fig. S7† provides the molecular structures for representative nitrate complexes. We can generally observe a dependence on the influence of the coordinating solvent molecule (water or methanol) on the molecules present in the crystal structure that are not coordinated to the lanthanide(III) ions (methanol or acetonitrile, respectively). However, such situations are not observed for complexes 22 (Er) and 23 (Tm) with formulae: [ErL(NO3)3H2O] and [TmL(NO3)3CH3OH]LH+NO3−, respectively. In the case of the complex with the erbium(III) ion, where the coordination sites are additionally completed by one water molecule, no other solvent is involved in the outer coordination sphere, despite using identical reaction conditions and similar crystallization process via slow diffusion methods in either MeOH or the MeOH, MeCN/iPr2O system – yielding different outcomes compared to analogous complexes 16, 17, 19 and 20. In those cases, the coordinated water molecule pushes out the methanol molecule, which is present in the crystal structure. In the complex with the thulium(III) ion, despite the observation of a coordinated methanol molecule, there is an absence of an acetonitrile molecule in the crystal structure. This contrasts with analogous complexes where one coordinated methanol molecule was present, even though the crystallization conditions for obtaining crystals suitable for X-ray analysis remained identical (via slow diffusion methods in the MeOH, MeCN/iPr2O system). Instead of an acetonitrile molecule, the crystal structure also contains the ionic pair LH+ and NO3−. This occurrence is unique compared to other nitrate complexes due to the reaction conditions applied, involving a higher amount of ligand. Consequently, this case differs significantly from all other complexes within the nitrate series. These two distinct complexes, differing significantly from the others, strongly indicate a particular preference for a specific coordination environment among the metal ions of the Ln(III) complexes in the nitrate series. In addition to the coordination of lanthanide(III) ions by nitrogen atoms from ligand molecules and oxygen atoms from different solvent molecules, the coordination sites in these assemblies are filled also by oxygen atoms from three molecules of counterions. The specific coordination of these counterions appears to be a variable factor affecting the achievable coordination number of the Ln(III) center for these systems. For nearly all systems, it is evident that each nitrate anion remains in a bidentate mode and consequently, the metal ion is coordinated by six oxygen atoms – two oxygen atoms from each of the three nitrate anions present in the structure of complexes 13–21 containing La(III), Pr(III), Nd(III), Sm(III), Eu(III), Gd(III), Tb(III), Dy(III) and Ho(III) rare earth cations, respectively. The coordination modes of nitrate anions in complexes 22–24 with Er(III), Tm(III) and Yb(III) metal ions differ significantly. These compounds exhibit similar geometries, involving three nitrates, but with only two in a bidentate mode and one in a monodentate mode. As a result, in these systems, the lanthanide(III) ion is coordinated by five oxygen atoms from nitrate anions, which is probably a result of their prolate electronic character. We can also observe such a situation in the case of the obtained crystal structure for complex 25 with a Lu(III) metal ion (Fig. S8†), where two of the nitrates remain in a bidentate mode, while one of them remains in a monodentate one. In this compound, beyond coordination by three nitrogen atoms from the ligand molecule, the lanthanide ion is coordinated by an oxygen atom from a dimethyl sulfoxide molecule, which is directly related to the synthetic conditions used. The coordination of the central atom in complexes 22–25 by one nitrate anion in a monodentate mode directly leads to lower coordination numbers (9 instead of 10) for these assemblies compared to the other supramolecular systems in this series, significantly influencing the coordination geometry. Over the whole nitrate series, the appearance of nitrate anions in bidentate mode is noticeable, a trend also observed in the literature referring to rare earth compounds with similar coordination sites.29,30,39,55 Generally, two isostructural groups of compounds can be identified where metal ions are coordinated by the same donor atoms and in the same way. This includes consideration of not only the solvent molecules not coordinated to the Ln(III) center, but also two examples of compounds differing solely in the solvent present in the outer coordination sphere, as well as an individual case of a complex that differs from any within these groups. This diversity is demonstrated in Fig. 6, illustrating various coordination modes exhibited by selected lanthanide ions as representative examples.
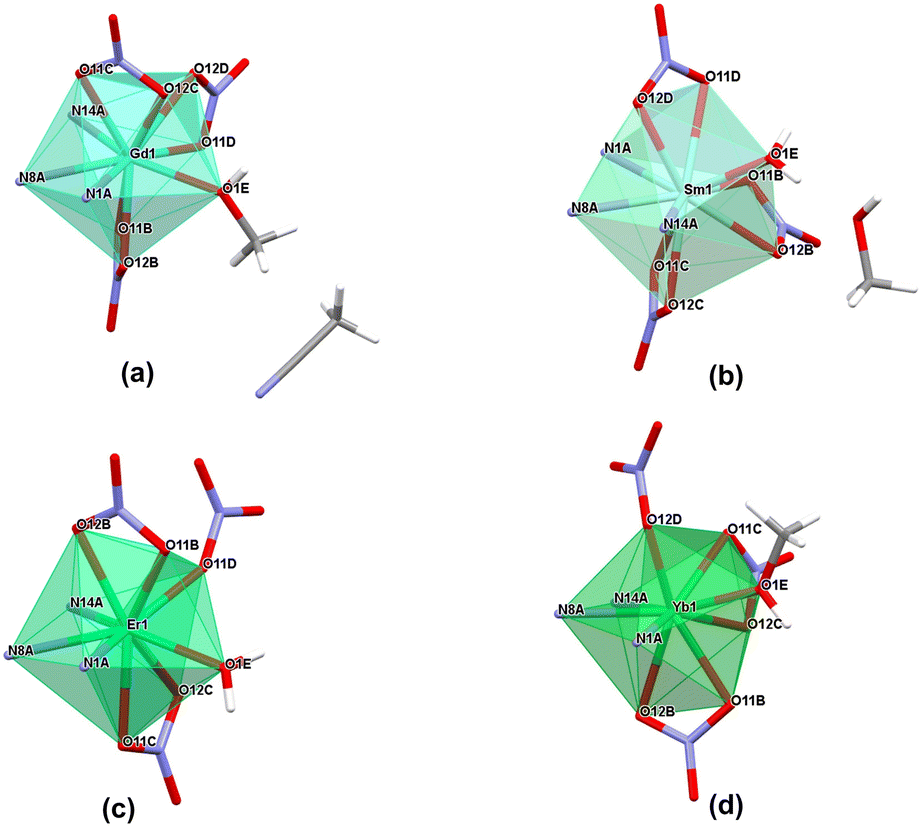 | ||
| Fig. 6 Coordination polyhedron representation for (a) Gd1 (18), (b) Sm1 (16), (c) Er1 (22) and (d) Yb1 (24) as representative examples of coordination modes present in monometallic lanthanide complexes in the nitrate series, considering coordinating to the lanthanide ions oxygen atoms from water, methanol or nitrate molecules, as well as nitrogen atoms from ligand molecules; for clarity, only coordinating nitrogen atoms (N1A, N8A and N14A) from the ligand molecule are shown, with the rest of the molecule omitted. | ||
Lanthanide complexes from the nitrate series display very similar geometries as they crystallize in the same space groups, and all of them are neutral species. However, despite this similarity, several differences are observed at the level of the crystal structure architecture. Notably, in complexes where a molecule of methanol is involved in coordination sites (complexes 13, 14, 15, 18, 21, 24), the formation of O–H⋯O hydrogen bonding between the OH group from the methanol molecule and the nitrate oxygen atom, coordinating with the lanthanide ions, leads to differences in the supramolecular structures formed by these molecules, illustrated in Fig. S9.† Examining the crystal packing in the nitrate complexes, we note a consistent distribution in the crystal lattice, wherein the unit cells of complexes contain, as a repetitive entity, two molecules of the individual lanthanide complex.
When considering complexes, where a water molecule is involved in the coordination sites of the Ln(III) center (complexes 16, 17, 19, 20), a similar trend is observed with the presence of an intramolecular hydrogen bond O–H⋯O formed between the coordinated water molecule and the molecule of methanol present in the outer coordination sphere. In particular, the specific arrangement of the created supramolecular structure in these complexes is significantly influenced by the additional hydrogen bond between a molecule of coordinated water from one single lanthanide complex and a coordinated nitrate anion from another (Fig. S10†).
:1 ligand-to-metal ratio is evident, similar to analogous complexes with triflate anions. During the synthesis of perchlorate complexes, we found some synthetic challenges, in particular, the tendency to form products in an oily form, leading to difficulties in purification and resulting in a very low yield of completed complexation reactions. Despite these challenges, we managed to obtain crystals suitable for X-ray analysis, exemplified by a complex named Tb-perchlorate. Analysis of the obtained crystal structure proved similarities between these complexes with lanthanide perchlorate counterions and triflate complexes. The Ln(III) center is coordinated by six nitrogen atoms from two ligand molecules and both perchlorate and triflate anions exhibit a preference not to coordinate with the metal ions, unlike the stronger coordination preferences observed for nitrate analogues. As a result of these supramolecular preferences in the crystal structures of the Tb-perchlorate complex (Fig. S11†), only one perchlorate anion is observed to coordinate, while the other two remain in the outer coordination sphere, not coordinated to the Tb(III) center. The coordination sites are filled by solvent molecules, in a similar relation found in complex 7 of the terbium(III) ion with triflate counterions.
The obtained crystal structure of the complex named Eu-hydrogen containing the europium(III) ion (Fig. 7), with an identical ligand molecule except for the substitution of the methyl group with a hydrogen atom on the imidazole ring, clearly demonstrates the significant influence of different groups in ligand molecules on the formed assemblies. Comparing this structure with the analogous complex 5 with Eu(III), we can conclude that: a) the lanthanide contraction may not exert as strong an effect as anticipated (larger lanthanide ions are not always coordinated by three triflate counterions) and b) the introduction of a hydrogen bond donor into the ligand encourages the displacement of one triflate in the outer coordination sphere, leading to observed effects on crystal packing. This change creates a network of hydrogen bonds in complete contrast to complex 5, where no hydrogen bonds are present (Fig. 7). These dependencies serve as excellent examples illustrating that these types of ligands play a crucial role and exert a strong influence on self-assembly and self-organization processes in supramolecular chemistry.
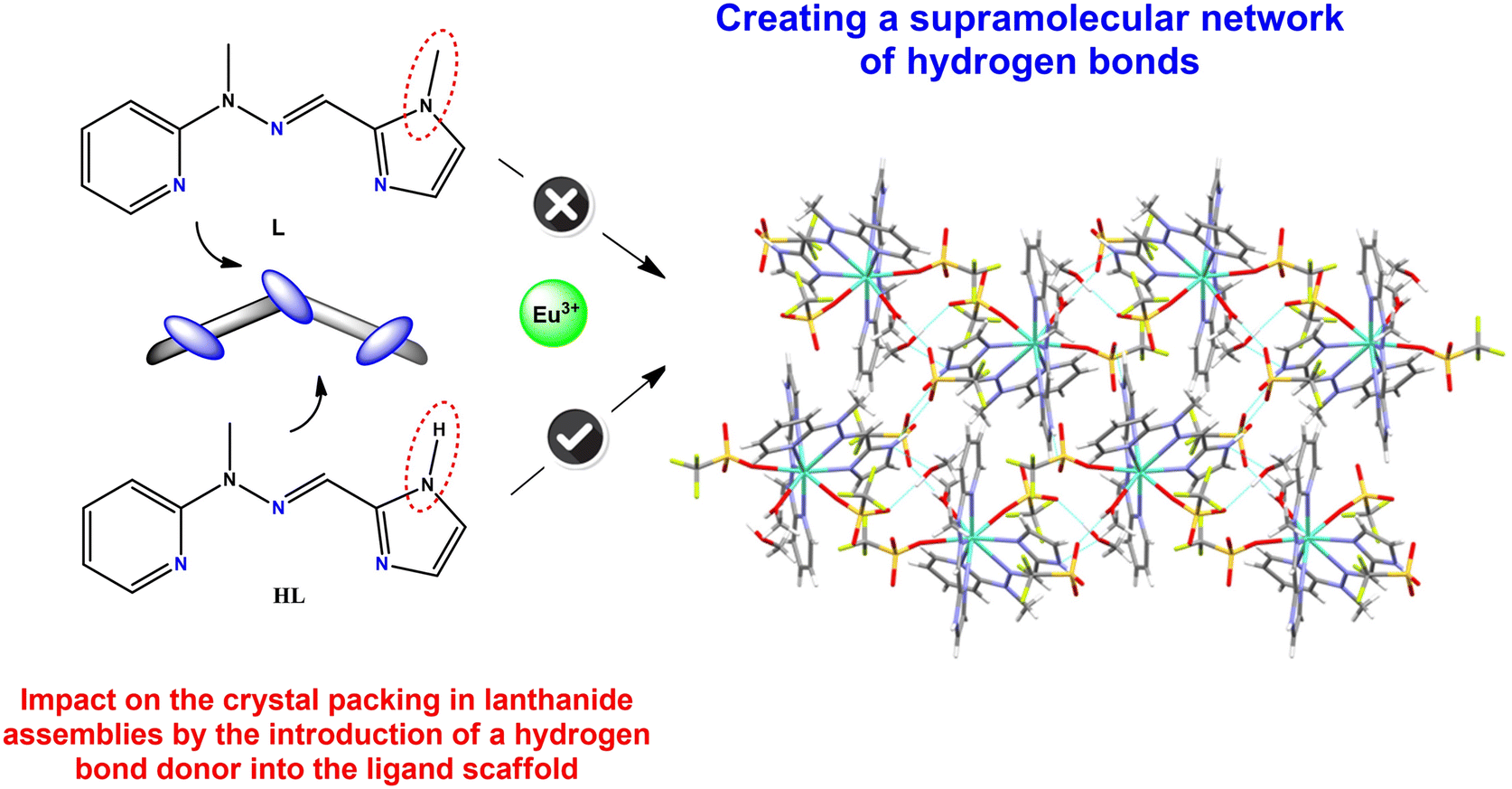 | ||
| Fig. 7 Formation of a supramolecular hydrogen bond network in complex Eu-hydrogen with the europium(III) ion based on a modified hydrazone ligand molecule, namely changing the methyl group from the imidazole moiety into a hydrogen atom. | ||
It is worth mentioning that complexes formed with rare earth elements (such as gadolinium and terbium metal ions) with nitrate counterions and a very similar N3-tridentate Schiff-base ligand with a benzimidazole moiety, previously obtained by us,55 are quite comparable to the complexes reported here. Lanthanide cations remain the same 10-coordinated, which we can see mainly in the nitrate series presented here. The coordination modes are also very similar – Gd(III) or Tb(III) ions are coordinated by three nitrogen atoms from the ligand molecule and six oxygen atoms from three bidentate nitrate anions. Additionally, the coordination sites are also filled by one molecule of the solvent, specifically an acetonitrile molecule in these cases. While differences can be observed at the level of the crystal structure architecture (such as crystallizing in different space groups), the series of nitrate complexes with ligand molecules like ligand L generally display very similar geometries. Interestingly, dissimilar dependencies are noted when considering complexes formed by various lanthanide ions with ligand L in the triflate series, as well as complexes with a similar ligand with a benzimidazole group published previously. When the mentioned ligand reacts with europium(III) trifluoromethanesulfonate – even with a ligand-to-metal ratio of 1:1 during the reaction – a monometallic complex is formed, where the coordination sphere of the central ion is occupied by 9 nitrogen atoms from three ligand molecules, and the charge is balanced by three triflate counterions that are not coordinated to the Ln(III) center. It is particularly interesting that in the case of complexes in the triflate series discussed in this work, even 1 mole of Ln(CF3SO3)3 reacts with 3 moles of Schiff-base L, an excess of ligand remains unreacted despite heating and the application of solvothermal reaction conditions. Consequently, the coordination sites are filled by six nitrogen atoms from two ligand molecules and oxygen or nitrogen atoms from triflate modes or solvent molecules. When comparing this situation with the synthesis of complexes from the nitrate series, we notice a similar correlation. Increasing the molar ratio of the ligand to metal ion during the synthesis does not result in more coordinated ligand molecules to the Ln(III) center, but it leads solely to the protonation of an additional ligand molecule. This observation is confirmed by the obtained crystal structure of the complex with the thulium(III) ion (23), wherein the excess ligand crystallizes as a protonated molecule associated with the lanthanide complex. In summary, for complexes synthesized with ligand L and those with a quite similar ligand featuring a benzimidazole group, both with triflate anions, several differences are observed. It appears that the presence of an H-bond donor can significantly influence the final metal-to-ligand ratio within these analogous supramolecular assemblies. Even a slight change in the ligand molecule can greatly affect the preferred stoichiometry and geometry of the formed complexes, which is important when considering the design of supramolecular systems with specific coordination sites and resulting properties. Hydrazone ligands, known for their high flexibility, exhibit a rich structural diversity, which is the result of possibilities for modifying ligand scaffolds through the selection of various building blocks during their synthesis.56–59 It appears that the introduction of additional groups with their variable character can significantly impact the structures formed within lanthanide supramolecular assemblies based on these types of ligands. When comparing the behavior of the assembly using the N3-tridentate ligand L studied here with similar hydrazone ligand scaffolds, where coordination sites are changed by substituting nitrogen donor atoms with oxygen atoms, several dependencies on the formed structure can be observed. It is evident that the character of the oxygen donor atoms notably influences their preferences towards complexity. Despite the preference of all mentioned ligands to coordinate via two nitrogen atoms and one oxygen atom, their assembly properties are quite different in the presence of nitrate or chloride lanthanide metal salts. Among them, only one demonstrates identical anion-driven tunability to the ligand L synthesized by us, in the presence of nitrate counterions. This highlights the importance of small structural differences in the design of supramolecular systems with specific coordination sites. When a phenol moiety, potentially a donor of an oxygen atom from a hydroxy group, is introduced into the ligand molecule, reactions in the presence of nitrate or chloride lanthanide salts consistently lead to obtaining 1:2 metal:ligand species.60,61 On the other hand, when the ligand scaffold contains a carboxyl group involved in the coordination of the Ln(III) center, assemblies formed with nitrate anions maintain a molar ratio of the metal to ligand of 1:1,30 as exactly observed in the synthesis of lanthanide nitrate complexes here. Moreover, with this type of N,N,O-hydrazone Schiff-base ligand, we also observe anion-driven self-assembly (utilization of chloride salts leads to formation of 1:2 metal:ligand complexes) and fine-tuning solvent characteristics are noted, showing structure-dependent magnetic properties.
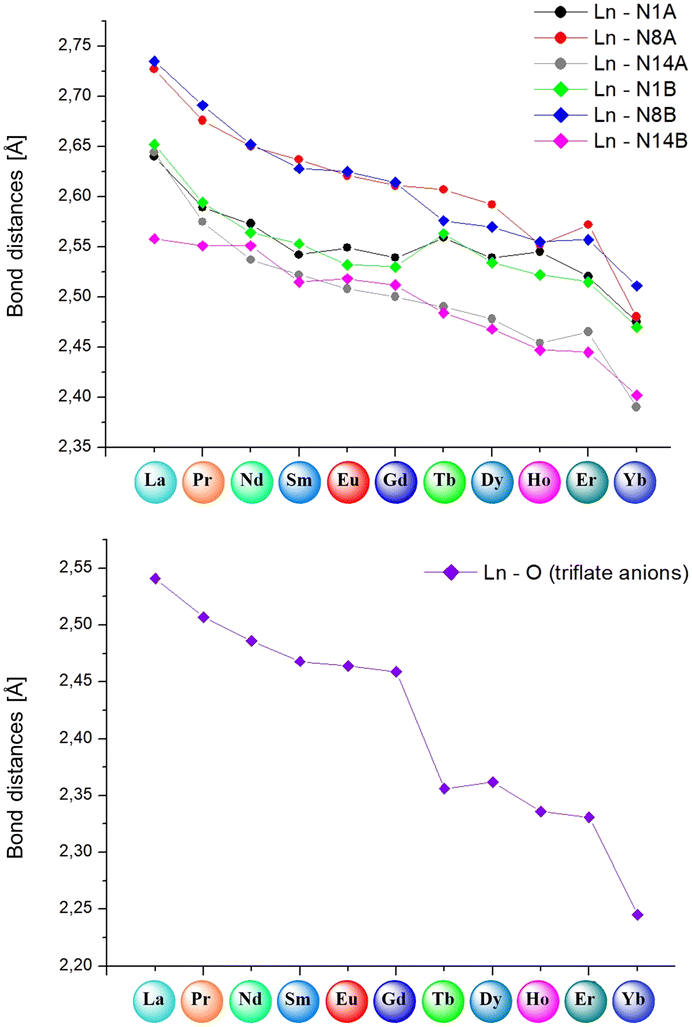 | ||
| Fig. 8 (Top) Comparison of bond lengths between lanthanide(III) ions and particular nitrogen atoms from ligand molecules (N1A/B, N8A/B and N14A/B); (bottom) comparison of average bond lengths between the Ln(III) center and oxygen atoms from triflate anions. | ||
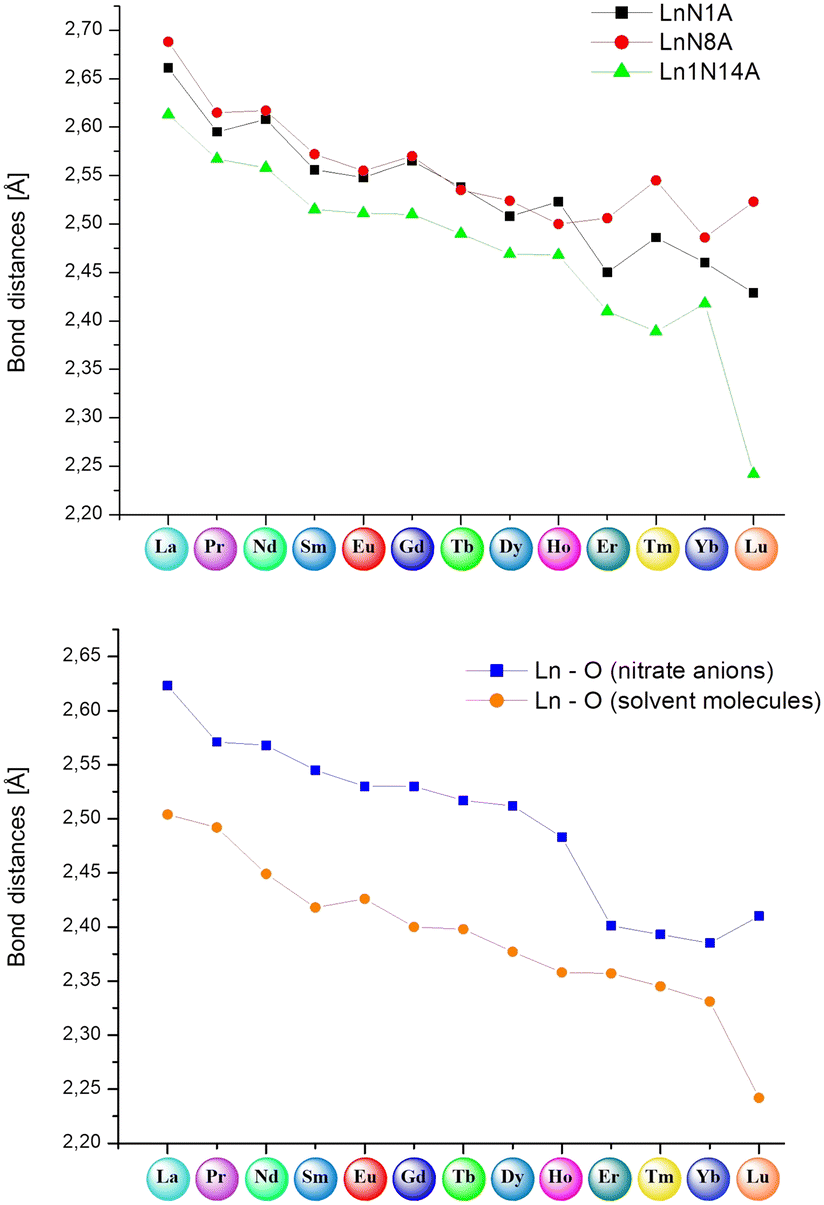 | ||
| Fig. 9 (Top) Comparison of bond lengths between lanthanide(III) ions and particular nitrogen atoms from ligand molecules (N1A, N8A and N14A); (bottom) comparison of average bond lengths between the Ln(III) center and oxygen atoms from nitrate anions and methanol, water or dimethyl sulfoxide molecules. | ||
Conclusions
We synthesized and structurally characterized series of monometallic complexes with a number of lanthanide metal ions – La, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, and Lu. It was found that the obtained compounds demonstrate anion-driven self-assembly, as the utilization of appropriate counterions allows for obtaining 1:1 (nitrate anions) or 2:1 (triflate anions) ligand-to-metal ratio systems, independent of the lanthanide(III) ion or the reaction conditions used. The obtained crystal structures of the complexes exhibit a diversity of coordination modes around the lanthanide ions, allowing the classification of complexes into a variety of isostructural groups. Structural dependencies within each series of complexes (triflate vs. nitrate) were examined to estimate the influence of the lanthanide contraction phenomenon, counterions or solvent molecules on the formed assemblies. Based on the rich structural diversity of the presented systems, we further aim to undertake studies related to molecular magnetism, biomimetics and absorption/emission characteristics.
Data availability
Crystallographic data for compounds in this paper (1–11, 13–25, LH·Br, Tb-perchlorate, Eu-hydrogen) have been deposited at the Cambridge Crystallographic Data Centre, no. CCDC-2338931 (LH·Br−), CCDC-1005948 (1), CCDC-1005949 (2), CCDC-1052303 (2a), CCDC-1052299 (3), CCDC-1005950 (4), CCDC-1005951 (5), CCDC-1005952 (6), CCDC-1052307 (7), CCDC-1052306 (7a), CCDC-1052308 (8), CCDC-1435578 (9), CCDC-1052304 (10), CCDC-1435579 (11), CCDC-1435580 (13), CCDC-1052302 (14) CCDC-1435581 (15), CCDC-1435582 (16), CCDC-1435583 (17), CCDC-1435584 (18), CCDC-1052300 (19), CCDC-1006326 (20), CCDC-1052301 (21), CCDC-1435585 (22), CCDC-1052305 (23), CCDC-1435586 (24), CCDC-2338933 (25), CCDC-2338932 (Tb-perchlorate) and CCDC-2338934 (Eu-hydrogen).Author contributions
Dominika Prętka: conceptualization, investigation, formal analysis, data curation, visualization, writing – original draft, writing – review & editing; Dawid Marcinkowski: investigation, formal analysis, funding acquisition; Agnieszka Siwiak: investigation, data curation; Maciej Kubicki: formal analysis, investigation, data curation, writing – review & editing, visualization; Giuseppe Consiglio: writing – review & editing, visualization, funding acquisition; Violetta Patroniak: supervision, resources, visualization, writing – review & editing; Adam Gorczyński: conceptualization, methodology, writing – review & editing, project administration, supervision, resources, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by: SONATA grant UMO-2020/39/D/ST4/01182 and PRELUDIUM grant UMO-2022/45/N/ST4/00344 from the National Science Centre, Poland, and by the University of Catania, PIACERI 2020/2022, Linea di Intervento 2.Notes and references
- Y. Lin and C. Mao, Front. Mater. Sci., 2011, 5, 247–265 CrossRef PubMed.
- Q. Zhang, Y.-X. Deng, H.-X. Luo, C.-Y. Shi, G. M. Geise, B. L. Feringa, H. Tian and D.-H. Qu, J. Am. Chem. Soc., 2019, 141, 12804–12814 CrossRef PubMed.
- B. H. Northrop, Y.-R. Zheng, K.-W. Chi and P. J. Stang, Acc. Chem. Res., 2009, 42, 1554–1563 CrossRef CAS PubMed.
- C. H. Chen, L. C. Palmer and S. I. Stupp, Soft Matter, 2021, 17, 3902–3912 RSC.
- S. Guo, Y. Song, Y. He, X.-Y. Hu and L. Wang, Angew. Chem., Int. Ed., 2018, 57, 3163–3167 CrossRef CAS PubMed.
- S. I. Stupp and L. C. Palmer, Chem. Mater., 2014, 26, 507–518 CrossRef.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef.
- J. Wankar, N. G. Kotla, S. Gera, S. Rasala, A. Pandit and Y. A. Rochev, Adv. Funct. Mater., 2020, 30, 1909049 CrossRef.
- Q. Wang, Q. Zhang, Q.-W. Zhang, X. Li, C.-X. Zhao, T.-Y. Xu, D.-H. Qu and H. Tian, Nat. Commun., 2020, 11, 158 CrossRef PubMed.
- G. Olivo, G. Capocasa, D. Del Giudice, O. Lanzalunga and S. Di Stefano, Chem. Soc. Rev., 2021, 50, 7681–7724 RSC.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- R. Lu, X. Zhang, X. Cheng, Y. Zhang, X. Zan and L. Zhang, Front. Chem., 2020, 8, 1–25 CrossRef CAS PubMed.
- M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
- K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2017, 345, 54–72 CrossRef CAS.
- P. Hobza and J. Řezáč, Chem. Rev., 2016, 116, 4911–4912 CrossRef CAS PubMed.
- F. B. Ilhami, Y.-T. Yang, A.-W. Lee, Y.-H. Chiao, J.-K. Chen, D.-J. Lee, J.-Y. Lai and C.-C. Cheng, Biomacromolecules, 2021, 22, 4446–4457 CrossRef CAS PubMed.
- D. A. Roberts, B. S. Pilgrim and J. R. Nitschke, Chem. Soc. Rev., 2018, 47, 626–644 RSC.
- S. Saha, I. Regeni and G. H. Clever, Coord. Chem. Rev., 2018, 374, 1–14 CrossRef CAS.
- S. Zhang, H. Wu, L. Sun, H. Ke, S. Chen, B. Yin, Q. Wei, D. Yang and S. Gao, J. Mater. Chem. C, 2017, 5, 1369–1382 RSC.
- S. Yu, Z. Chen, H. Hu, B. Li, Y. Liang, D. Liu, H. Zou, D. Yao and F. Liang, Dalton Trans., 2019, 48, 16679–16686 RSC.
- W.-Y. Zhang, Y.-Q. Zhang, S.-D. Jiang, W.-B. Sun, H.-F. Li, B.-W. Wang, P. Chen, P.-F. Yan and S. Gao, Inorg. Chem. Front., 2018, 5, 1575–1586 RSC.
- J. Wu, X.-L. Li, L. La Droitte, O. Cador, B. Le Guennic and J. Tang, Dalton Trans., 2021, 50, 15027–15035 RSC.
- K. Zhang, C. Yuan, F.-S. Guo, Y.-Q. Zhang and Y.-Y. Wang, Dalton Trans., 2017, 46, 186–192 RSC.
- W.-B. Sun, P.-F. Yan, S.-D. Jiang, B.-W. Wang, Y.-Q. Zhang, H.-F. Li, P. Chen, Z.-M. Wang and S. Gao, Chem. Sci., 2016, 7, 684–691 RSC.
- S.-M. Xu, Z.-W. An, W. Zhang, Y.-Q. Zhang and M.-X. Yao, CrystEngComm, 2021, 23, 2825–2834 RSC.
- R. B. Jordan, Inorg. Chem., 2023, 62, 3715–3721 CrossRef PubMed.
- S. A. Cotton and P. R. Raithby, Coord. Chem. Rev., 2017, 340, 220–231 CrossRef.
- X.-Z. Li, C.-B. Tian and Q.-F. Sun, Chem. Rev., 2022, 122, 6374–6458 CrossRef PubMed.
- F. Ma, Q. Chen, J. Xiong, H.-L. Sun, Y.-Q. Zhang and S. Gao, Inorg. Chem., 2017, 56, 13430–13436 CrossRef PubMed.
- L. Sun, S. Zhang, C. Qiao, S. Chen, B. Yin, W. Wang, Q. Wei, G. Xie and S. Gao, Inorg. Chem., 2016, 55, 10587–10596 CrossRef CAS PubMed.
- R. Li, B. Moubaraki, K. S. Murray and S. Brooker, Dalton Trans., 2008, 6014–6022 RSC.
- A. Bocian, M. Szymańska, D. Brykczyńska, M. Kubicki, M. Wałęsa-Chorab, G. N. Roviello, M. A. Fik-Jaskółka, A. Gorczyński and V. Patroniak, Molecules, 2019, 24, 3173 CrossRef CAS PubMed.
- V. Vrdoljak, G. Pavlović, N. Maltar-Strmečki and M. Cindrić, New J. Chem., 2016, 40, 9263–9274 RSC.
- R. S. Bhaskar, C. A. Ladole, N. G. Salunkhe, J. M. Barabde and A. S. Aswar, Arabian J. Chem., 2020, 13, 6559–6567 CrossRef CAS.
- A. Bocian, A. Gorczyński, D. Marcinkowski, S. Witomska, M. Kubicki, P. Mech, M. Bogunia, J. Brzeski, M. Makowski, P. Pawluć and V. Patroniak, J. Mol. Liq., 2020, 302, 112590 CrossRef CAS.
- A. Bocian, M. Skrodzki, M. Kubicki, A. Gorczyński, P. Pawluć and V. Patroniak, Appl. Catal., A, 2020, 602, 117665 CrossRef CAS.
- Ł. Banach, D. Brykczyńska, A. Gorczyński, B. Wyrzykiewicz, M. Skrodzki and P. Pawluć, Chem. Commun., 2022, 58, 13763–13766 RSC.
- K. M. Ayers, N. D. Schley and G. Ung, Chem. Commun., 2019, 55, 8446–8449 RSC.
- I. Pospieszna-Markiewicz, M. A. Fik-Jaskółka, Z. Hnatejko, V. Patroniak and M. Kubicki, Molecules, 2022, 27(23), 8390 CrossRef CAS PubMed.
- A. Gorczyński, D. Marcinkowski, M. Kubicki, M. Löffler, M. Korabik, M. Karbowiak, P. Wiśniewski, C. Rudowicz and V. Patroniak, Inorg. Chem. Front., 2018, 5, 605–618 RSC.
- A. Gorczyński, M. Zaranek, S. Witomska, A. Bocian, A. R. Stefankiewicz, M. Kubicki, V. Patroniak and P. Pawluć, Catal. Commun., 2016, 78, 71–74 CrossRef.
- Agilent Technologies, CrysAlis PRO, Agilent Technologies Ltd, 2011 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- H. D. Flack and G. Bernardinelli, Chirality, 2008, 20, 681–690 CrossRef CAS PubMed.
- J. Wu, J. Jung, P. Zhang, H. Zhang, J. Tang and B. Le Guennic, Chem. Sci., 2016, 7, 3632–3639 RSC.
- S. Ghosh, S. Kamilya, M. Das, S. Mehta, M.-E. Boulon, I. Nemec, M. Rouzières, R. Herchel and A. Mondal, Inorg. Chem., 2020, 59, 7067–7081 CrossRef CAS PubMed.
- J. Long, A. N. Selikhov, N. Y. Rad'kova, A. V. Cherkasov, Y. Guari, J. Larionova and A. A. Trifonov, Eur. J. Inorg. Chem., 2021, 2021, 3008–3012 CrossRef CAS.
- A. Mondal, M. Díaz-Ruiz, F. Deufel, F. Maseras and M. van Gemmeren, Chem, 2023, 9, 1004–1016 CAS.
- J. A. de Gracia Triviño and M. S. G. Ahlquist, Top. Catal., 2022, 65, 383–391 CrossRef.
- N. Geue, R. E. P. Winpenny and P. E. Barran, Chem. Soc. Rev., 2022, 51, 8–27 RSC.
- C. L. Lewis, K. Stewart and M. Anthamatten, Macromolecules, 2014, 47, 729–740 CrossRef CAS.
- K. Dong, S. Zhang and J. Wang, Chem. Commun., 2016, 52, 6744–6764 RSC.
- D. Marcinkowski, M. Wałęsa-Chorab, A. Bocian, J. Mikołajczyk, M. Kubicki, Z. Hnatejko and V. Patroniak, Polyhedron, 2017, 123, 243–251 CrossRef CAS.
- J. Galindo, J. Castaño, R. Visbal and M. Chaur, Eur. J. Org. Chem., 2022, 2022, e202200228 CrossRef.
- X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981 RSC.
- L. A. Tatum, X. Su and I. Aprahamian, Acc. Chem. Res., 2014, 47, 2141–2149 CrossRef CAS PubMed.
- K.-s. Kim, H. J. Cho, J. Lee, S. Ha, S. G. Song, S. Kim, W. S. Yun, S. K. Kim, J. Huh and C. Song, Macromolecules, 2018, 51, 8278–8285 CrossRef.
- S. Roy, J. Du, E. M. Manohar, L. Sun, N. Ahmed, A. Dey and S. Das, Cryst. Growth Des., 2023, 23, 1066–1075 CrossRef.
- P. Kalita, A. Malakar, J. Goura, S. Nayak, J. M. Herrera, E. Colacio and V. Chandrasekhar, Dalton Trans., 2019, 48, 4857–4866 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedure details and characterization of the lanthanide complexes. Crystallographic data and structure refinement details of the ligand and complexes. Representative mass and IR spectra of the complexes. 1H NMR spectra. X-ray representation of selected isostructural monometallic complexes from triflate and nitrate series. Selected bond distances (Å) for complexes 1–25. Crystal packing and supramolecular structure representation of selected lanthanide(III) complexes. CCDC 1005948–1005952, 1435578–1435584, 1052299–1052308, 1006326, 1435585, 1435586 and 2338931–2338934. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00450g |
| This journal is © The Royal Society of Chemistry 2024 |