
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dynamic metal–ligand bonds in MFM-300: the use of semi-open metal sites to “teach an old dog new tricks”
Ricardo A.
Peralta
 *a and
Ilich A.
Ibarra
*b
*a and
Ilich A.
Ibarra
*b
aDepartamento de Química, Universidad Autónoma Metropolitana-Iztapalapa, Av. Ferrocarril San Rafael Atlixco 186, Col. Leyes de Reforma 1A Sección, Iztapalapa, Ciudad de México, Mexico. E-mail: rperalta@izt.uam.mx
bLaboratorio de Fisicoquímica y Reactividad de Superficies (LaFReS), Instituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior s/n, CU, Del. Coyoacán, 04510, Ciudad de México, Mexico. E-mail: argel@unam.mx
First published on 25th September 2024
Abstract
Developing a deep understanding of the fascinating properties of MFM-300 (MFM = Manchester framework material) opens up a world of new and thrilling applications. The unique dynamic metal–ligand bonding for MFM-300(Sc) and MFM-300(In) is chronologically presented here, highlighting the exacting applications that were associated with such special behaviour (i.e., description of complex NH3 adsorption mechanisms, conversion of toxic H2S to polysulfides for promising fabrication of lithium–sulphur batteries and heterogeneous catalysis). This highlight aims to showcase the current advances in semi-open metal sites inherent to MFM-300(Sc) and MFM-300(In) to inspire new investigations to be carried out to develop new applications of this promising family of MOF materials.
1. Introduction
Metal–organic frameworks (MOFs), or porous coordination polymers (PCPs), are a promising family of hybrid materials constructed by metal ions coordinated to organic ligands.1 MOFs were first reported back in the 1989s, and since then they have drawn significant attention due to their chemical variability, high crystallinity, extraordinary apparent surface area, and, in some specific cases, high chemical stability.2 Also, their integrated design and ordered lattice permit chemical, spatial and dynamic properties to be carefully adjusted in ways not available in other porous materials (e.g., carbon molecular sieves, zeolites and porous metal oxides). Thus, not surprisingly, these appealing properties have postulated MOFs as attractive contenders for applications such as gas storage/separation,3 catalysis,4 drug delivery,5 environmental air and water remediation,6,7 and molecular sensing.8 Interestingly, more than 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 reported MOF structures constitute a rapidly expanding collection of chemical functionalities originating from their organic ligands, inorganic metal centres and different post-synthetic modifications.9,10 Such an appealing set of characteristics enhances the crystal engineering of MOFs as one of the most researched fields in the past decades.
000 reported MOF structures constitute a rapidly expanding collection of chemical functionalities originating from their organic ligands, inorganic metal centres and different post-synthetic modifications.9,10 Such an appealing set of characteristics enhances the crystal engineering of MOFs as one of the most researched fields in the past decades.
The classic perception of MOFs as static crystalline structures has transited to flexible (i.e., dynamic metal–ligand interactions or hemilabile coordinate bonds), which has been recognised for contributing to unusual physiochemical properties.11–13 Although MOFs can typically generate functionality from their building blocks (i.e., functional groups incorporated into the organic ligands or with access to open metal sites), functionality is progressively derived from such dynamic processes (flexibility around the coordination bonds).14–16
One of the most relevant concepts in coordination chemistry is ligand lability, and, for example, labile ligands provide access to the active metal sites in catalysis.17,18 Hemilabile ligands are multidentate ligands (i.e., “hard” and “soft” donors) in which one donor is labile and can be reversibly disrupted to generate an open metal site to which substrates bind.19,20 In MOF chemistry, labile metal–ligand coordination can be dislocated by different stimuli without compromising the crystal structure of the material (e.g., framework connectivity), in other words, the generation of transient open metal sites.21
Similarly, the assembly of crystallinity in extended metal–organic lattices (such as MOFs, metal–organic cages and HOFs) is based on the reversible exchange of coordinate bonds.22,23 Thus, such reversible metal–ligand bonding, which can be exemplified by stimuli-responsive, adaptable, dynamic, and self-healing bonding, has been lately presented to elucidate key phenomena in MOFs, from their crystal growth to their phase transitions.24 Typically, the perception of metal–ligand dynamics has been exclusively attributed to temperature increase as the external stimulus. However, recently, it has been demonstrated that guest molecules can trigger such reversible bond reorganisation.20,25 Such studies on metal–ligand dynamics in MOFs have demonstrated that it is fundamental to recognise phase transitions to discover uncommon adsorption mechanisms.26
This highlight aims to establish the role of dynamic bonding in a particular family of MOF materials (MFM-300, MFM = Manchester framework material), which has been observed so far only for MFM-300(Sc) and MFM-300(In), by presenting the principal contributions related to such special behaviour and its associated exciting applications while providing relevant perspectives on the future of MOF chemistry in this field, particularly with the aim to encourage the scientific community to investigate the other members of the MFM-300 family (i.e., MFM-300(Al), MFM-300(Cr,) MFM-300(V), MFM-300(Ga) and MFM-300(Fe)).
2. Structural description
The MFM-300 family is synthesised using a tetracarboxylic ligand H4BPTC (biphenyl-3,3′,5,5′-tetracarboxylic acid) and different trivalent metal centres (Sc(III),27 Al(III),28 In(III),29 Cr(III),30 V(III),31 Ga(III)32 and Fe(III)33) under different reaction conditions; Schröder and Yang have elegantly reported this family. The constituents (BPTC4− and metal ions) of the MFM-300 family are arranged such that every ligand (BPTC4−) is coordinated to eight metal centres via four carboxylate groups (Fig. 1a), affording an infinite chain of [MO4(OH)2] which constituted the secondary building unit (SBU) (Fig. 1b). The metal centres display an octahedral geometry associated by two mutually cis-μ2-OH (hydroxo groups) anions that comprise the helical SBUs which are connected by the BPTC ligands to form a one-dimensional cylindrical channel (Fig. 1c).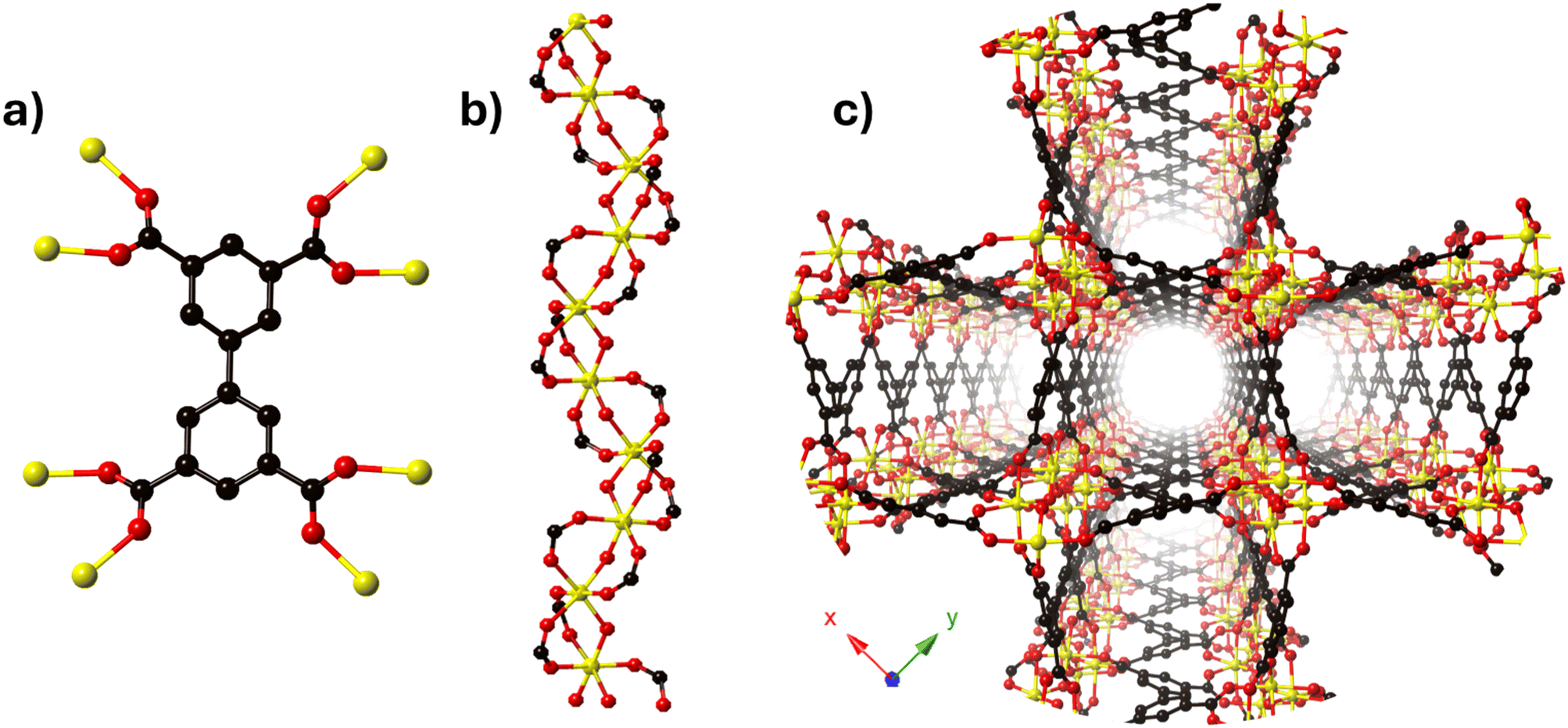 | ||
| Fig. 1 MFM-300(M) (M = Sc(III), Al(III), In(III), Cr(III), V(III), Ga(III) and Fe(III)) ligand–metal framework structure constitution. (a) Coordination arrangement of BPTC4− ligand (C atoms in grey, O atoms in red, hexacoordinated metal ions in yellow (Sc, In)); (b) M(OH)(CO2)2, 1D chiral chain; (c) MFM-300(Sc, In) cylindrical channels viewed along the c axis. | ||
3. The first reversible guest-induced evidence: NH3 adsorption
The first reported material of the MFM-300 family was described by Champness and Schröder in 2011 (MFM-300(Sc), also known as NOTT-400), and H2 was the first molecule (not considering N2 at 77 K to estimate the surface area of this material) to be investigated inside the pores of MFM-300(Sc).27 From that point, adsorption of many molecules was investigated (CO2, CH4, H2O, I2 C6H6 and SO2), showing interesting properties such as the high chemical stability of this Sc(III)-based material, high CO2 adsorption and CO2/CH4 selectivity, facile adsorption–desorption, H2O adsorption for promising heat-pump applications, very high I2 uptake for relevant radioactive decontamination, benzene stabilization and effective SO2 capture with remarkable CO2/SO2 selectivity.33–37 However, none of these molecules provided any evidence of dynamic bonding behaviour.Then, in 2021, Dincă and Maurin investigated an unusual ammonia adsorption behaviour for MFM-300(Sc).26 First, upon the adsorption of NH3 (at 298 K and 1 bar) on MFM-300(Sc) a very unusual isotherm was shown (see Fig. 2), which was not correlated to any of the isotherms previously collected for other gases under the same conditions (e.g., H2, CH4 and SO2).27,35,37 Moreover, this particular adsorption/desorption behaviour was preserved over multiple cycles (corroborating the chemical stability of the material and confirmed by PXRD), which strongly suggested a fully reversible capture/release mechanism. In particular, the presence of an abnormal low-pressure step change (see Fig. 2) with an uptake of approximately 1 NH3 molecule per μ2-OH group provided the clue to a possible “partial” coordination of the NH3 molecule to the Sc(III) metal centre. Thus, force field-based Monte Carlo simulations, periodic DFT calculations and ab initio molecular dynamics (AIMD) simulations demonstrated a Sc-NH3 coordinative mode that remained stable over time, corresponding to an energetically favourable configuration (Fig. 3). Interestingly, AIMD calculations also showed that once NH3 is disconnected from the metal centre (Sc(III)), the MOF structure self-heals, and the initial crystal structure configuration is restored. Thus, such NH3-triggered dynamic metal–ligand bonding is demonstrated to be fully reversible, explaining the characteristic experimentally observed adsorption/desorption behaviour for MFM-300(Sc).
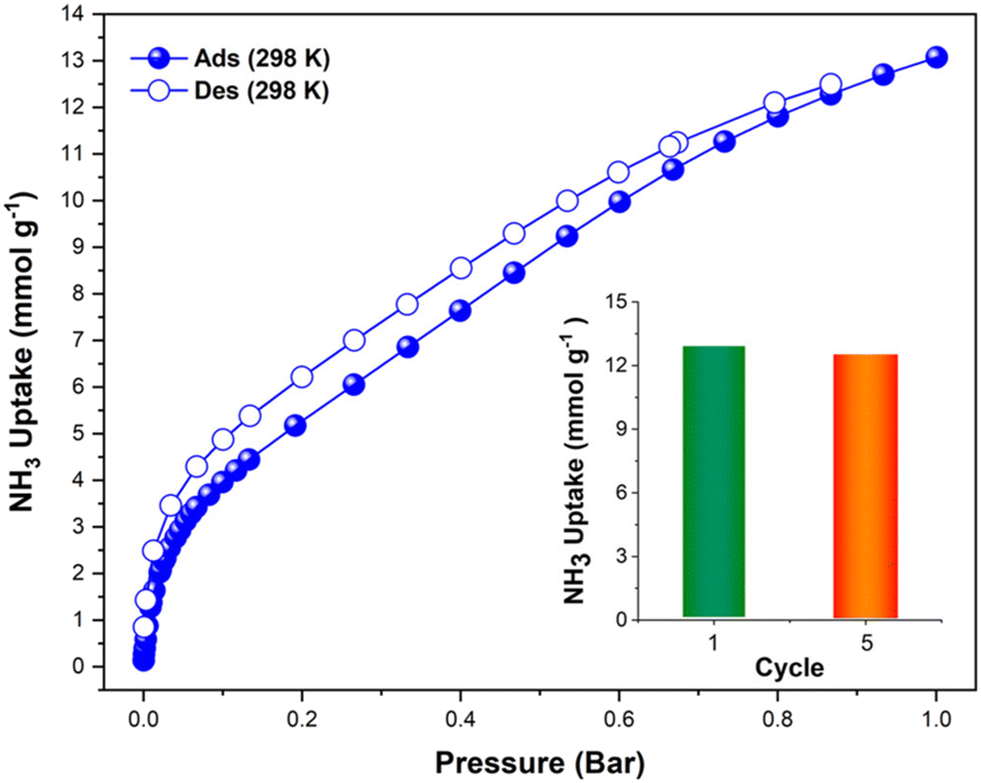 | ||
| Fig. 2 Experimental NH3 adsorption–desorption isotherms of MFM-300(Sc) (filled blue circles = adsorption; open blue circles = desorption) at 298 K and up to 1 bar. Inset: comparison of NH3 uptake of MFM-300(Sc) from the first and fifth adsorption cycles (13.1 and 12.6 mmol g−1, respectively, 3.8% decrease) at 298 K and up to 1 bar. Reproduced with permission from ref. 26. Copyright 2021, American Chemical Society. | ||
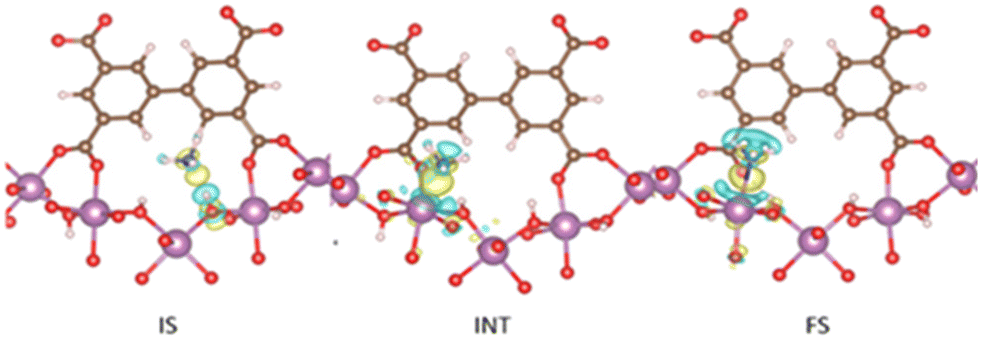 | ||
| Fig. 3 DFT-derived charge differences for the adsorption of NH3 in IS, INT, and FS with an isovalue of ±0.002 e Å−3; yellow and cyan colours represent the charge accumulation and depletion regions, respectively. Colour code: carbon (brown), oxygen (red), nitrogen (blue), hydrogen (white), and scandium (purple). Reproduced with permission from ref. 26. Copyright 2021, American Chemical Society. | ||
Finally, in situ DRIFT spectroscopy experiments were performed at 298 K, (by increasing the amount of pressure of NH3, see Fig. 4), which corroborated the computational predicted NH3 adsorption mechanism. We evidenced that when starting with a sample loaded with 0.24 bar NH3, upon applying vacuum (1.7 × 10−3 Torr) it is possible to fully recover the IR spectrum collected for the pristine sample (see Fig. 4), confirming the reversibility of the NH3 adsorption process. Although the NH3 adsorption on MFM-300(Sc) was the first concrete evidence of metal–ligand bonding (or semi-open metal sites), this interesting phenomenon was almost caught some time ago by the adsorption of H2S (vide infra).
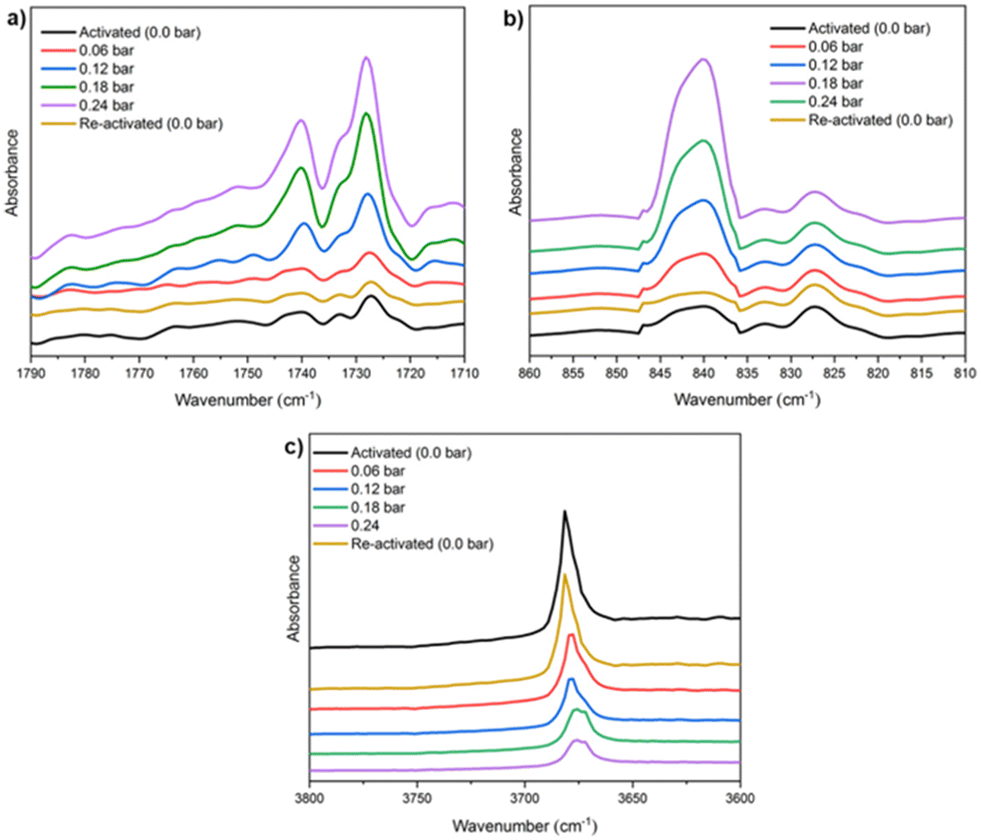 | ||
| Fig. 4 DRIFT spectra of NH3 adsorbed at different pressures (from 0 to 0.24 bar) over activated MFM-300(Sc) at 298 K, split into three wavelength regions: (a) 1790–1710 cm−1; (b) 860–810 cm−1, and (c) 3800–3600 cm−1. Reproduced with permission from ref. 26. Copyright 2021, American Chemical Society. | ||
4. H2S adsorption: the unexpected formation of polysulfides
The H2S capture on MFM-300(Sc) was investigated years before the capture of NH3, and finally, in 2020, these unusual results were published by Gutierrez-Alejandre and Maurin.38 Thus, by a set of H2S breakthrough experiments, it was consistently found that the total H2S uptake in MFM-300(Sc) was approximately 16.55 mmol g−1 (Fig. 5).38 Interestingly, when trying to reuse MFM-300(Sc) after the first adsorption of H2S by reactivating the material (at high temperatures and under vacuum), it was not possible to get back to the original uptake; instead, such total uptake went down to approximately 10 mmol g−1, even for five consecutive cycles (see Fig. 5). Although PXRD experiments exhibited the retention of the crystalline structure of MFM-300(Sc), after the exposure to H2S, N2 adsorption experiments at 77 K showed a reduction of the pore volume of approximately 34%, which suggested a partial blockage of the pores by the remaining molecules, possibly associated with a chemical reaction inside the pores of the material (i.e., chemisorption). Interestingly, PXRD experiments showed the retention of the crystalline structure (Fig. 5) after every H2S adsorption cycle, discarding a chemical reaction with MFM-300(Sc), which can partially or totally decompose the structure of the material.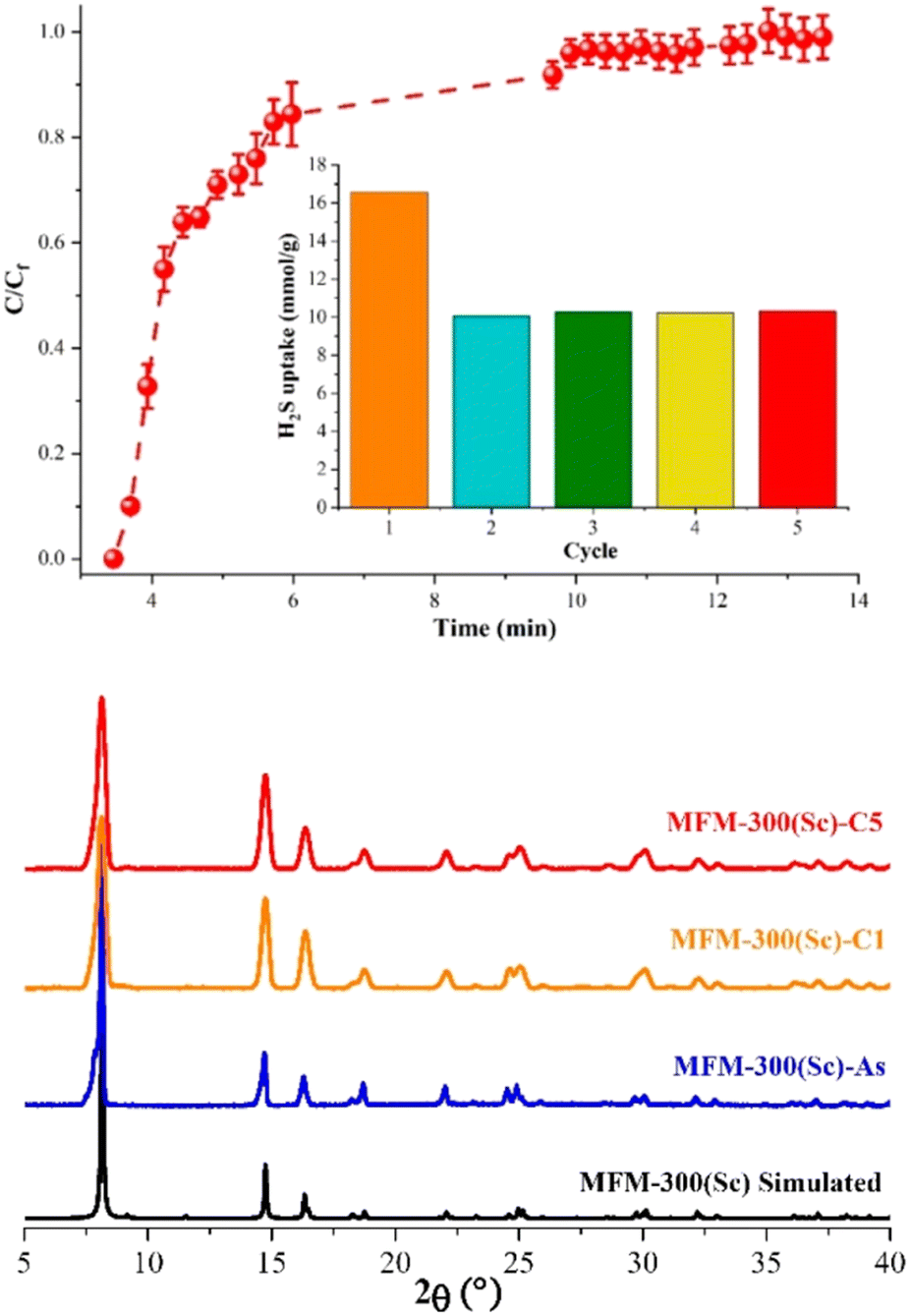 | ||
| Fig. 5 (Top) Breakthrough curves of H2S capture on MFM-300(Sc) at 25 °C and 1 bar with a H2S feed concentration of 10 vol%. The total H2S/N2 flow rate was 25 cm3 min−1. The inset shows the proportional H2S adsorption capacities for each cycle. (Bottom) PXRD patterns for MFM-300(Sc): simulated (black), as-synthesized (blue), after the first H2S adsorption–desorption cycle (cycle 1, MFM-300(Sc)-C1, orange), and after 5 H2S adsorption–desorption experiments (cycle 5, MFM-300(Sc)-C5, red). Reproduced with permission from ref. 38. Copyright 2020, American Chemical Society. | ||
Thus, puzzled by the unusual performance of MFM-300(Sc) upon H2S adsorption, cyclic H2S adsorption experiments were carried out on the isostructural material MFM-300(In), showing a total H2S uptake of 9.10 mmol g−1 for the first cycle. When performing a second cycle (after reactivating this material after the first capture of H2S), the total uptake dramatically decreased to almost 0.2 mmol g−1, and upon estimating the pore volume, a drastic reduction to 0.02 cm3 g−1 was observed. However, PXRD demonstrated the retention of the crystallinity for MFM-300(In), similarly to MFM-300(Sc). Thus, for both materials, it was possible to suggest an unexpected chemisorption process, which presumably produced unknown sulphur chemical species inside the pores without decomposing the crystal structure of MFM-300(Sc) and MFM(In). Raman spectroscopy on both “sulphur” samples was employed to uncover the nature of these chemisorbed sulphur species. Thus, the characteristic signals observed in the range 350 to 520 cm−1 are associated with the S–S stretching vibrational modes of different chain length (n = 8, 6, 4, 2, 1) polysulfides. Finally, the key approach to distinguish such polysulfides was based on the implementation of electrochemical experiments, which demonstrated that low-order polysulfides (n = 2) were produced in MFM-300(Sc), while high-order polysulfides (n = 6 and 8) are formed in MFM-300(In). Open-circuit potential (OCP) experiments exhibited a relatively small difference in the potential of the materials (0.36 V): MFM-300(Sc) (2.29 V) and MFM-300(In) (2.65 V) (see Fig. 6), which is responsible for the formation of different polysulfides.
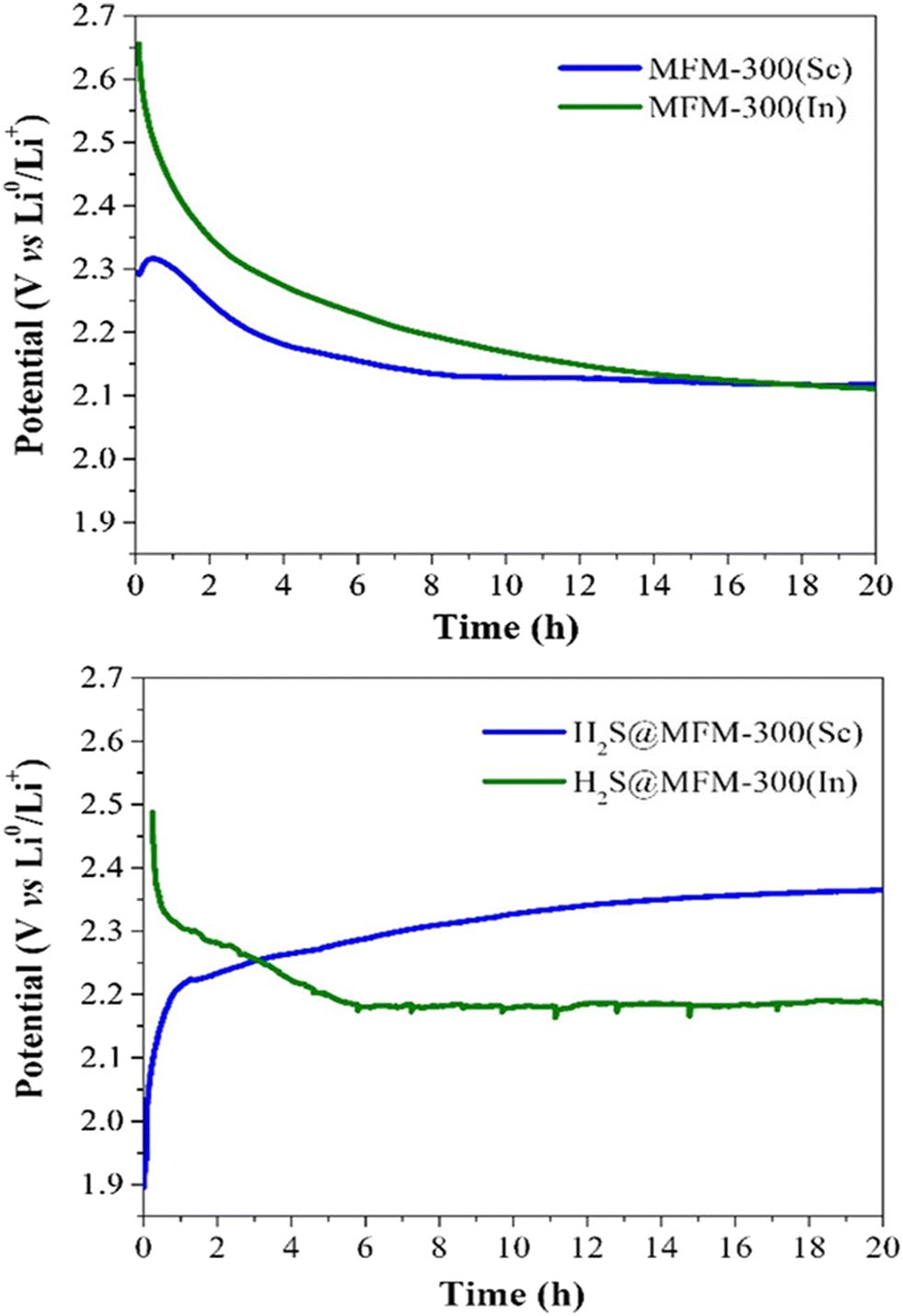 | ||
| Fig. 6 Open-circuit potential (OCP) monitoring of MFM-300(M) as active materials in cathodes for lithium-ion batteries to describe the cell stabilisation processes. (Top) Pristine materials MFM-300(Sc) and MFM-300(In); (bottom) H2S saturated materials H2S@MFM300(Sc) and H2S@MFM-300(In). Reproduced with permission from ref. 38. Copyright 2020, American Chemical Society. | ||
At that time, in 2020, the proposed catalytic mechanism for the formation of polysulfides was not fully understood, and it was postulated based on the effect of the pore confinement within MFM-300, where a H2S molecule can spontaneously react with another H2S molecule to form polysulfides.38 However, thanks to the understanding related to the NH3-triggered dynamic metal–ligand bonding, in 2021,26 a different catalytic mechanism was proposed by Gutiérrez-Alejandre.39 Thus, the generation of polysulfides is feasible due to the direct interaction of Sc(III) metal centres (semi-open metal sites) with H2S to establish a “classic catalytic reaction” (see Fig. 7), concluding that the inclusion of H2S inside the pores of MFM-300(Sc) and MFM-300(In) triggers the dynamic metal–ligand bonding. This fascinating and unexpected catalytic discovery provided the incommensurable curiosity to explore a “classic catalytic reaction” (Strecker hydrocyanation reaction) by MFM-300 (vide infra).
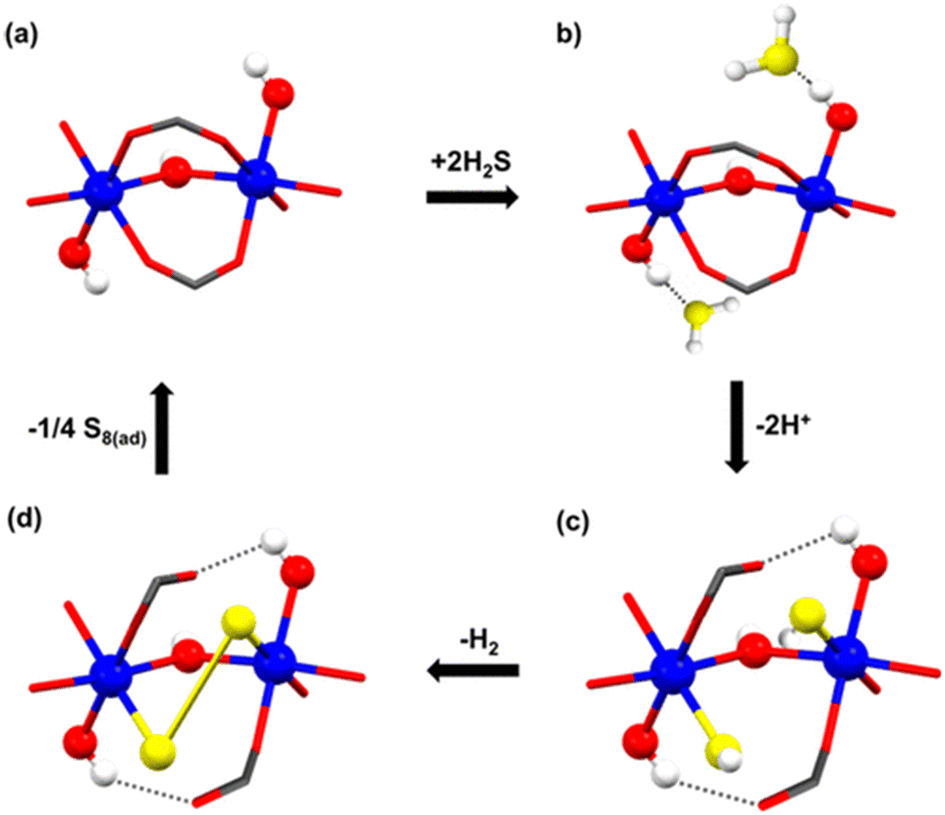 | ||
| Fig. 7 Schematic representation of the proposed catalytic steps (sequence) for the transformation of H2S to polysulfides in MFM-300(Sc). Scandium in blue, oxygen in red, hydrogen in white, and sulphur in yellow. Reproduced with permission from ref. 39. Copyright 2021, American Chemical Society. | ||
5. Heterogeneous catalysis: a highly desired application for MOFs
MOFs have been envisioned as excellent candidates for heterogeneous catalysis due to their structural tunability, vast diversity in the incorporation of functional groups, high internal surface areas, access to active metal sites, and highly crystalline structures.40–42 Typically, MOFs show access to active metals by the removal of labile terminal ligands or molecules (e.g., H2O, DMF and EtOH), affording what is known as uncoordinated metal centres or open metal sites (OMS).43,44 By this approach, countless examples of MOFs with OMS for remarkable catalytic applications have been reported in the literature.45–48However, the introduction of the new concept in MOFs related to dynamic metal–ligand bonding or semi-open metal sites was only recently reported in 2022 by Cheon-Jeong and Maurin, where MFM-300(Sc) was investigated as a catalyst for the well-known Strecker hydrocyanation reaction.20 Typically, the Strecker reaction is performed under homogeneous conditions, proceeding by the nucleophilic addition of cyanide ions to imines using different Lewis acid catalysts, such as Sc(III) metal centres (as dissolved cations).49 Thus, MFM-300(Sc) was employed to heterogeneously catalyse the formation of 2-phenyl-2-(phenylamino)acetonitrile from the reaction between N-benzylideneaniline and TMSCN,20 by taking advantage of the metal–hemilabile ligand bond dynamics in MFM-300(Sc), as was demonstrated for the conversion of H2S to polysulfides.39 Continuous 1H NMR monitoring showed that MFM-300(Sc) was an effective catalyst for this Strecker reaction with a conversion of approximately 45% (see Fig. 8). Five catalytic cycles showed a consistent catalytic performance (see Fig. 8), and PXRD experiments demonstrated the retention of the crystalline structure after the catalytic cycling (Fig. 8).
 | ||
| Fig. 8 (a) Illustration of the catalytic mechanism for the production of 2-phenyl-2-(phenylamino)acetonitrile driven by the metal–hemilabile linker dynamics in MFM-300(Sc) (C, gold; H, white; N, blue; O, red; Sc, pink). (b) PXRD data for pre- and post-catalysis of MFM-300(Sc). (c) Catalytic activity of MFM-300(Sc) for 5 cycles, in which significant loss of activity was not detected. Reproduced with permission from ref. 20. | ||
The molecule responsible for the dynamic metal–ligand bonding to have access to the Sc(III) metal centres (semi-open metal sites) is HCN, which was corroborated by DFT calculations, finding a similar mechanism observed for the NH3 adsorption.20 The proposed Strecker mechanism (see Fig. 8) begins with the formation of a bond between the C(HCN) and the Sc(III) semi-open metal site, followed by a proton transfer from the coordinated HCN to the N(imine), and the −CN group remained coordinated to the unsaturated Sc(III). Then, the second step continues with a change of the −CN to −NC, which is perfectly fitting for a nucleophilic attack on the intermediate imine species, affording the formation of the cyanylated product.
Later, in 2023, Cheon-Jeong and Maurin evaluated this same reaction with an isostructural material to MFM-300(Sc) based on In(III): MFM-300(In).50 This material also showed access to semi-open metal sites, which was examined by variable temperature resonance Raman spectroscopy (VT-RRS) and variable temperature diffuse reflectance infrared Fourier-transform spectroscopy (VT-DRIFTS), showing that at higher temperatures, more available active In(III) semi-open metal sites are formed (see Fig. 9). Thus, the Strecker reaction performed with MFM-300(In) afforded a 41% conversion to 2-phenyl-2-(phenylamino)acetonitrile, which was similar to that obtained with MFM-300(Sc). Remarkably, since active In(III) sites can be obtained by simply increasing the temperature, the same Strecker reaction was carried out at a higher temperature (323 K), achieving a higher conversion of 93%.
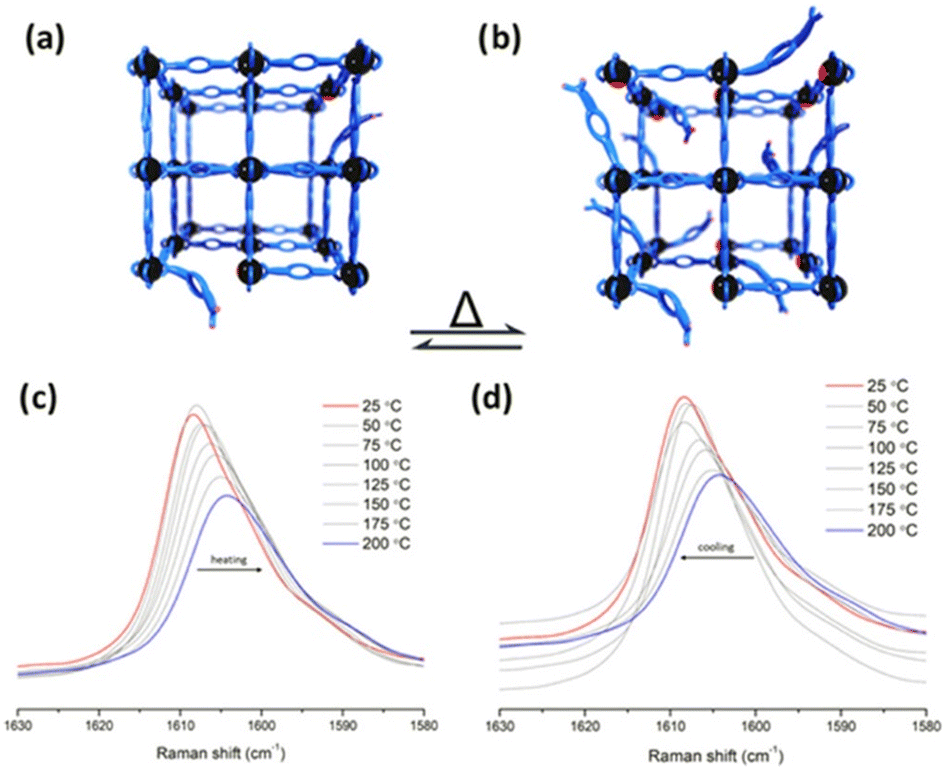 | ||
| Fig. 9 Representation of transient open metal sites generated by hemilabile metal–ligand bonds and poised for catalysis at (a) lower temperatures and (b) higher temperatures. VT-RRS studies of InOF-1 (c) from 200 to 25 °C and (d) from 25 to 200 °C. Reproduced with permission from ref. 50. Copyright 2023, American Chemical Society. | ||
Finally, MFM-300(Sc) was investigated for the acetone hydrogenation reaction, finding that the semi-open metal sites were active for this gas-phase reaction, affording the formation of methyl isobutyl ketone.51 This reaction was successfully performed at 453 K for 4 hours, yielding a conversion of approximately 28%. After two additional catalytic cycles, the catalytic performance was maintained, and PXRD experiments corroborated the retention of the crystalline structure of MFM-300(Sc) with no significant changes in the BET surface. The proposed catalytic reaction mechanism involves all three steps that occur at the Sc(III) semi-open metal sites (Fig. 10).
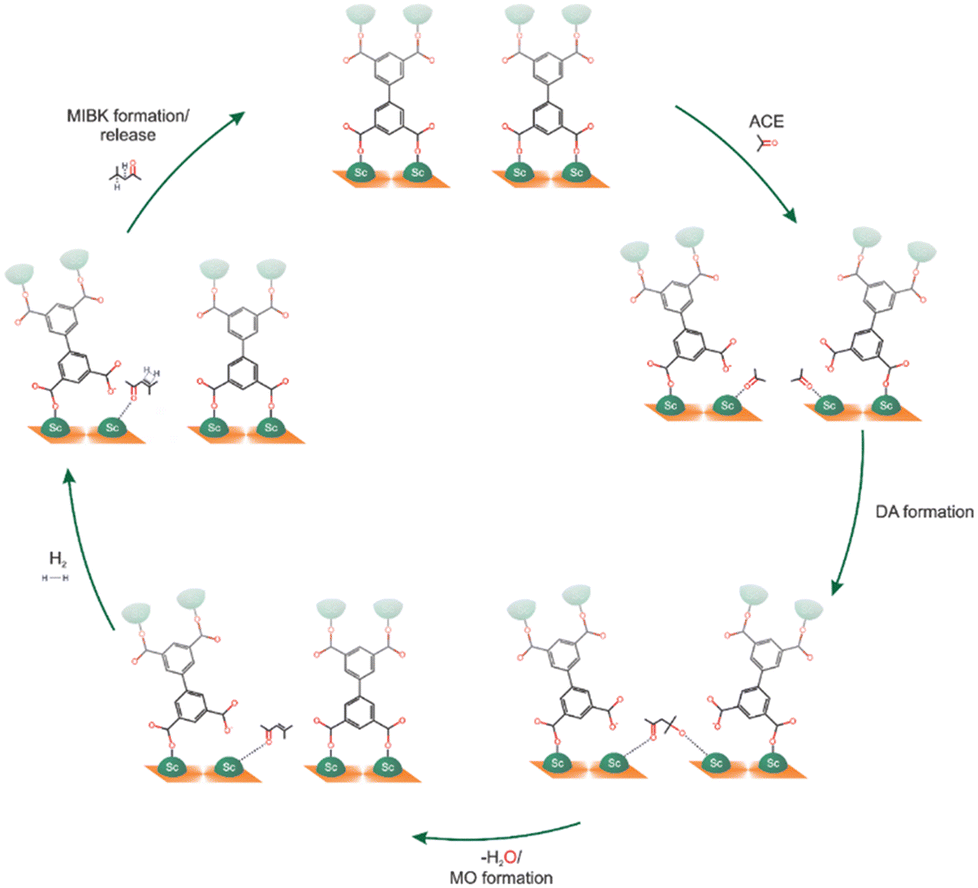 | ||
| Fig. 10 Scheme showing the proposed acetone hydrogenation mechanism in MFM-300(Sc). Reproduced with permission from ref. 51. Copyright 2023, the Royal Society of Chemistry. | ||
These catalytic results for MFM-300(Sc) and MFM-300(In) demonstrate that due to the access to semi-open metal sites (i.e., Sc(III) and In(III)), these chemically stable materials can effectively work as a heterogeneous catalyst in either aqueous or gaseous medium, highlighting the dynamic metal–ligand bonding as the key role in high performances for novel heterogenous catalysts with remarkable regeneration properties.
6. Conclusions
This highlight provides a chronological account of the experimental and computational observations that revealed the unique dynamic metal–ligand bonding for MFM-300(Sc) and MFM-300(In). Remarkably, such dynamic binding, in the case of MFM-300(Sc), provided access to temporary open metal sites (semi-open metal sites), opening new portals for exciting and unexpected chemistry, such as the description of complex adsorption mechanisms (i.e., NH3 adsorption), paving the way toward advance developments of MOFs for the capture of highly corrosive molecules.The unexpected in situ chemical transformation of H2S inside the pores of MFM-300(Sc) and MFM-300(In) to generate polysulfides not only is an exciting discovery but also can signify the permanent sequestration and conversion of toxic H2S for new environmental remediation technologies against air pollution. In addition, the homogeneous insertion of polysulfides (the homogeneous sulphur distribution affords higher electrochemical performance) within MOFs can provide new energy platforms for the development of novel MOF–sulphur battery electrodes, as recently demonstrated by Marquez and Mullins, who showed how SU-101 (a Bi(III)-based MOF with “classic” open metal sites) transformed H2S into polysulfides to fabricate lithium–sulphur batteries.52 Thus, exploring MFM-300 for the investigation of how toxic H2S emissions can be directly converted and applied in lithium–sulphur batteries, considering that the different metals that constitute the MFM-300 family can provide different polysulfides, opens exciting options for different capacities to power novel lithium–sulphur batteries.
MFM-300(Sc) and MFM-300(In) demonstrated promising results in developing novel “intelligent” catalysts, taking advantage of how molecule inclusion can trigger dynamic metal–ligand bonding (semi-open metal sites). Thus, understanding that selected examples of MOFs (a great and fascinating task to investigate) are dynamic rather than static environments can dramatically expand the reservoir of candidate MOF materials which can be contemplated for classic and new heterogenous catalytic applications.
Finally, the fundamental understanding of MOFs remains developing. In particular, the discovery of dynamic metal–ligand bonding in a wide range of MOF materials (starting with the MFM-300 family and a remarkable example recently reported by Farha and co-workers where a Zr(IV)-based MOF demonstrated unexpected SO2-induced modulator-node dynamics53) turns out to be crucial for discovering new horizons with exciting applications.
Data availability
All data are available in the main text.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I. A. I. thanks PAPIIT-UNAM (IN201123). We thank U. Winnberg (Euro Health) for scientific discussions and G. Ibarra-Winnberg for scientific encouragement.References
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1989, 111, 5962–5964 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- S. Liu, R. Dupuis, D. Fan, S. Benzaria, M. Bonneau, P. Bhatt, M. Eddaoudi and G. Maurin, Chem. Sci., 2024, 15, 5294–5302 RSC.
- R. A. Peralta, M. T. Huxley, J. D. Evans, T. Fallon, H. Cao, M. He, X. S. Zhao, S. Agnoli, C. J. Sumby and C. J. Doonan, J. Am. Chem. Soc., 2020, 142, 13533–13543 CrossRef CAS PubMed.
- M. Moharramnejad, A. Ehsani, M. Shahi, S. Gharanli, H. Saremi, R. E. Malekshah, Z. S. Basmenj, S. Salmani and M. Mohammadi, J. Drug Delivery Sci. Technol., 2023, 81, 104285 CrossRef CAS.
- J. L. Obeso, D. R. Amaro, C. V. Flores, A. Gutiérrez-Alejandre, R. A. Peralta, C. Leyva and I. A. Ibarra, Coord. Chem. Rev., 2023, 485, 215135 CrossRef CAS.
- E. Martínez-Ahumada, M. L. Díaz-Ramírez, M. d. J. Velásquez-Hernández, V. Jancik and I. A. Ibarra, Chem. Sci., 2021, 12, 6772–6799 RSC.
- T. K. Pal, Mater. Chem. Front., 2023, 7, 405–441 RSC.
- J. L. Obeso, M. T. Huxley, J. A. de los Reyes, S. M. Humphrey, I. A. Ibarra and R. A. Peralta, Angew. Chem., Int. Ed., 2023, 62, e202309025 CrossRef CAS PubMed.
- J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Soc. Rev., 2014, 43, 5933–5951 RSC.
- A. B. Andreeva, K. N. Le, L. Chen, M. E. Kellman, C. H. Hendon and C. K. Brozek, J. Am. Chem. Soc., 2020, 142, 19291–19299 CrossRef CAS PubMed.
- A. B. Andreeva, K. N. Le, K. Kadota, S. Horike, C. H. Hendon and C. K. Brozek, Chem. Mater., 2021, 33, 8534–8545 CrossRef CAS.
- A. Halder and D. Ghoshal, CrystEngComm, 2018, 20, 1322–1345 RSC.
- P. K. Allan, K. W. Chapman, P. J. Chupas, J. A. Hriljac, C. L. Renouf, T. C. A. Lucas and R. E. Morris, Chem. Sci., 2012, 3, 2559–2564 RSC.
- M. Wahiduzzaman, D. Lenzen, G. Maurin, N. Stock and M. T. Wharmby, Eur. J. Inorg. Chem., 2018, 2018, 3626–3632 CrossRef CAS.
- D. Fairen-Jimenez, S. A. Moggach, M. T. Wharmby, P. A. Wright, S. Parsons and T. Düren, J. Am. Chem. Soc., 2011, 133, 8900–8902 CrossRef CAS PubMed.
- A. J. Martínez-Martínez, C. G. Royle, S. K. Furfari, K. Suriye and A. S. Weller, ACS Catal., 2020, 10, 1984–1992 CrossRef PubMed.
- S. D. Pike, A. L. Thompson, A. G. Algarra, D. C. Apperley, S. A. Macgregor and A. S. Weller, Science, 2012, 337, 1648–1651 CrossRef CAS PubMed.
- K. Leus, I. Muylaert, M. Vandichel, G. B. Marin, M. Waroquier, V. Van Speybroeck and P. Van Der Voort, Chem. Commun., 2010, 46, 5085–5087 RSC.
- R. A. Peralta, P. Lyu, A. Lopez-Olvera, J. L. Obeso, C. Leyva, N. C. Jeong, I. A. Ibarra and G. Maurin, Angew. Chem., Int. Ed., 2022, 61, e202210857 CrossRef CAS PubMed.
- C.-C. Cao, C.-X. Chen, Z.-W. Wei, Q.-F. Qiu, N.-X. Zhu, Y.-Y. Xiong, J.-J. Jiang, D. Wang and C.-Y. Su, J. Am. Chem. Soc., 2019, 141, 2589–2593 CrossRef CAS PubMed.
- E. Sánchez-González, M. Y. Tsang, J. Troyano, G. A. Craig and S. Furukawa, Chem. Soc. Rev., 2022, 51, 4876–4889 RSC.
- B. Wang, R.-B. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef CAS PubMed.
- A. Półrolniczak and A. Katrusiak, Mater. Adv., 2021, 2, 4677–4684 RSC.
- J. L. Obeso, M. T. Huxley, C. Leyva, J. Gabriel Flores, N. Martín-Guaregua, M. Viniegra, J. Aguilar-Pliego, J. A. de los Reyes, I. A. Ibarra and R. A. Peralta, Coord. Chem. Rev., 2023, 496, 215403 CrossRef CAS.
- P. Lyu, A. M. Wright, A. López-Olvera, P. G. M. Mileo, J. A. Zárate, E. Martínez-Ahumada, V. Martis, D. R. Williams, M. Dincă, I. A. Ibarra and G. Maurin, Chem. Mater., 2021, 33, 6186–6192 CrossRef CAS.
- I. A. Ibarra, S. Yang, X. Lin, A. J. Blake, P. J. Rizkallah, H. Nowell, D. R. Allan, N. R. Champness, P. Hubberstey and M. Schröder, Chem. Commun., 2011, 47, 8304–8306 RSC.
- X. Han, H. G. W. Godfrey, L. Briggs, A. J. Davies, Y. Cheng, L. L. Daemen, A. M. Sheveleva, F. Tuna, E. J. L. McInnes, J. Sun, C. Drathen, M. W. George, A. J. Ramirez-Cuesta, K. M. Thomas, S. Yang and M. Schröder, Nat. Mater., 2018, 17, 691–696 CrossRef CAS PubMed.
- J. Qian, F. Jiang, D. Yuan, M. Wu, S. Zhang, L. Zhang and M. Hong, Chem. Commun., 2012, 48, 9696–9698 RSC.
- L. Briggs, R. Newby, X. Han, C. G. Morris, M. Savage, C. P. Krap, T. L. Easun, M. D. Frogley, G. Cinque, C. A. Murray, C. C. Tang, J. Sun, S. Yang and M. Schröder, J. Mater. Chem. A, 2021, 9, 7190–7197 RSC.
- Z. Lu, H. G. W. Godfrey, I. da Silva, Y. Cheng, M. Savage, F. Tuna, E. J. L. McInnes, S. J. Teat, K. J. Gagnon, M. D. Frogley, P. Manuel, S. Rudić, A. J. Ramirez-Cuesta, T. L. Easun, S. Yang and M. Schröder, Nat. Commun., 2017, 8, 14212 CrossRef CAS PubMed.
- C. P. Krap, R. Newby, A. Dhakshinamoorthy, H. García, I. Cebula, T. L. Easun, M. Savage, J. E. Eyley, S. Gao, A. J. Blake, W. Lewis, P. H. Beton, M. R. Warren, D. R. Allan, M. D. Frogley, C. C. Tang, G. Cinque, S. Yang and M. Schröder, Inorg. Chem., 2016, 55, 1076–1088 CrossRef CAS PubMed.
- X. Zhang, I. da Silva, H. G. W. Godfrey, S. K. Callear, S. A. Sapchenko, Y. Cheng, I. Vitórica-Yrezábal, M. D. Frogley, G. Cinque, C. C. Tang, C. Giacobbe, C. Dejoie, S. Rudić, A. J. Ramirez-Cuesta, M. A. Denecke, S. Yang and M. Schröder, J. Am. Chem. Soc., 2017, 139, 16289–16296 CrossRef CAS PubMed.
- M. Rivera-Almazo, E. Perez-Sanchez, E. Martínez-Ahumada, A. Martínez, J. Garza, I. A. Ibarra and R. Vargas, J. Phys. Chem. C, 2022, 126, 6465–6471 CrossRef CAS.
- M. R. Gonzalez, J. H. González-Estefan, H. A. Lara-García, P. Sánchez-Camacho, E. I. Basaldella, H. Pfeiffer and I. A. Ibarra, New J. Chem., 2015, 39, 2400–2403 RSC.
- I. A. Ibarra, A. Mace, S. Yang, J. Sun, S. Lee, J.-S. Chang, A. Laaksonen, M. Schröder and X. Zou, Inorg. Chem., 2016, 55, 7219–7228 CrossRef CAS PubMed.
- J. A. Zárate, E. Sánchez-González, D. R. Williams, E. González-Zamora, V. Martis, A. Martínez, J. Balmaseda, G. Maurin and I. A. Ibarra, J. Mater. Chem. A, 2019, 7, 15580–15584 RSC.
- J. G. Flores, J. A. Zarate-Colin, E. Sanchez-Gonzalez, J. R. Valenzuela, A. Gutierrez-Alejandre, J. Ramirez, V. Jancik, J. Aguilar-Pliego, M. C. Zorrilla, H. A. Lara-Garcia, E. Gonzalez-Zamora, G. Guzman-Gonzalez, I. Gonzalez, G. Maurin and I. A. Ibarra, ACS Appl. Mater. Interfaces, 2020, 12, 18885–18892 CrossRef CAS PubMed.
- A. López-Olvera, J. G. Flores, J. Aguilar-Pliego, C. K. Brozek, A. Gutiérrez-Alejandre and I. A. Ibarra, Chem. Mater., 2021, 33, 6269–6276 CrossRef.
- J. L. Obeso, J. G. Flores, C. V. Flores, M. T. Huxley, J. A. de los Reyes, R. A. Peralta, I. A. Ibarra and C. Leyva, Chem. Commun., 2023, 59, 10226–10242 RSC.
- A. Dhakshinamoorthy, A. M. Asiri and H. García, Trends Chem., 2020, 2, 454–466 CrossRef CAS.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed.
- Ü. Kökçam-Demir, A. Goldman, L. Esrafili, M. Gharib, A. Morsali, O. Weingart and C. Janiak, Chem. Soc. Rev., 2020, 49, 2751–2798 RSC.
- J. Bae, E. J. Lee and N. C. Jeong, Chem. Commun., 2018, 54, 6458–6471 RSC.
- J. L. Obeso, J. G. Flores, C. V. Flores, V. B. López-Cervantes, V. Martínez-Jiménez, J. A. de los Reyes, E. Lima, D. Solis-Ibarra, I. A. Ibarra, C. Leyva and R. A. Peralta, Dalton Trans., 2023, 52, 12490–12495 RSC.
- J. G. Flores, J. L. Obeso, V. Martínez-Jiménez, N. Martín-Guaregua, A. Islas-Jácome, E. González-Zamora, H. Serrano-Espejel, B. Mondragón-Rodríguez, C. Leyva, D. A. Solís-Casados, I. A. Ibarra, R. A. Peralta, J. Aguilar-Pliego and J. A. de los Reyes, RSC Adv., 2023, 13, 27174–27179 RSC.
- S. H. Park, R. A. Peralta, D. Moon and N. C. Jeong, J. Mater. Chem. A, 2022, 10, 23499–23508 RSC.
- J. L. Obeso, J. Gabriel Flores, C. V. Flores, R. Rios-Escobedo, J. Aguilar-Pliego, A. Ken Inge, J. A. de los Reyes, R. A. Peralta, I. A. Ibarra and C. Leyva, ChemCatChem, 2023, 15, e202300471 CrossRef CAS.
- S. Kobayashi and T. Busujima, Chem. Commun., 1998, 981–982, 10.1039/A801464G.
- R. A. Peralta, M. T. Huxley, P. Lyu, M. L. Diaz-Ramirez, S. H. Park, J. L. Obeso, C. Leyva, C. Y. Heo, S. Jang, J. H. Kwak, G. Maurin, I. A. Ibarra and N. C. Jeong, ACS Appl. Mater. Interfaces, 2023, 15, 1410–1417 CrossRef CAS PubMed.
- J. L. Obeso, A. Lopez-Olvera, C. V. Flores, R. A. Peralta, I. A. Ibarra and C. Leyva, Chem. Commun., 2023, 59, 3273–3276 RSC.
- R. A. Marquez, J. L. Obeso, R. R. Vaidyula, V. B. López-Cervantes, R. A. Peralta, P. Marín Rosas, J. A. de los Reyes, C. B. Mullins and I. A. Ibarra, J. Mater. Chem. A, 2024 10.1039/D4TA03620D.
- W. Gong, Y. Xie, A. Yamano, S. Ito, X. Tang, E. W. Reinheimer, C. D. Malliakas, J. Dong, Y. Cui and O. K. Farha, J. Am. Chem. Soc., 2023, 145, 26890–26899 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |