
Photo-oxidation of methanol in complexes with pyrido[2,3-b]pyrazine: a nonadiabatic molecular dynamics study†
Joanna
Jankowska
 *a and
Andrzej L.
Sobolewski
b
*a and
Andrzej L.
Sobolewski
b
aFaculty of Chemistry, University of Warsaw, Pasteura 1, Warsaw 02-093, Poland. E-mail: jjankowska@chem.uw.edu.pl
bInstitute of Physics, Polish Academy of Sciences, al. Lotników 32/46, Warsaw 02-668, Poland
First published on 30th November 2023
Abstract
Excited-state Proton-Coupled Electron Transfer (PCET) constitutes a key step in the photo-oxidation of small, electron-rich systems possessing acidic hydrogen atoms, such as water or alcohols, which can play a vital role in green hydrogen production. In this contribution, we employ ab initio quantum-chemical methods and on-the-fly nonadiabatic molecular dynamics simulations to study the mechanism and the photodynamics of PCET in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 pyrido[2,3-b]pyrazine complexes with methanol. We find the process to be ultrafast and efficient when the intramolecular hydrogen bond is formed with one of the β-positioned nitrogen atoms. The complex exhibiting a hydrogen bond with an isolated nitrogen site, on the contrary, shows much lower reactivity. We explain this effect with the stabilization of the reactive ππ* charge-transfer electronic state in the former case.
:1 pyrido[2,3-b]pyrazine complexes with methanol. We find the process to be ultrafast and efficient when the intramolecular hydrogen bond is formed with one of the β-positioned nitrogen atoms. The complex exhibiting a hydrogen bond with an isolated nitrogen site, on the contrary, shows much lower reactivity. We explain this effect with the stabilization of the reactive ππ* charge-transfer electronic state in the former case.
Introduction
Photooxidation of organic matter via the excited-state Proton-Coupled Electron Transfer (PCET) reaction, involving the transfer of both a proton and an electron, is a fundamental process that occurs in natural and engineered environments. PCET plays a crucial role in various environmental, biological, and technological systems.1–4 Understanding the mechanisms and dynamics of this reaction is of great significance for advancing renewable energy technologies and developing sustainable methods for fuel production. Investigation of the intermolecular PCET process in hydrogen-bonded complexes is widely used in theoretical modeling of hydrogen-evolution photocatalysis (for recent reviews, see5,6).The information that can be obtained with static ab initio electronic structure calculations of PCET, such as reaction energies or reaction barriers, is only indirectly related to the data measured in experiment. A more substantial support of the experiments can be provided by first-principles simulations of the photoinduced reaction dynamics. This includes simulating the ultrafast nonadiabatic relaxation of the initially populated excited singlet state of the complex. Such simulations were recently performed for several hydrogen-bonded complexes of azaaromatic chromophores with water molecules.7–10 and with phenol.11
It has been shown both experimentally and theoretically that an increase in the electron-donating strength of the substrate lowers its oxidation potential, which, in turn, lowers the barrier of the PCET reaction on the S1 potential energy surface of the complex.12,13 Nonadiabatic dynamic simulations performed for such complexes support this observation, too.12,14 A similar experiment, performed on hexaazatrinaphthylene (HATN) dissolved in water and in methanol, confirms the relation between the electron-donating strength of the substrate and the yield of the PCET reaction.15
The photooxidation of alcohols is generally representative of the photooxidation of organic matter,16,17 with potential applications in various fields, such as organic synthesis and renewable energy conversion.18–20 Understanding the mechanisms and factors influencing the photooxidation of organic matter via PCET is essential for developing strategies to control and optimize these processes. By harnessing the power of light and PCET, researchers aim to design innovative and sustainable solutions for environmental remediation and the degradation of organic pollutants,21,22 with potential applications in various fields, such as organic synthesis,23–25 and renewable energy conversion.4
The goal of this work is to elaborate a qualitative ab initio-based picture of the PCET reaction mechanism and dynamics in a photoexcited pyrido[2,3-b]pyrazine complex with methanol (PrdPyr-MeOH, Fig. 1), with the PrdPyr molecule representing in this study the reactive center of a HATN-like system.
 | ||
| Fig. 1 Geometrical structures and ZPE-corrected relative energies of the optimized PrdPyr-MeOH S0 minima, with indicated HB length values. | ||
Methods
Ab initio electronic structure calculations
Structural optimizations, potential energy profile scans, and harmonic vibrational frequencies in the ground electronic state were obtained with the Møller–Plesset second order perturbation theory (MP2),26 while in excited-state calculations (i.e., structural optimizations, UV-Vis absorption properties calculations, and reaction-profiles scans) the second order algebraic diagrammatic construction method (ADC(2)) was used.27 Minimum-energy S1/S0 conical intersection structures were optimized at the XMS-CASPT2 level of theory,28 and with use of the penalty functional method combined with ADC(2), as proposed by Levine et al.29 In additional, benchmark calculations we also employed the quasidegenerate n-electron valence state perturbation theory (QD-NEVPT) approach,30 and the spin-component scaled (SCS) variant of ADC(2).31 The active space in the multireference calculations consisted of 8 electrons in 6 orbitals, and the state averaging over two states with equal weights was performed. All calculations employed the Dunning correlation consistent double-zeta (cc-pVDZ) basis set.32The structural optimizations and evaluation of the excited-state properties at the MP2, ADC(2), and SCS-ADC(2) level were performed with the TURBOMOLE V7.1 software,33 while the conical-intersection optimizations were run with the Bagel,34 and CIOpt29 suite of programs. The QD-NEVPT2 calculations were done with ORCA version 5.0.3.35,36 The charge-transfer (CT) properties, and the natural transition orbitals (NTO) were obtained with the TheoDORE 3.0 program.37
Nonadiabatic molecular dynamics simulations
Quantum-classical nonadiabatic molecular dynamics (NAMD) simulations within the Tully surface hopping (TSH) approach were employed in the dynamic part of the study, with the electronic structure of the system treated at the ADC(2)/cc-pVDZ level. Two electronic states were explicitly included in the dynamics: S2 and S1. Due to limitations of the single-reference ADC(2) method near the S1/S0 intersection, no nonadiabatic couplings to the ground state were computed, and the dynamics was terminated when the energy difference between the currently-occupied and the ground electronic state dropped below 0.20 eV. Reaching this degeneracy threshold was taken as an indicator of internal conversion to S0.The ab initio on-the-fly NAMD simulations included 75 surface-hopping trajectories per each of the studied complexes. The simulations covered up to 300 fs of the dynamics, with 0.5 fs timestep set for the nuclear motion, for which the velocity Verlet algorithm was applied to solve Newtons equations. The electronic equations were solved with a 0.025 fs timestep, using interpolated energies and wavefunctions between consecutive nuclear configurations. The initial conditions were generated by sampling coordinates and momenta obtained from the zero-temperature Wigner distribution,38 with subsequent S0 → S2 photon absorption probability filtering.39 The decoherence correction of Grannucci and Persico was employed to account for the quantum decoherence effects.40
The dynamic simulations were performed with the Newton X software (Version 2.0),41,42 coupled to the Turbomole program33 for the electronic-structure evaluation.
Results and discussion
PrdPyr-MeOH structure and energetics in the S0 state
Optimization of the PrdPyr-MeOH geometry in the ground electronic state led to three local minima: PPMet-1, PPMet-6, and PPMet-4, whose structures and zero-point (ZPE) corrected relative energies are shown in Fig. 1. In all cases, the MeOH molecule acts as a proton donor in a single hydrogen bond (HB) formed with one of nitrogen atoms of the PrdPyr moiety. In Table 1, one can find selected geometrical parameters of the obtained structures. Upon inspection, a similar N–H distance (i.e., the HB length) of ca. 2.0 Å is noticed in all three conformers, which falls into a range characteristic for medium-strength HBs in organic systems.43 Interestingly, the MeOH molecule orients itself (the CONC dihedral angle) with different level of perpendicularity with respect to the PrdPyr plane, which is likely driven by interplay between steric repulsion between PrdPyr and the methyl group, and the attractive interaction between the methyl hydrogens and the adjacent electronegative center. This orientation directly impacts the electronic coupling between the proton-donor molecule and the proton-acceptor moiety, making an important difference as compared to analogous water complexes formed with multi-center, electron-rich organic molecules, in which the water molecule usually adopts an all-planar orientation.9,10,44 For the complete data on the S0PrdPyr-MeOH molecular structures, please refer to Cartesian coordinates included in the ESI.†| PPMet-1 | PPMet-6 | PPMet-4 | |
|---|---|---|---|
| NH | 1.96 | 1.97 | 2.00 |
| OH | 0.98 | 0.97 | 0.97 |
| OX | 3.21 | 3.61 | 3.56 |
| CNC | 117 | 116 | 116 |
| CONC | 96 | 58 | 50 |
UV-vis absorption at the Franck–Condon region
Vertical UV-Vis absorption properties of PPMet-1, PPMet-6, and PPMet-4 are summarized in Table 2; for the plots of key Natural Transition Orbitals (NTOs) for the PPMet-4 system, check Fig. 2, while the NTOs for the remaining transitions and other complexes can be found in Fig. S1–S3 in the ESI.† The vertical electronic excitation properties, calculated up to the S5 state, reveal several characteristic optical features of the PrdPyr-MeOH system. Firstly, in all cases the S1 state is dark, of nπ* character, and is mostly localized on the PrdPyr molecule, with just a minor MeOH → PrdPyr charge transfer (CT) contribution noticeable for PPMet-4. Secondly, the absorbing state, S2, originates from a pure ππ* transition within the PrdPyr moiety. The S2 state, separated from the S1 state by almost 1.0 eV in the Franck–Condon region, is followed by a manifold of closer-lying states of mostly nπ* character and negligible oscillator strengths. Hence, in the low-energy excitation regime, the photochemistry of the PrdPyr-MeOH system is expected to depend primarily on the S2 and S1 states′ properties.| State | E/eV | f | Character | |
|---|---|---|---|---|
| PPMet-1 | S1 | 3.53 | 0.000 | LE (nπ*) |
| S2 | 4.43 | 0.144 | LE (ππ*) | |
| S3 | 4.59 | 0.005 | LE (nπ*) | |
| S4 | 4.82 | 0.000 | LE (nπ*) | |
| S5 | 5.16 | 0.054 | LE (ππ*) | |
| PPMet-6 | S1 | 3.57 | 0.001 | LE (nπ*) |
| S2 | 4.56 | 0.145 | LE (ππ*) | |
| S3 | 4.72 | 0.004 | LE/CT (nπ*) | |
| S4 | 4.86 | 0.001 | LE (nπ*) | |
| S5 | 4.99 | 0.000 | CT (ππ*) | |
| PPMet-4 | S1 | 3.56 | 0.001 | LE/CT (nπ*) |
| S2 | 4.44 | 0.146 | LE (ππ*) | |
| S3 | 4.68 | 0.005 | LE/CT (nπ*) | |
| S4 | 4.85 | 0.001 | LE (nπ*) | |
| S4 | 4.82 | 0.000 | LE (nπ*) | |
| S5 | 4.97 | 0.000 | CT (ππ*) |
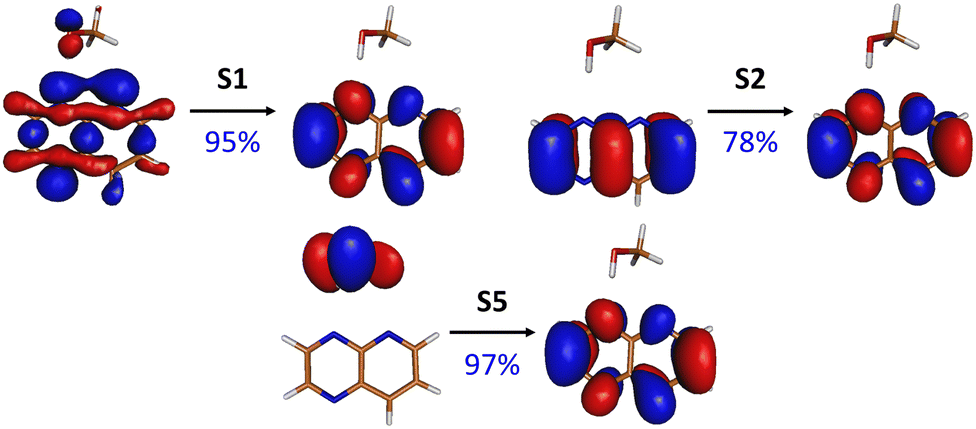 | ||
| Fig. 2 NTOs (and their respective contributions) for selected electronic transitions of the PPMet-4 complex in its S0-optimized geometry; the NTOs for remaining transitions and NTOs for other complexes can be found in Fig. S1–S3 in the ESI.† | ||
It is worth to note, however, the ππ* transition to S5 at about 5.0 eV. While in PPMet-1 this transition is localized at PrdPyr and posses some oscillator strength, it reveals pure CT character in the case of PPMet-6 and PPMet-4, in which it corresponds to an electron transfer from MeOH to PrdPyr. The important difference between S5 and lower-lying dark states is its ππ* character, involving the oxygen lone electron pair coupled to the C–H bonding orbitals, interacting with the π density at the PrdPyr moiety (Fig. 2 and Fig. S1–S3 in the ESI†). This transition, in contrast to nπ* excitations, exhibits character observed previously in systems undergoing efficient EDPT transformations.6,9
Finally, one may have a look at overlapped absorption spectra calculated for the Wigner distribution of vibrationally distorted molecular structures,45 shown in Fig. 3. For each system, 150 points were included, with Lorentzian peak broadening of 0.05 eV at the half maximum. In general, for all three systems, the low-energy absorption is dominated by a strong peak centered around 4.4 eV which can be primarily assigned to the S0 → S2 transition, with gradually growing contributions from the S0 → S3 and higher transitions at increasing excitation energy. The low-energy shoulder visible around 3.2–3.7 eV originates from the very weak S0 → S1 absorption.
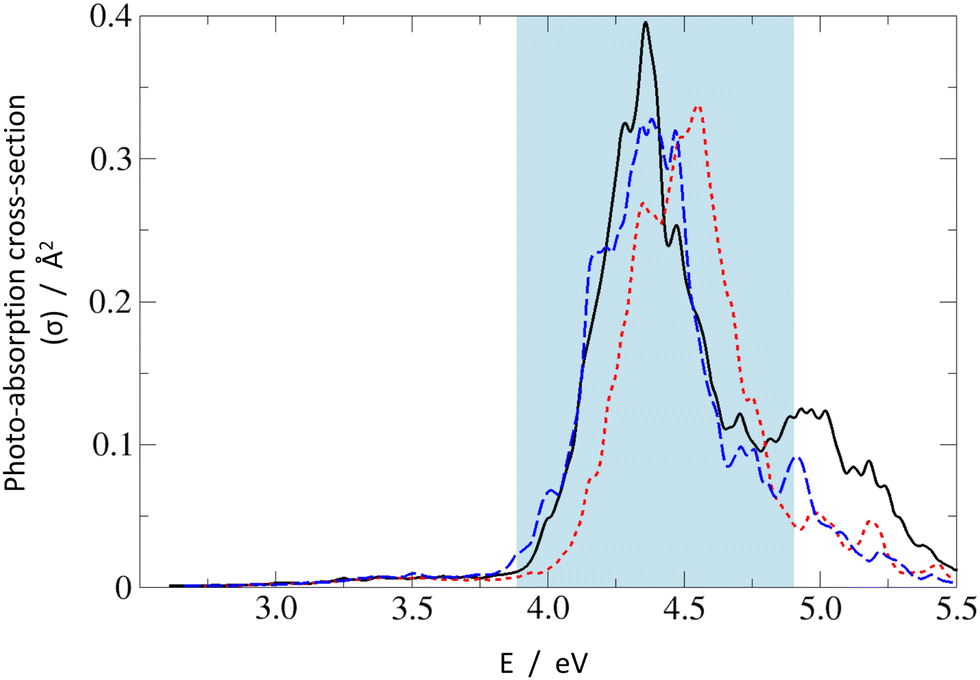 | ||
| Fig. 3 UV-Vis absorption spectra of PPMet-1 (black solid line), PPMet-6 (red dotted line), and PPMet-4 (blue dashed line) calculated with the Wigner distribution. The shadowed area marks the excitation energy window applied in the NAMD simulations. | ||
Static exploration of the mechanism of the EDPT reaction
To get a notion of photochemical properties of the PrdPyr-MeOH system, before calculating the potential energy profiles (PEPs), we performed unconstrained structural optimization of all complexes, firstly in the S2 electronic state and, subsequently, in the S1 state, with the latter optimization starting from the respective S2 minimum. All resulting structures and their energies are shown in Fig. 4. In the course of these optimizations, a local S2 minimum positioned in the Franck–Condon region has been found for each complex. In all cases, the obtained geometries resembled closely the starting S0 structures, with almost unchanged OH and NH interatomic distances. Following reoptimization of thus-obtained structures in the S1 electronic state resulted in two other minima, obtained respectively for PPMet-1 and PPMet-6. In both cases, the identified structures were similar to their S2 counterparts, differing by slight opening of the CNC angle in the pyridine ring, up to 128°/124° for PPMet-1 and PPMet-6, respectively. On the contrary, the unconstrained S1 optimization of PPMet-4 resulted in a barrierless PCET reaction leading to an S1/S0 crossing (final optimization convergence was not achieved due to numerical instability of the single-reference ADC(2) method). In the course of the optimization process, initially the O–N6 distance decreased from 3.56 Å to ca. 2.15 Å, resulting in reduction of the S1/S0 energy gap to about 0.8 eV. Subsequently, the hydrogen atom from the OH group was transferred to the N4 center of the pyrazine ring, which was eventually followed by dissociation of the methoxy radical, upon which the SCF convergence failed. | ||
| Fig. 4 Geometrical structures and adiabatic relative energies of the optimized PrdPyr-MeOH S2 and S1 minima, with indicated HB length values. The shown PPMet-4-S1 structure corresponds to an approximate geometry extracted from the unconverged optimization. | ||
Taking into account the local excitation character of the identified S2 and S1 minima (with the exception of the S1 state of PPMet-4), in the next step, we calculated relaxed adiabatic reaction profiles along the proton-transfer coordinate, i.e., the O–H distance, in the S1 state. For the PPMet-1 and PPMet-6 complexes, the O–H bond length was the only parameter kept fixed, while for the PPMet-4 isomer a more complex approach was undertaken, to prevent the immediate S1 → S0 degeneration due to the PCET process. Namely, in the latter case, two different paths were calculated: (i) one following the PT reaction but with the ON4N6 angle fixed at its S0-optimized value for O–H distances shorter than 1.20 Å (the direct path), and (ii) a two-part sequential scan, including, firstly, an OH-fixed scan along the descending O–N6 distance and, secondly, the O–H bond elongation from the final obtained structure (the indirect path). Results of these scans are presented in Fig. 5.
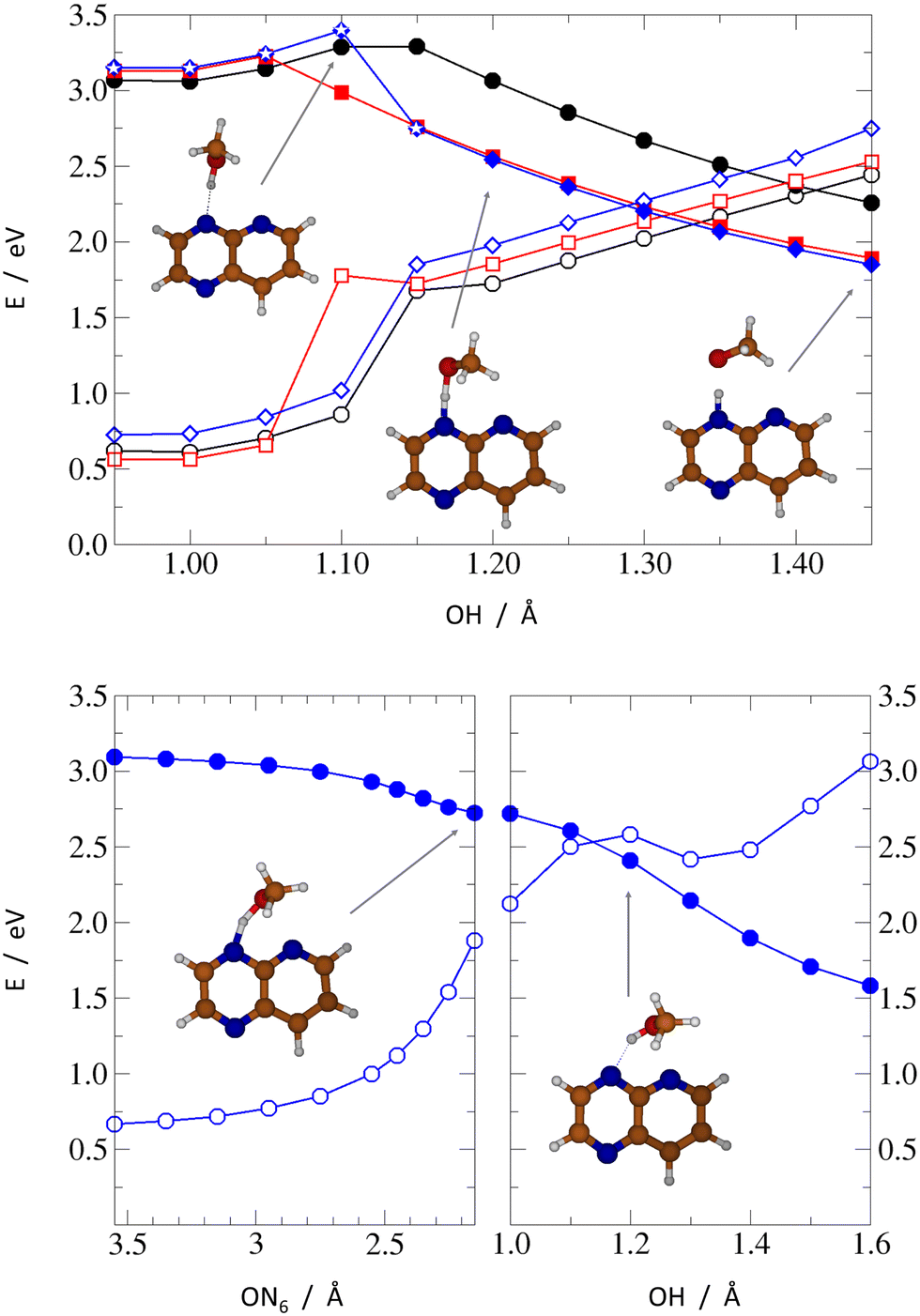 | ||
| Fig. 5 Upper panel: Relaxed S1-PEP scans along the PT reaction coordinate: PPMet-1 – black circles, PPMet-6 – red squares, PPMet-4 – blue diamonds. The full/empty symbols mark the S1/S0 energy, respectively, with the latter energy calculated vertically at the S1-optimized geometry. White stars mark the points along the PPMet-4 profile for which the ON4N6 angle was kept fixed at its S0-optimized value. Lower panel: A two-step relaxation path of the PPMet-4 complex: an OH-fixed scan along the descending ON6 distance (left), and the following OH bond elongation scan (right). The full/empty symbols mark the S1/S0 energy, respectively. | ||
The direct relaxed scans, shown in the upper panel of Fig. 5, illustrate the general photorection mechanism of PrdPyr-MeOH. After the initial S0 → S2 excitation and the subsequent ultrafast internal conversion to the S1 state, the system is likely to reach energy minimum of the S1 surface, with the O–H bond length almost unchanged with respect to its ground-state equilibrium value. The barrier separating the minimum from a downhill PT reaction exhibits similar height for all three isomers (about 0.1 eV for PPMet-6 and 0.2 eV for PPMet-1 and PPMet-4). However, its position, that is the necessary O–H elongation to move past the barrier, gradually shifts away from the Franck–Condon region in the PPMet-6 → PPMet-4 → PPMet-1 sequence. This shift could translate to a kinetic effect, enhancing the relaxation mechanism requiring the least distorted geometry. Additionally, at the barrier a change of the character of the S1 state occurs. The oxygen lone-pair alignment with the aromatic π orbitals at the PrdPyr fragment changes after crossing the barrier, resembling more the orientation observed in the Franck–Condon region for the S0→S5 transition, with strongly enhanced charge-transfer character (see Fig. S4 and Table S1 in the ESI† for the NTOs plots and CT coefficients calculated along the PT path). This change, accompanied by weak, yet observable interaction between the oxygen and the neighbouring-N atom (i.e., the N atom not participating in the HB formation) lone electronic pairs, is expected to facilitate the intermolecular electron transfer process.
The second, indirect path calculated for the PPMet-4 complex adds new insights, concerning the immediate N–O interactions in the PPMet-4 Franck–Condon region. As indicated by the bottom-left panel of Fig. 5, a weak attraction between the MeOH oxygen and the nitrogen atom of the pyridine ring drives the system in a barrierless manner towards a potential energy surface (PES) region of rather small S1/S0 energy difference (<1.0 eV). From this conformation, if one allows gradual OH distance relaxation (bottom-righ panel of Fig. 5), the S1/S0 degeneracy advances even further, almost immediately leading to an energy crossing, as the proton is transferred from MeOH to PrdPyr. The topography of the indirect path is in full agreement with the result of unconstrained S1 optimization discussed before. It should be noted that, having the generally rapid OH bond oscillations on the one hand (promoting the direct PT path), and relatively small attractive O–N force on the other hand (promoting the indirect path), one should expect an interplay of the two relaxation mechanisms in the PPMet-4 system, which could be sensitive to the system environment.
To get a notion of the credibility of the discussed ADC(2) reaction profiles,46 we performed additional test scans at the QD-NEVPT2 level of theory (single-point calculations at the ADC(2)-optimized geometries), and with the SCS-ADC(2) method (fully relaxed scans), obtaining generally very good agreement between the tested methods. In this stage we also checked the effect of the molecular orbital active space choice on the multireference calculations results. Data obtained in these additional calculations can be found in the ESI† (Fig. S5–S7).
In order to have a more complete view of the nonadiabatic S1/S0 interaction during the EDPT process, we performed an XMS-CASPT2 optimization of the minimum-energy conical intersection (MECI) points in all complexes. The resulting structures and their relative energies calculated with respect to the corresponding S0 minima are shown in Fig. 6. One can notice that all identified MECIs represent EDPT structures: in all cases, the proton is fully transferred to the PrdPyr moiety, with the universal N--H bond length of ca. 1.025 Å, and the O--H distances of 1.865 Å (PPMet-4), 1.964 Å (PPMet-6), and 2.000 Å (PPMet-1). To provide most accurate comparison between MECI points obtained in the multi-reference calculations, and the interstate crossing points determined at the ADC(2) level, we performed additional MECI optimization employing the penalty functional method combined with ADC(2): results of these calculations are shown in Fig. S8 in the ESI.† MECIs determined with the penalty-functional method lie very close to S1/S0 crossing points at the adiabatic scans reported in Fig. 5. Moreover, a connection between the ADC(2) and the corresponding data from the XMS-CASPT2 calculations can be observed, with systematic energy-up-shift of ca. 0.6 eV, and with elongation of the O–H bond length of 0.50 Å in the latter case. At the same time, the order of the PPMet isomers with respect to the O–H distances at the MECI points is consistent between both approaches. It should be also noted that the MECI energy rise is accompanied by a similar up-shift of the S0 → S1 excitation energy at each isomers Franck–Condon region. More details regarding the discussed MECI calculations, including visualization of employed molecular orbital active spaces, can be found in the ESI† (Fig. S9 and Table S2).
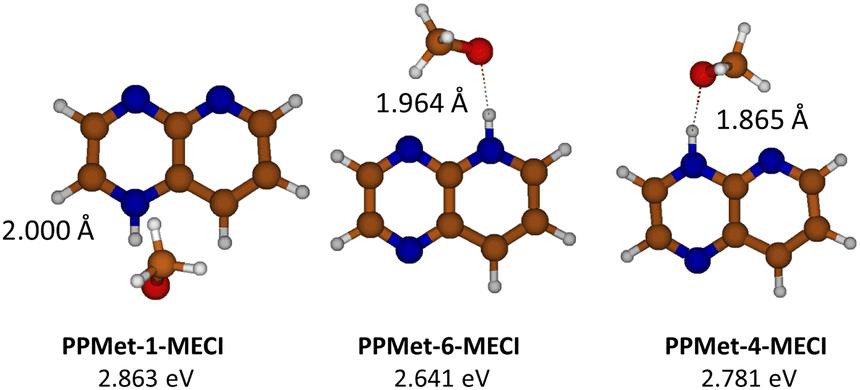 | ||
| Fig. 6 Geometrical structures and adiabatic relative energies of the optimized PrdPyr-MeOH MECI points, with indicated HB length values. | ||
PrdPyr-MeOH photo-relaxation dynamics
To gain insights about the process timescale and efficiency, and to get a notion about possible impact of nonadiabatic effects on the EDPT process, we investigated the PrdPyr-MeOH photo-relaxation by means of on-the-fly nonadiabatic molecular dynamics simulations. The initial excitation in all three systems targeted the bright S2 state, with the excitation energy window set to 4.40 ± 0.50 eV, as marked in Fig. 3. In Fig. 7, one can see the mean electronic states’ population evolution in time. While all systems start with the pure S2 state population, a much more efficient relaxation can be observed for PPMet-6 and PPMet-4, than for PPMet-1: firstly, to S1 (on an ultrafast timescale) and, later, to S0. If a monoexponential S0 population growth is assumed, the characteristic times at which the 50% S0 population is achieved are 74 fs and 89 fs, respectively for PPMet-4 and PPMet-6. It should be noted that, given the applied Eocc − E0 ≤ 0.20 eV stopping condition, these estimated values represent the lower bound for the process timescale. At the same time, the predicted final S0 state populations, i.e., populations in the limit of t → ∞, are 77%, 65% and 24% for PPMet-4, PPMet-6 and PPMet-1. The obtained results, on the one hand, give clear manifestation of the much higher reactivity of the reaction center consisting of two neighboring N atoms, than of a single N atom in the pyrazine ring. On the other hand, the final S0 populations, predicted from the monoexponential fits, suggest that other relaxation mechanisms may play a role on larger timescales. In this respect, radiative or inter-system-crossing induced processes could be considered, which are, however, beyond the scope of the present study. For more details on the performed fitting procedure, including the fitted functions’ plots (Fig. S10), please refer to the ESI.†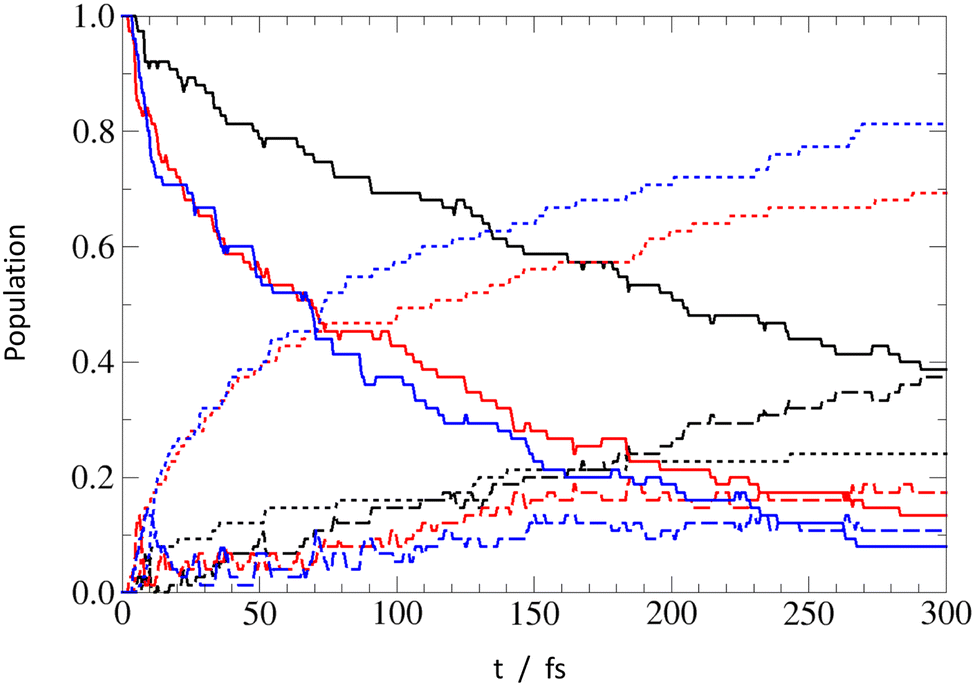 | ||
| Fig. 7 Mean electronic states' population evolution in time. The full/dashed/dotted lines correspond to S2/S1/S0 state populations, respectively. Results for PPMet-1, PPMet-6, and PPMet-4 are respectively marked with black, red, and blue. | ||
To obtain additional mechanistic insights into the relaxation process in the NAMD simulations, in Fig. 8 the final O–H distance values were plotted against the corresponding final trajectory time (i.e., either the final time of 300 fs set for the dynamics, or the time at which the applied stopping condition was satisfied). While the collapsed group of points at t = 300 fs corresponds to the unreactive trajectories, all other points, positioned at t < 300 fs, mark events of successful excited state relaxation to S0. It can be seen that all the reactive points, except two at t ≈ 250 fs, show strong elongation of the original O–H bond, indicating the PT from MeOH to PrdPyr as the deactivation mechanism. The mean value of the O–H distance at the moment of the S1 → S0 hopping is similar in all systems, increasing slightly from PPMet-4 (1.40 ± 0.01 Å), through PPMet-6 (1.47 ± 0.01 Å), to PPMet-1 (1.53 ± 0.01 Å) which is in overall agreement with the predictions formulated on the grounds on the PEPs analysis. Moreover, if one has a look at the correlation between initial velocities of atoms involved in the HB formation (Fig. S11–S13 in the ESI†), and the individual trajectories outcome, it can be observed that large kinetic energy initially accumulated in the HB-proton vibrations effectively supports the EDPT process.
 | ||
| Fig. 8 Final O–H distance in individual trajectories: black circles – PPMet-1, red empty diamonds – PPMet-6, blue crosses – PPMet-4. | ||
The two points standing out in Fig. 8 with their short O–H distance at the moment of deactivation demonstrate strong O–N-interaction and can be associated with the indirect relaxation path mechanism discussed above. Indeed, in these two cases the O–N distance at the moment of hopping was found to be as low as 2.00 Å. Interestingly, while the points originate from two different dynamics (i.e., PPMet-6 and PPMet-4), both structures adopt the HB pattern characteristic for PPMet-4 (for molecular structures visualization, see Fig. S14 in the ESI†). Although this is only a single event, it stays in line with the preference for the photo-relaxation through the PPMet-4 channel. At the same time it should be noted that, in other cases, the final relaxation to the ground electronic state follows almost exclusively the original system configuration; for more details on this aspect, please refer to Fig. S15 in the ESI.†
Conclusion
In this contribution, we investigated the photo-induced EDPT process in isolated PrdPyr-MeOH complexes by means of ab initio electronic-structure and nonadiabatic molecular dynamics calculations. We found that the methanol molecule acts as a proton-donor in the hydrogen-bonded complex with PrdPyr, adopting perpendicular orientation with respect to the aromatic plane. Three stable structures differing by the nitrogen atom acting as the proton-acceptor were identified. Upon photo-excitation to the lowest-energy bright state, S2 (ππ*), all systems undergo rapid relaxation, firstly to S1 (nπ*) and, subsequently, to the S0 state. The rate of the process is visibly higher when the hydrogen bond is formed with one of the neighbouring N atoms and not with the single nitrogen of the pyrazine ring. This effect is rationalized by interaction between the methanol oxygen and the neighboring nitrogen atom, not directly involved in the HB formation. Our findings point to beneficial effect of the β-position arrangement of electronegative centers for photo-oxidation reactions, providing further clues for future design of efficient organic catalysts for water splitting applications.Author contributions
Joanna Jankowska: methodology, investigation, formal analysis, writing. Andrzej L. Sobolewski: conceptualization, writing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Authors would both like to thank Prof. Wolfgang Domcke for the great support and fruitful discussion upon preparation of the manuscript. J. J. thanks the Polands high-performance computing infrastructure PLGrid (HPC Centers: ACK Cyfronet AGH) for providing computer facilities and support within computational grant no. PLG/2023/016166. A. S. acknowledges funding through the National Science Centre project (#2020/39/B/ST4/01723).Notes and references
- J. W. Darcy, B. Koronkiewicz, G. A. Parada and J. M. Mayer, Acc. Chem. Res., 2018, 51, 2391–2399 CrossRef CAS PubMed.
- V. R. I. Kaila, Acc. Chem. Res., 2021, 54, 4462–4473 CrossRef CAS.
- E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef CAS PubMed.
- D. G. Nocera, J. Am. Chem. Soc., 2022, 144, 1069–1081 CrossRef CAS PubMed.
- W. Domcke and A. L. Sobolewski, J. Phys. Chem. B, 2022, 126, 2777–2788 CrossRef CAS PubMed.
- N. Ullah, S. Chen and R. Zhang, J. Chin. Chem. Soc., 2023, 70, 195–208 CrossRef CAS.
- X. Pang, C. Jiang, W. Xie and W. Domcke, Phys. Chem. Chem. Phys., 2019, 21, 14073–14079 RSC.
- F. Weber, J. C. Tremblay and A. Bande, J. Phys. Chem. C, 2020, 124, 26688–26698 CrossRef CAS.
- X. Huang, J. P. Aranguren, J. Ehrmaier, J. A. Noble, W. Xie, A. L. Sobolewski, C. Dedonder-Lardeux, C. Jouvet and W. Domcke, Phys. Chem. Chem. Phys., 2020, 22, 12502–12514 RSC.
- X. Huang and W. Domcke, J. Phys. Chem. A, 2021, 125, 9917–9931 CrossRef CAS PubMed.
- X. Huang and W. Domcke, Phys. Chem. Chem. Phys., 2022, 24, 15925–15936 RSC.
- E. J. Rabe, K. L. Corp, X. Huang, J. Ehrmaier, R. G. Flores, S. L. Estes, A. L. Sobolewski, W. Domcke and C. W. Schlenker, J. Phys. Chem. C, 2019, 123, 29580–29588 CrossRef CAS.
- E. R. Sayfutyarova and S. Hammes-Schiffer, J. Am. Chem. Soc., 2020, 142, 487–494 CrossRef CAS PubMed.
- E. R. Sayfutyarova and S. Hammes-Schiffer, J. Phys. Chem. Lett., 2020, 11, 7109–7115 CrossRef CAS PubMed.
- O. Morawski, P. Gawryś, J. Sadło and A. L. Sobolewski, ChemPhysChem, 2022, 23, 1–9 Search PubMed.
- F. López-Tenllado, A. Marinas, F. Urbano, J. Colmenares, M. Hidalgo, J. Marinas and J. Moreno, Appl. Catal., B, 2012, 128, 150–158 CrossRef.
- D. Hwang, L. M. Wrigley, M. Lee, A. L. Sobolewski, W. Domcke and C. W. Schlenker, J. Phys. Chem. B, 2023, 127, 6703–6713 CrossRef CAS.
- X. M. C. Ta, R. Daiyan, T. K. A. Nguyen, R. Amal, T. Tran-Phu and A. Tricoli, Adv. Energy Mater., 2022, 12, 18–21 Search PubMed.
- M. Rahman, H. Tian and T. Edvinsson, Angew. Chem., Int. Ed., 2020, 59, 16278–16293 CrossRef CAS PubMed.
- Z. Shen, Y. Hu, B. Li, Y. Zou, S. Li, G. Wilma Busser, X. Wang, G. Zhao and M. Muhler, J. Energy Chem., 2021, 62, 338–350 CrossRef CAS.
- X. Zhang, Z. Wang, Z. Li, S. Shaik and B. Wang, ACS Catal., 2023, 13, 1173–1185 CrossRef CAS.
- Q. Zheng, E. Xu, E. Park, H. Chen and D. Shuai, Appl. Catal., B, 2019, 240, 262–269 CrossRef CAS.
- V. Augugliaro and L. Palmisano, ChemSusChem, 2010, 3, 1135–1138 CrossRef CAS PubMed.
- P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. McLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Chem. Rev., 2022, 122, 2017–2291 CrossRef CAS PubMed.
- A. Galushchinskiy, Y. Zou, J. Odutola, P. Nikačević, J. Shi, N. Tkachenko, N. López, P. Farràs and O. Savateev, Angew. Chem., Int. Ed., 2023, 62, e202301815 CrossRef CAS PubMed.
- M. Head-Gordon, J. A. Pople and M. J. Frisch, Chem. Phys. Lett., 1988, 153, 503–506 CrossRef CAS.
- J. Schirmer and A. B. Trofimov, J. Chem. Phys., 2004, 120, 11449–11464 CrossRef CAS PubMed.
- T. Shiozaki, W. Györffy, P. Celani and H.-J. Werner, J. Chem. Phys., 2011, 135, 081106–081111 CrossRef PubMed.
- B. G. Levine, J. D. Coe and T. J. Martínez, J. Phys. Chem. B, 2008, 112, 405–413 CrossRef CAS PubMed.
- C. Angeli, S. Borini, M. Cestari and R. Cimiraglia, J. Chem. Phys., 2004, 121, 4043–4049 CrossRef CAS PubMed.
- C. M. Krauter, M. Pernpointner and A. Dreuw, J. Chem. Phys., 2013, 138, 044107 CrossRef PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- TURBOMOLE v7.1, TURBOMOLE GmbH, 2016 Search PubMed.
- T. Shiozaki, WIREs Comput. Mol. Sci., 2018, 8, 1–7 Search PubMed.
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- F. Neese, WIREs Comput. Mol. Sci., 2018, 8, e1327 CrossRef.
- F. Plasser, J. Chem. Phys., 2020, 152, 084108 CrossRef CAS PubMed.
- M. Barbatti and K. Sen, Int. J. Quantum Chem., 2016, 116, 762–771 CrossRef CAS.
- M. Barbatti, G. Granucci, M. Persico, M. Ruckenbauer, M. Vazdar, M. Eckert-Maksić and H. Lischka, J. Photochem. Photobiol., A, 2007, 190, 228–240 CrossRef CAS.
- G. Granucci, M. Persico and A. Zoccante, J. Chem. Phys., 2010, 133, 134111 CrossRef CAS PubMed.
- M. Barbatti, M. Ruckenbauer, F. Plasser, J. Pittner, G. Granucci, M. Persico and H. Lischka, WIREs Comput. Mol. Sci., 2014, 4, 26–33 CrossRef CAS.
- M. Barbatti, G. Granucci, M. Ruckenbauer, F. Plasser, R. Crespo-Otero, J. Pittner, M. Persico and H. Lischka, NEWTON-X: Version 2, 2016 Search PubMed.
- F. Hibbert and J. Emsley, Adv. Phys. Org. Chem., 1990, 26, 255–379 CrossRef CAS.
- M. J. Janicki, R. Szabla, J. Šponer and R. W. Góra, Phys. Chem. Chem. Phys., 2022, 24, 8217–8224 RSC.
- R. Crespo-Otero and M. Barbatti, Theor. Chem. Acc., 2012, 131, 1237 Search PubMed.
- F. Plasser, R. Crespo-Otero, M. Pederzoli, J. Pittner, H. Lischka and M. Barbatti, J. Chem. Theory Comput., 2014, 10, 1395–1405 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Natural Transition Orbitals (NTO) plots, Charge Transfer (CT) analysis, ab initio benchmark energy scan, structural correlation plots, Cartesian Coordinates of studied complexes. See DOI: https://doi.org/10.1039/d3cp04148d |
| This journal is © the Owner Societies 2024 |