
Research on structural strengthening technology for regenerative denitration catalysts
Dongliang
Ji
a,
Dongxue
Jiang
b,
Yang
Li
c,
Huan
Zhang
d,
Haiyun
Zhou
e,
Zhaoqin
Huang
 *a and
Jianzhong
Zhu
*b
*a and
Jianzhong
Zhu
*b
aCollege of Environment and Ecology, Jiangsu Open University, Nanjing, 210036, China
bCollege of Environment, Ho Hai University, Nanjing, 210024, China. E-mail: zhuhhai2010@hhu.edu.cn
cJiangsu Longking- Coalogix Environmental Protection Technology Co. Ltd, Yancheng, 224000, China
dSchool of Chemical Engineering and Materials, Changzhou Institute of Technology, Changzhou, 213032, China
eJiangsu Environmental Engineering Technology Co., Ltd, Nanjing, 210009, China
First published on 11th December 2023
Abstract
The cost of replacing failed selective catalytic reduction (SCR) catalysts and their disposal as hazardous solid waste is high. If failed catalysts are recovered and regenerated into new SCR denitration catalysts, the cost of flue gas denitration can be effectively reduced. However, regenerated SCR catalysts have relatively low structural strength and activity and cannot yet form an effective replacement. In this study, aluminum dihydrogen phosphate, aluminum nitrate, and aluminum sulfate were used as structural strengthening agents in the regeneration of SCR catalysts, and over-impregnation, drumming-assisted impregnation, and ultrasonic-assisted preparation techniques were compared. The corresponding regenerated SCR catalysts were then prepared and analyzed for compressive strength, wear strength, H2-TPR, NH3-TPD, and in situ IR. Factors influencing the structural strength, physical properties, and catalytic activity of the regenerated catalysts were investigated. The best results were obtained as follows: compressive strength of 4.57 MPa, wear rate of 0.088% kg−1, and denitration of 58% after 10 min of drumming-assisted impregnation in an aluminum sulfate solution with a concentration of 16%. Based on this, a synergistic method for catalyst activity and structural strengthening was explored to support the design of better SCR catalysts for regeneration.
1. Introduction
NOx emissions are a global problem, and flue gases emitted from various industrial processes seriously pollute the environment.1,2 With China's improved flue gas emission standards3,4 and the strengthening of air pollution regulations,3 the focus of NOx treatment has shifted to non-electric industries, mainly the steel, cement, and coking industries. The requirements for the denitration of boiler exhaust gases in various industries are becoming increasingly stringent. SCR denitration catalysts are a mature and low-cost technology that are rapidly gaining popularity in China, with significant emission reduction effects.5,6 However, the replacement costs of failed SCR catalysts and their disposal as a hazardous solid waste are high. If failed catalysts are recycled and regenerated into new SCR denitration catalysts, the cost of flue gas denitration can be effectively reduced. However, the regeneration of domestic SCR denitration catalysts has just begun, and the poor quality and complexity of domestic coal have led to a complex deactivation mechanism and various deactivation causes.7,8 SCR catalysts are affected by dust and water vapor in flue gas during operation, leading to wear and a lower compressive strength of the spent SCR catalyst surface.9,10 Long-term operation at high temperatures can easily cause clogging and sintering of vanadium and titanium SCR catalysts, thus severely wearing the catalyst surface and further degrading the catalyst strength.11 Regeneration techniques for deactivated SCR catalysts include water-wash regeneration,12,13 thermal reduction regeneration,14,15 acid (alkali)-wash regeneration,12,16 and compound regeneration.5,17,18 Regeneration is also subject to ultrasonic cleaning, which leads to wear and tear of the catalyst surface and a reduction in the compressive strength, thereby affecting the performance of the regenerated SCR catalyst. Many current studies on improving the mechanical properties of catalysts have focused on the mechanical strength of industrial catalysts during molding, and few studies have focused on improving the mechanical properties of the molded or regenerated catalysts.5,12,13,19 However, the regenerated catalysts must satisfy the mechanical strength requirements of GB/T 31587-2015 (compressive strength ≥2.0 Mpa, wear rate ≤0.1% kg−1). Researchers13–15,20–23 conducted regeneration experiments on SCR catalysts using a microwave-ethanol solution and found that a microwave-ethanol solution not only increased the specific surface area of denitration catalysts but also improved their mechanical properties. However, the compressive strength of the catalysts increased only by 1.612 MPa after three reaming cycles, because of the absence of a structural strengthening process, and could not meet practical requirements. In this study, based on the conventional catalyst regeneration process, aluminum dihydrogen phosphate, aluminum nitrate, and aluminum sulfate were used as structural strengthening agents to compare over-impregnation, drumming-assisted impregnation, and ultrasonic-assisted preparation techniques. Through these different regeneration processes, the corresponding regenerated SCR catalysts were prepared and analyzed for their compressive strength, wear strength, H2-TPR, NH3-TPD, and in situ IR. The effects of the structural strength, physical properties, and catalytic activity of the regenerated catalysts were investigated to optimize the structural strengthening techniques for the regenerated SCR catalyst, and the best results were obtained as follows: after 10 min of drumming-assisted impregnation in an aluminum sulfate solution with a concentration of 16%, the compressive strength reached 4.57 MPa, the wear rate was 0.088% kg−1, and an excellent catalytic performance was attained. Based on the traditional catalyst regeneration process, organic and inorganic auxiliaries were also used to improve the compressive and wear resistance of the catalyst, while ensuring that the denitration activity of the denitration catalyst was not affected and extending the service life of the regenerated catalyst as well as the regeneration time.21 Based on this, a synergistic approach for catalyst activity and structure enhancement was explored to provide support for the design of better-regenerated SCR catalysts.2. Experimental materials and methods
2.1. Waste SCR catalyst regeneration
2.2. Test characterization
| Name | Index |
|---|---|
| Fume flow | 73 m3 h−1 |
| Space velocity | 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 000 h−1 |
| NO concentration | 300 μL L−1 |
| Ammonia nitrogen molar ratio | 1:1 |
| Temperature | 380 °C |
| O2 | 3% |
| H2O | 8% |
| SO2 concentration | 500 μL L−1 |
The denitration efficiency of catalyst η was calculated using the following equation.
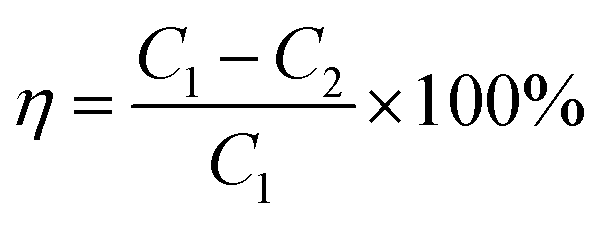 | (1) |
Programmed temperature rise reduction (H2-TPR): Approximately 50 mg of the catalyst was weighed and raised from 50 °C to 500 °C in a stream of ultra-high-purity He (30 mL min−1) for 30 min after pretreatment. The catalyst was then cooled to room temperature and then adsorbed by passing a mixture of 5% NH3 and He at a flow rate of 20 mL min−1. The catalyst was purged again with He to stabilize the baseline and then tested at a ramp rate of 5 °C min−1 from 30 °C to 800 °C.
| P = F/(L × W) | (2) |
 | (3) |
3. Results and discussion
3.1. Analysis and optimization of factors influencing the structural strengthening of regenerative catalysts
| Samples | 26% aluminum dihydrogen phosphate | 16% aluminum sulfate | 28% aluminum nitrate | |||
|---|---|---|---|---|---|---|
| Loading rate (%) | Compressive strength (MPa) | Loading rate (%) | Compressive strength (MPa) | Loading rate (%) | Compressive strength (MPa) | |
| Note: the above dates are an average. | ||||||
| Blank samples | 0 | 2.69 | 0 | 2.69 | 0 | 2.69 |
| Static impregnation | 25.03 | 7.67 | 5.66 | 4.465 | 10.10 | 4.71 |
| Ultrasonic-assisted impregnation | 21.41 | 7.42 | 5.91 | 4.288 | 10.44 | 4.57 |
| Drum-bubble-assisted impregnation | 19.19 | 7.76 | 5.73 | 4.577 | 10.14 | 4.68 |
 | ||
| Fig. 1 Effect of impregnation time on the loading rate of different reinforcing agents. And these dates are an average. | ||
 | ||
| Fig. 2 The loading rate (a), compressive strength (b and d) and wear strength (c) of structurally reinforced catalysts. And these dates are average. | ||
Meanwhile, the compressive strength of the catalyst increased rapidly after impregnation with an aluminum dihydrogen phosphate solution. When the density of the impregnated solution reached 20%, the compressive strength of the catalyst increased to 7.515 MPa, which was 179% higher after impregnation compared with that of the original regenerated catalyst (2.69 MPa). As the density of the aluminum dihydrogen phosphate solution continued to increase, the compressive strength began to level off. At a solution concentration of 16–20%, aluminum sulfate enhanced the compressive strength of the catalyst better than aluminum nitrate did, reaching 4.465 MPa and increasing the compressive strength by 66% compared to the blank sample, which was even lower than the blank sample. In addition, when the impregnation solution concentration exceeded 24%, the compressive strength of aluminum nitrate catalyst started to increase rapidly, and when the solution concentration was 28%, the compressive strength of the catalyst reached the highest (4.71 MPa) (Fig. 2(b) and Table 1). In conclusion, the enhancement of compressive strength by aluminum dihydrogen phosphate was much higher than that by aluminum nitrate and aluminum sulfate, and the compressive strengths after strengthening by aluminum nitrate and aluminum sulfate were relatively close.
Fig. 2(c) and Table 1 show that the wear rate of the catalyst without aluminum nitrate impregnation was 0.177% kg−1. In the low-concentration aluminum nitrate impregnation solution, the wear rate of the catalyst did not vary significantly, and the wear rate did not decrease significantly, whereas in the 16% impregnation solution concentration, the wear rate started to decrease until the impregnation solution reached saturation, and the wear rate decreased to 0.082% kg−1, compared to the blank sample. Compared with the blank sample, the wear rate was reduced by 53.7%. When impregnated with the aluminum sulfate solution, the wear rate of the catalyst decreased sharply, and with an increase in the impregnation solution concentration, the wear rate decreased. When the mass fraction of the impregnating solution was 16%, the wear rate reached 0.093% kg−1, which was already lower than 0.1% kg−1, and the wear rate was reduced by 47% compared with the blank sample. However, the most significant decrease was for aluminum dihydrogen phosphate, which was reduced to 0.04303% kg−1 at 13%, after which the decrease was not particularly significant with an increase in concentration and dropped to a minimum of 0.0317% kg−1 at 26%. After the structural strength increased to a certain level, the decrease lessened as the impregnation solution concentration continued to increase.
Fig. 2(d), Exhibit 1, and Fig. 1 show that the compressive strength increased and the wear rate decreased as the loading rate of the reinforcing agent increased, but the compressive strength of aluminum dihydrogen phosphate was significantly higher and the wear rate was significantly lower at the same loading rate, indicating that aluminum dihydrogen phosphate was the best in terms of structural strengthening. Although the compressive strength increased and the wear rate decreased as the loading rate of the aluminum nitrate and aluminum sulfate impregnated solutions increased, aluminum sulfate had a higher compressive strength and lower wear rate at the same loading rate. The compressive strength increased more slowly and the wear rate decreased more slowly as the loading rate increased. Aluminum dihydrogen phosphate reached an inflection point of 7.515 MPa after reaching a loading rate of 13.34%, while the inflection point of aluminum nitrate was approximately 8.19% (4.675 MPa) and that of aluminum sulfate was approximately 7.96% (4.43 MPa), corresponding to impregnating solution concentrations of 20%, 24% and 24%, respectively. Moreover, the wear rate did not always increase with increases in the loading rate and compressive strength. The compressive strength of aluminum dihydrogen phosphate reached an inflection point of 6.365 MPa at a concentration of 13% (7.01% loading rate), and the wear rate reached an inflection point of 0.04332% kg−1, after which the wear rate only fluctuated within a small range. The wear rate of aluminum sulfate was significantly lower than that of aluminum nitrate at the same loading rate. When the concentration of aluminum sulfate impregnation increased to 28%, the loading rate reached 8.01%, and the wear rate dropped to the lowest point of 0.057% kg−1 (compressive strength 4.5 MPa); when the concentration of aluminum nitrate impregnation increased to 28%, the loading rate reached 10.10%, and the wear rate dropped to the lowest point of 0.08192% kg−1 (compressive strength of 4.705 MPa). Furthermore, it appeared as though the wear rate of aluminum nitrate and aluminum sulfate had not reached the inflection point yet. In conclusion, the loadings corresponding to different reinforcing agents were significantly positively correlated with the compressive strength, but the reinforcement effect was more disparate, with a higher compressive strength and wear rate for aluminum dihydrogen phosphate at the same loading rate. However, aluminum dihydrogen phosphate had a significant negative effect on the catalyst activity, which is discussed in detail in the subsequent section.
3.2. Effect of the structural strengthening process on catalyst performance
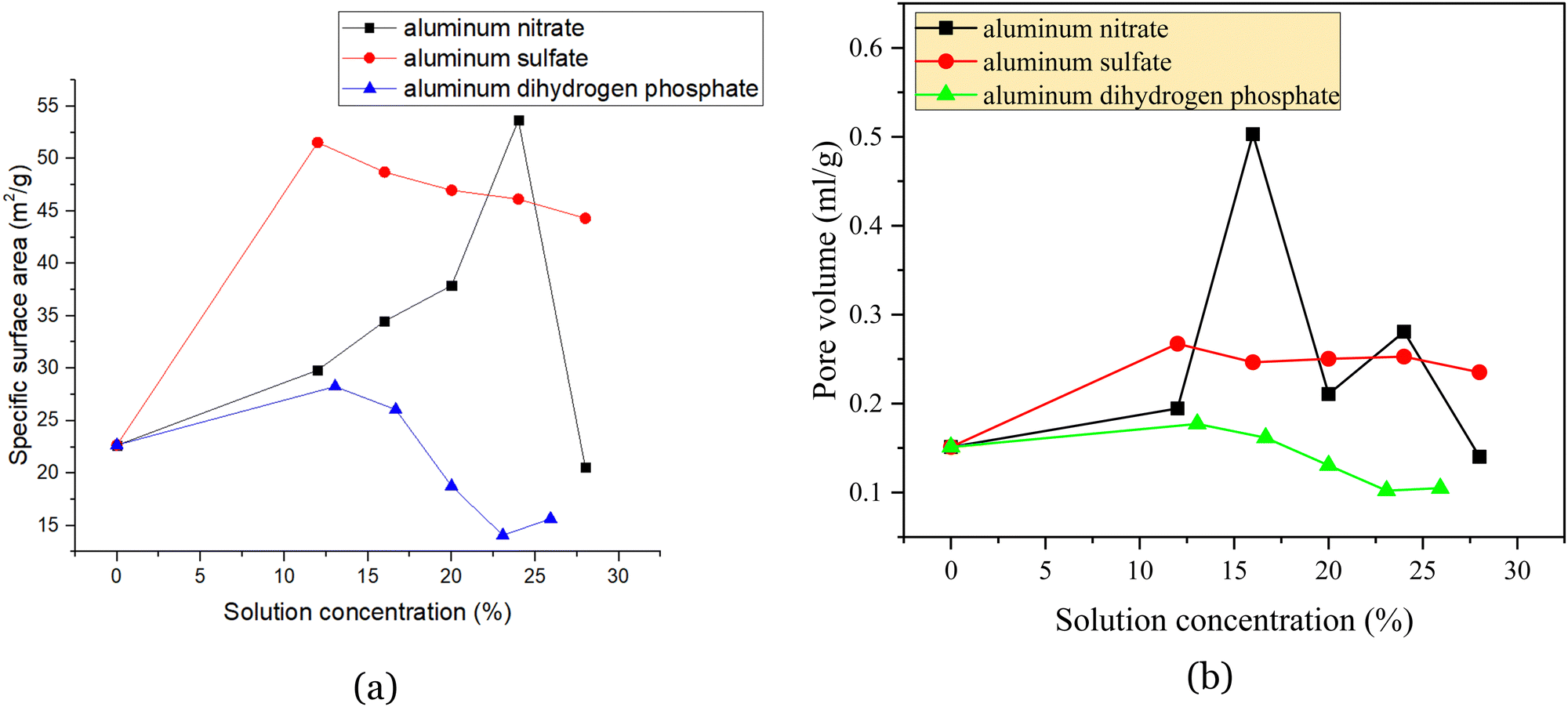 | ||
| Fig. 3 Effect of three reinforcing agents and their concentrations on the specific surface area (a) and pore volume (b) of regenerated catalysts. | ||
Fig. 3 shows that the catalyst with 12% aluminum nitrate had a small specific surface area. Furthermore, the specific surface area of the catalyst impregnated with aluminum nitrate increased, and the pore volume increased compared to the blank. Both the pore volume and specific surface area increased with the concentration. However, from 28%, the specific surface area suddenly decreased rapidly to 20.5708 m2 g−1, and the pore volume also decreased to lower than that of the blank. Aluminum sulfate significantly increased both the specific surface area and pore volume of the catalyst relative to the blank, reaching a maximum of 51.5405 m2 g−1 at 12%, but slowly decreased the specific surface area of the catalyst as the mass fraction of the aluminum sulfate solution increased. Accordingly, it can be concluded that aluminum salt curing agents can provide part of the pore structure itself, i.e., increase the specific surface area and pore volume; however, they can also plug the pore structure of the spent catalyst itself.25 In conclusion, aluminum dihydrogen phosphate is more severely clogged and can provide less of the pore structure, aluminum sulfate is less clogged and can provide more of the pore structure by itself, and aluminum nitrate also provides a higher pore structure but has a strong clogging ability.
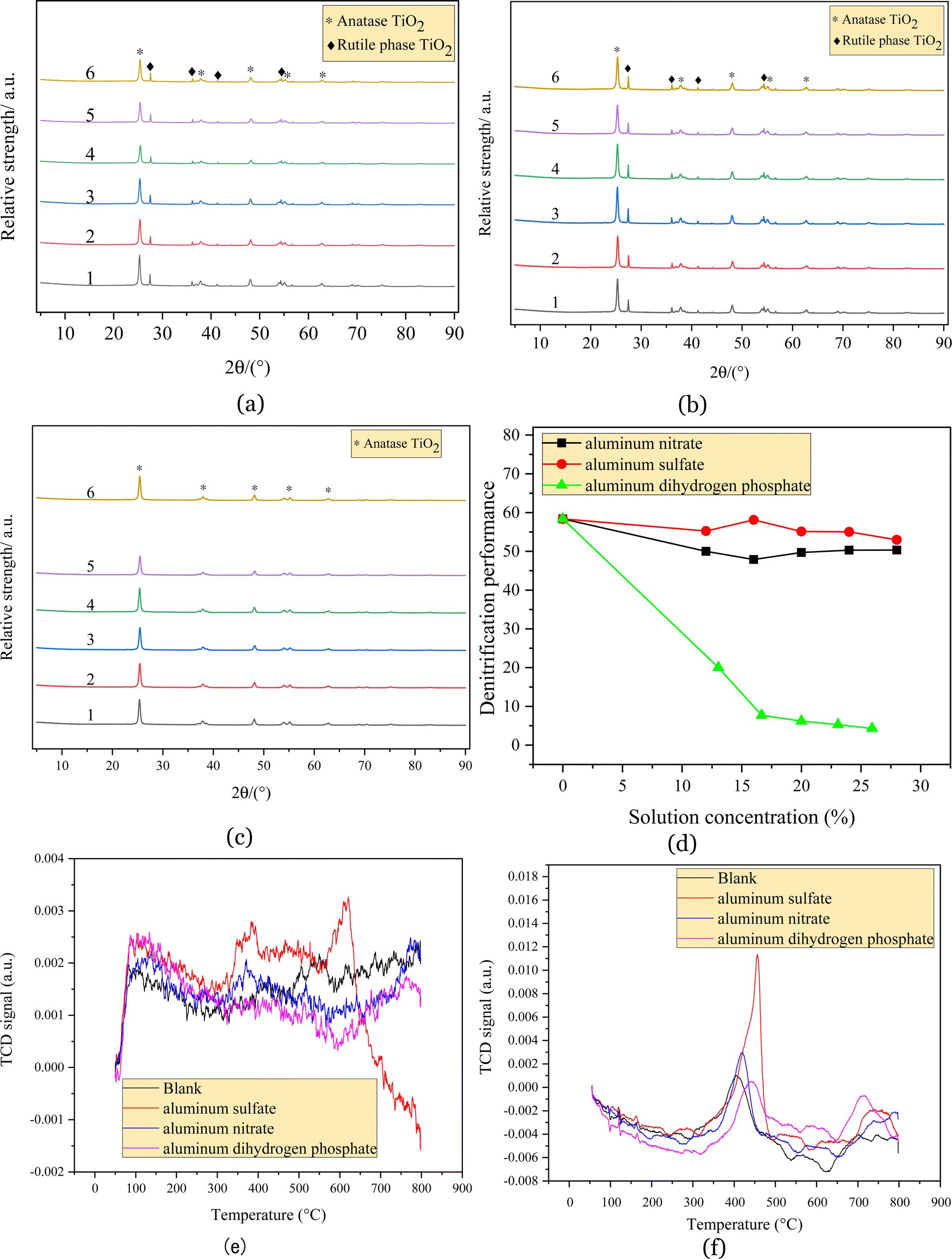 | ||
| Fig. 4 Composition of catalysts with denitration efficiency at different concentrations. (a) Aluminum dihydrogen phosphate impregnation, 1–6 represent 0%, 13%, 17%, 20%, 23%, and 26%, respectively; (b) aluminum nitrate impregnation, 1–6 represent 0%, 12%, 16%, 20%, 24%, and 28%, respectively; (c) aluminum sulfate impregnation, 1–6 represent 0%, 12%, 16%, 20%, 24%, and 28%, respectively. Where 1: blank; 2: impregnation solution mass fraction of 12%; 3: impregnation solution mass fraction of 16%; 4: impregnation solution mass fraction of 20%; 5: impregnation solution mass fraction of 24%; 6: impregnation solution mass fraction of 28%. (d) Denitration performance of catalysts with different resource-based technologies; (e) NH3-TPD spectra of catalysts with different resource-based technologies; (f) H2-TPR spectra for different resource-based technologies. | ||
Fig. 4(d) shows that all the regenerated catalysts decreased relative to the blank samples after impregnation with aluminum salts. The denitration efficiency of the catalyst impregnated with aluminum nitrate gradually decreased and reached its lowest denitration efficiency of 48% at an aluminum nitrate solution concentration of 16%. The effect of aluminum sulfate on the catalyst activity was minimal. At an impregnating solution mass fraction of 16%, the denitration efficiency of the catalyst was 58%, which did not differ significantly from the denitration efficiency of the blank sample, but the denitration efficiency of the catalyst decreased gradually as the concentration of the impregnating solution continued to increase. The denitration efficiency of the catalyst reached a minimum of 53% at a concentration of 28%. The denitration efficiency of the catalyst was greatly influenced by aluminum dihydrogen phosphate, which rapidly decreased to 20% after impregnation with aluminum dihydrogen phosphate and continued to decrease as the density of the impregnation solution increased, with the catalyst almost deactivating at a 16.7% density of the aluminum dihydrogen phosphate solution.
Fig. 4(e) shows that the blank regenerated catalyst samples showed the desorption peaks of NH3 at 430 °C, 500 °C and 550 °C.26 The shoulder peak located at approximately 430 °C represents the medium-to-strong acid desorption peak. The desorption peaks at 500 °C and 550 °C represent strong acid desorption peaks. The catalyst loaded with aluminum dihydrogen phosphate exhibited almost no desorption peaks, which showed that the catalyst surface had a reduced number of adsorbed species, suggesting that the impregnation of aluminum dihydrogen phosphate reduced the number of acid sites on the catalyst surface. The catalysts impregnated with aluminum nitrate showed the desorption peaks of NH3 at 370 °C and 520 °C. The slightly lower number of adsorption peaks for the aluminum nitrate impregnated catalysts compared to those of the blank regenerated catalysts indicated that the number of species that can be adsorbed on the catalyst surface was reduced and that the acidic sites of the aluminum nitrate impregnated catalysts were mainly medium-to-strong acids, whereas the original regenerated catalysts exhibited stronger acids. The TPD desorption peak area also showed that the NH3 adsorption area of the aluminum nitrate-impregnated catalyst was smaller than that of the blank regenerated catalyst, and the number of acid sites on the catalyst surface was slightly reduced. The catalysts impregnated with aluminum sulfate showed strong NH3 desorption peaks from 345 °C to 397 °C and from 580 °C to 620 °C. The area of the TPD desorption peak also showed that the NH3 adsorption area of the catalyst impregnated with aluminum sulfate was larger than those of the blank regenerated catalyst and the aluminum nitrate catalyst; thus, the catalyst sample impregnated with aluminum sulfate was theoretically more active.27 However, according to the results of the catalyst denitration tests, the denitration rates exhibited by the original regenerated catalysts impregnated with aluminum sulfate in the denitration activity tests were not significantly different, and the denitration performance of the catalysts was not only influenced by surface acidity.
Fig. 4(f) shows that the blank catalyst sample showed a hydrogen reduction peak at 400 °C, and 400–700 °C corresponds to the reduction peak of vanadium oxide,28 which is lower than the reduction temperature of 600–800 °C for pure V2O5. With an increase in the content of aluminum dihydrogen phosphate, the hydrogen reduction peak moved to near 438 °C, and a reduction peak appeared at 712 °C, which belonged to the reduction peak of tungsten oxide. After the impregnation of aluminum nitrate, the hydrogen reduction peak moved toward 419 °C, while the hydrogen reduction peak of the aluminum sulfate catalyst sample moved to near 455 °C, and both belonged to the reduction peak of vanadium oxide.29 This indicated that the structural strengthening process used in this experiment was not conducive to the SCR reaction on the catalyst surface. This may have been due to wrapping, which results in less contact with Ti and reduces the overall catalytic effect. The peak areas of the four catalysts were compared, and the reduction peak area of the catalyst impregnated with aluminum sulfate was the largest, indicating the optimal redox characteristics of the aluminum sulfate catalyst.
 | ||
| Fig. 5 In situ IR detection of catalysts after intensive regeneration of spent denitration catalysts. (a) and (c) and e: NO + O2 atmosphere conditions. (b), (d) and (f): NH3 + NO + O2 atmosphere conditions. (a) and (b): impregnation of 26% aluminum dihydrogen phosphate catalyst reaction. (c) and (d): impregnation of 28% aluminum nitrate catalyst reaction. (e) and (f): impregnation of 16% aluminum sulfate catalyst reaction. | ||
Fig. 5(b) shows that, for the catalyst impregnated with 26% aluminum dihydrogen phosphate in the reaction of NH3 + NO + O2 at 380 °C, with increasing adsorption time, after 10 min of adsorption, absorption peaks appeared at 1247, 1444, 1616, 2325, 3267, 3323, and 3735 cm−1. The absorption peak at 1247 cm−1 represents δ(N–H), and the broader absorption band at 1444 cm−1 represents the asymmetric deformation vibration δ(NH4+) of the amino group (NH4+) adsorbed at the Brønsted acidic site. The absorption band at 1616 cm−1 represents the superposition of the absorption peaks of ammonia and nitrate, which originates from the intermediate product33 generated by the dehydrogenation reaction of NH3 adsorbed in the ligand state at the Lewis acidic site; the absorption peak at 2325 cm−1 represents the combined vibration of nitric acid; the absorption peaks at 3267 cm−1 and 3323 cm−1 are assigned to the antisymmetric stretching vibration ν of N–H (N–H); and the band at 3735 cm−1 represents the stretching vibration ν(O–H) of hydroxyl groups. A comparison with Fig. 5(a) shows that the absorption peaks of both nitrate and ammonium salts appeared after 10 min of adsorption and were relatively weak, indicating that the active sites of the catalyst were masked, thereby reducing the acidity and redox properties of the catalyst and inhibiting the adsorption of NH3 and NOx.
Fig. 5(c) shows that the catalyst impregnated with 28% aluminum nitrate had several distinctive absorption peaks in the range of 4000–1000 cm−1 in the reaction of NO + O2 flue gas at 380 °C, located at 1381, 1519, 2378, 3316 and 3630 cm−1. Among them, the two strong and sharp peaks at 1381 cm−1 and 1519 cm−1 originated from nitrate contained in the catalyst sample itself or from nitrite produced by the reaction. The stronger peak at 2378 cm−1 is attributed to the combined vibration of nitrate-group substances, the absorption peak at 3316 cm−1 represents the antisymmetric stretching vibration ν(N–H) of N–H, and the band at 3630 cm−1 represents the stretching vibration peak of the hydroxyl group (–OH), which are weaker peaks formed by the interaction of hydroxyl groups with nitrate or nitrite. When the time reached 5 min, the absorption peaks of nitrate and nitro compounds appeared and increased with increasing adsorption time, indicating gradual adsorption of NOx on the catalyst surface.
Fig. 5(d) shows that the absorption peaks at 1254, 1311, 1454, 1623, 2192, 3177, 3265, and 3332 cm−1 appeared for the catalyst impregnated with 28% aluminum nitrate in the reaction of NH3 +NO + O2 at 380 °C for the flue gas. Among them, the strong absorption peak at 1254 cm−1 represents δ(N–H); the stronger absorption band at 1311 cm−1 is assigned to the symmetric stretching vibration peak ν(N–O) of nitrate; the broader absorption peak at 1454 cm−1 represents the asymmetric deformation vibration δ(NH4+) of the amino group adsorbed at the Brønsted acidic site, the occurrence of which is due to the interaction of ammonia molecules with the Al(NO)3 catalyst surface, resulting in the formation of NH4+ groups.34 The weaker absorption peak at 1623 cm−1 represents the superposition of the absorption peaks of ammonia and nitrate, which originates from the intermediate product generated by the dehydrogenation of NH3 in the ligand state adsorbed in the Lewis acidic site. The absorption band at 2192 cm−1 represents the combined vibration of nitrate; the absorption peaks at 3177, 3265 and 3332 cm−1 represent the antisymmetric stretching vibration ν(N–H)35 of N–H. These peaks are stronger and sharper and represent the abundant N–H bonds in the product. It can be seen from the figure that both B-acid and L-acid sites existed on the catalyst surface, and the peaks of NH4+ representing the B-acid site were relatively strong, indicating that NH4+ gradually accumulates with an increase in adsorption time, while the NH3 of the ligand state adsorbed on the Lewis acidic site appeared to be dehydrogenated, and therefore the NH3 of the ligand state adsorbed on the L-acid site was more active.
Fig. 5(e) shows that there were several distinct absorption peaks at 1075, 1384, 1535, 2241, 3429, and 3667 cm−1 for the catalyst impregnated with 16% aluminum sulfate in the reaction of NO + O2 flue gas at 380 °C. Among them, the absorption band located at 1075 cm−1 represents the stretching vibration of sulfate from the catalyst, or the sulfate or bisulfate adsorbed on the surface.36 The presence of sulfate increased the number of acidic sites on the catalyst surface, thus promoting the adsorption of ammonia and the formation of intermediate NH3+. The three strong peaks at 1230, 1384, and 1535 cm−1 originated from nitrite or nitrate produced by the reaction of the catalyst sample, indicating that NO is easily oxidized to NO2.37 The stronger absorption peak located at 2241 cm−1 represents the combined vibration of nitrate group substances; the band located at 3667 cm−1 is the characteristic peak of water, which is proof of the reaction of hydroxyl groups on the surface of the catalyst sample to produce water. From the figure, we find that the absorption peaks representing nitrate and nitro compounds appeared within 1 min after the passage of NO + O2, and the characteristic peak of NO + O2 adsorption on the catalyst surface gradually increased with an increase in the reaction time, indicating that the catalyst impregnated with aluminum sulfate had a strong redox property.
Fig. 5(f) shows that absorption peaks appeared at 1302, 1429, 3392, and 3682 cm−1 for the reaction of NH3 + NO + O2 at 380 °C for the flue gas impregnated with the 16% aluminum sulfate catalyst. Among them, the stronger absorption at 1302 cm−1 represents the symmetric stretching vibration peak ν(N–O) of nitrate, which is the most important reactive reactant under SCR reaction conditions. The broader absorption peak at 1429 cm−1 represents the asymmetric deformation vibration δ(NH4+) of the amine group adsorbed at the Brønsted acidic site, which appeared to be due to the interaction of ammonia molecules with the catalyst surface. The very weak absorption peak at 1609 cm−1 originated from the dehydrogenation of liganded NH3 adsorbed at the Lewis acidic site to produce a small number of intermediates. The NH4+ ion and liganded NH3 are the active intermediates of the SCR catalytic reaction. The band at 3392 cm−1 represents the anti-symmetric stretching vibration of N–H, which is weak and represents less N–H bonding in the product, while the band at 3682 cm−1 is the stretching vibration of –OH, which may be due to the vibration of water molecules generated by the interaction of hydroxyl groups with NH3. From the figure, it can be seen that, after 5 min of adsorption, the adsorption species of NOx and NH3 appeared on the catalyst surface, the absorption peak of NH4+ adsorbed by the catalyst at the acidic site of Brønsted was stronger, and only the absorption peak of the intermediate product generated by the dehydrogenation reaction of NH3 in the ligand state appeared on the catalyst surface. It can be deduced from the above phenomenon that the NH3 ligand on the catalyst surface was very active and reacted rapidly as the reaction proceeded, which also indicated to some extent that aluminum sulfate could improve the activity of the catalyst.
The results of the comprehensive analysis showed that both aluminum nitrate and aluminum sulfate could greatly increase the specific surface area of the catalysts, in which the specific surface area of the aluminum sulfate catalysts could reach 48.7119 m2 g−1 after impregnation with 16% aluminum sulfate, which provided more active sites for the reactants, while aluminum dihydrogen phosphate would have a great negative impact on the catalysts, reducing their specific surface area. The three aluminum salts did not affect the crystalline shape of the catalyst, Al2O3 and V2O5 were still distributed on the surface of the carrier in an amorphous or highly dispersed form, and the catalyst maintained a good anatase crystalline structure; comparing the NH3-TPR and H2-TPD results of the three aluminum salts, aluminum sulfate could increase the surface acidic sites of the catalyst, significantly improving the performance of NH3-SCR, and promoting the adsorption and activation of NH3, which greatly altered the redox properties on the surface of catalyst, while aluminum nitrate and aluminum dihydrogen phosphate inhibited the properties of the catalyst to different degrees, which might imply inhibition of the catalytically active anion; according to FT-IR analysis, both L and B acidic sites are present in the catalyst in the NH3-SCR reaction. The ligand NH3 adsorbed at the L-acidic site was very active and reacted rapidly with increasing adsorption time, indicating that the reaction of NH3 in the ligand state on the Lewis acidic site was the main reaction. Based on the results obtained in this study, it is hypothesized that a synergistic approach for further catalyst activity and structure enhancement is to use a small amount of the reinforcing agent acting on the critical fragile part while reducing the effect on the pore structure of the active ingredient that can be implanted and increasing the amount of active ingredient implanted.
4. Conclusion
The analysis revealed that the use of aluminum dihydrogen phosphate reinforcement caused catalyst P poisoning, which not only blocked the internal pore structure of the catalyst, reduced the specific surface area of the catalyst, masked the active sites on the catalyst surface, significantly reduced the number of acid sites on the catalyst surface, and greatly affected the redox performance, but also inhibited the adsorption of NOx, thus causing catalyst poisoning and deactivation. The best result was achieved by static impregnation of aluminum nitrate solution at 28% for 10 min; when the loading rate of aluminum nitrate was 10.1%, the compressive strength of the catalyst was 4.71 MPa, which was 45% higher than that of the original regenerated catalyst, and the wear rate was 0.082% kg−1, which was 53.7% lower than that of the original regenerated catalyst. However, the denitration efficiency of the regenerated catalyst was 50.35%, which was still relatively low compared to the 58% of the original regenerated catalyst, indicating that aluminum nitrate still had a certain inhibitory effect on the catalyst. The analysis revealed that the acidic site on the catalyst changed from being a strong acid to a medium strong acid, and the redox performance of the active center of the catalyst was reduced. A synergistic approach for further catalyst activity and structural strengthening is postulated to have a small amount of reinforcing agent acting on the critically vulnerable part, while reducing the impact on the pore structure of the active ingredient that can be implanted and increasing the amount of active ingredient implanted to avoid the introduction of toxic anions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Jiangsu Industry-University-Research Cooperation Project (BY2022669), the National Natural Science Foundation of China Project (51979077), the Jiangsu Province Science and Technology Program Project (BE2019121), the State Grid Jiangsu Province Electric Power Engineering Consulting Co., Ltd. Research Project (821105016), the Jiangsu Province Social Science Federation Research Project (22XTB-59), and the Jiangsu Province Youth and Blue Project (2021SZJS-003).References
- B. Ye, B. Jeong, M.-J. Lee, T. H. Kim, S.-S. Park, J. Jung and S. Lee, Nano Convergence, 2022, 9, 51 CrossRef CAS.
- H. Bai, Z. Zhang, Z. Li, X. Wu, X. Guo, J. Zhang and D. Bi, ACS Omega, 2022, 7, 11853–11861 CrossRef CAS.
- B. Chu, Q. Ma, J. Liu, J. Ma, P. Zhang, T. Chen, Q. Feng, C. Wang, N. Yang, H. Ma, J. Ma, A. G. Russell and H. He, Environ. Sci. Technol. Lett., 2020, 7(10), 695–700 CrossRef CAS.
- L. Wang, Ecol. Environ. Sci., 2019, 28, 90–96 Search PubMed.
- X. Lan and L. Zhao, Environ. Chem., 2022, 41, 3778–3788 CAS.
- W. Lia, X. Dua, Z. Lia, Y. Taoa, J. Xuea, Y. Chena, Z. Yanga, J. Rana, V. Racb and V. Rakićb, Process Saf. Environ., 2022, 159, 213–220 CrossRef.
- X. Zhang, S. Liu, K. Ma, Y. Chen, S. Jin, X. Wang and X. Wu, Catalysts, 2021, 11, 1360 CrossRef CAS.
- S. Feng, Z. Li, B. Shen, P. Yuan, J. Ma, Z. Wang and W. Kong, J. Environ. Manage., 2022, 317, 115457 CrossRef CAS PubMed.
- M. Kapkowski, T. Siudyga, R. Sitko, A. Niemczyk-Wojdyl, T. Zelenka, G. Zelenková, S. Golba, A. Smolinski and J. P. Kapkowski, J. Cleaner Prod., 2021, 316, 128291 CrossRef CAS.
- X. Geng, C. Xie, B. Zhu, J. Chen, Y. Sun and M. Xu, Environ. Sci. Pollut. Res., 2022, 29, 88256–88268 CrossRef CAS.
- J. Kim, D. H. Kim and H. P. Ha, J. Hazard. Mater., 2020, 397, 122671 CrossRef CAS.
- P. Wu, X. Tang, Z. He, Y. Liu and Z. Wang, Energy Fuels, 2022, 36, 5622–5646 CrossRef CAS.
- J. Liu, X. Shi, Z. Lv, Y. Yu and H. He, Catal. Sci. Technol., 2022, 12, 2471–2481 RSC.
- Y. Tian, J. Yang, L. Liu, Q. Liu, B. Kong, F. Lin, M. Kong and G. Hu, J. Energy Inst., 2020, 93, 387–394 CrossRef CAS.
- H. Yin. and Y. Wang, Mod. Chem. Ind., 2019, 39, 16–20 Search PubMed.
- X. Liu, Y. Jia, Z. Tang, P. Zhang, S. Wu, G. Liu, J. Wang and Y. Yang, Appl. Chem. Ind., 2020, 49, 1839–1844 Search PubMed.
- X. Wang, H. Ma, Y. Shi, Q. Wang, P. Xu, W. Li and S. Li, Fuel, 2021, 285, 119069 CrossRef.
- P. Gong, B. Zhang and S. H. Liu, Inorg. Chem. Ind., 2021, 53, 14–17 Search PubMed.
- K. Cheng, Y. Yu, B. Mei, Y. Li and L. Xu, Sustainability, 2022, 14, 10284 CrossRef.
- L. Song, H. Yue, K. Ma, W. Liu, W. Tian, C. Liu, S. Tang and B. Liang, ACS Sustainable Chem. Eng., 2020, 8, 16878–16888 CrossRef CAS.
- Q. Lu, X. Pei, M. Xu, H. Wang, Y. Wu and H. OuYang, Chem. Ind. Eng. Prog., 2021, 40, 2365–2374 CAS.
- S. Cimino, C. Ferone, R. Cioffi, G. Perillo and L. Lisi, Catalysts, 2019, 9, 464 CrossRef CAS.
- T. Zhu, et al., Research progress on regeneration of waste scr catalyst, Environ. Eng., 2018, 36, 92 Search PubMed.
- W. Shen, Y. Liu, B. Yan, J. Wang, P. He, C. Zhou, X. Huo, W. Zhang, G. Xu and Q. Ding, Renewable Sustainable Energy Rev., 2017, 75, 618–628 CrossRef CAS.
- L. Zhao, C. Li, S. Li, Y. Wang, J. Zhang, T. Wang and G. Zeng, Appl. Catal., B, 2016, 198, 420–430 CrossRef CAS.
- B. Zhao, R. Ran, X. Guo, L. Cao, T. Xu, Z. Chen, X. Wu, Z. Si and D. Weng, Appl. Catal., A, 2017, 545, 64–71 CrossRef CAS.
- M. Guo, Q. Liu, P. Zhao, J. Han, X. Li, Y. Ha, Z. Fu, C. Song, N. Ji, C. Liu, D. Ma and Z. Li, Chem. Eng. J., 2019, 361, 830–838 CrossRef CAS.
- D. W. Kwon, K. H. Park and S. C. Hong, Appl. Catal., A, 2013, 451, 227–235 CrossRef CAS.
- S. Zhao, J. Peng, R. Ge, K. Yang, S. Wu, Y. Qian, T. Xu, J. Gao, Y. Chen and Z. Sun, Process Saf. Environ., 2022, 168, 971–992 CrossRef CAS.
- I. Dahlan, K. T. Lee, A. H. Kamaruddin and A. R. Mohamed, J. Hard Mater., 2009, 166, 1556–1559 CrossRef CAS.
- R. Jin, Y. Liu, Z. Wu, H. Wang and T. Gu, Chemosphere, 2010, 78, 1160–1166 CrossRef CAS PubMed.
- F. Cao, J. Xiang, S. Su, P. Wang, L. Sun, S. Hu and S. Lei, Chem. Eng. J., 2014, 243, 347–354 CrossRef CAS.
- T. Gu, R. Jin, Y. Liu, H. Liu, X. Weng and Z. Wu, Appl. Catal., B, 2013, 129, 30–38 CrossRef CAS.
- N. Y. Topsoe, H. Topsoe and J. A. Dumesic, J. Catal., 1995, 151, 226–240 CrossRef CAS.
- M. A. Centeno, I. Carrizosa and J. A. Odriozola, Appl. Catal., B, 2001, 29, 307–314 CrossRef CAS.
- Y. Yu, C. Chen, M. Ma, M. Douthwaite, C. He, J. Miao, J. Chen and C. Li, Chem. Eng. J., 2019, 361, 820–829 CrossRef CAS.
- F. Liu, K. Asakura, H. He, W. Shan, X. Shi and C. Zhang, Appl. Catal., B, 2011, 103, 369–377 CrossRef CAS.
| This journal is © the Owner Societies 2024 |