
Boosting the oxygen reduction reaction activity of dual-atom catalysts on N-doped graphene by regulating the N coordination environment†
Lei
Li
 *ab,
Xiaoxia
Wu
ab,
Qiuying
Du
ab,
Narsu
Bai
ab and
Yuhua
Wen
*c
*ab,
Xiaoxia
Wu
ab,
Qiuying
Du
ab,
Narsu
Bai
ab and
Yuhua
Wen
*c
aModern Physics Research Center, College of Physics and Electronic Information, Inner Mongolia Normal University, Hohhot 010022, China. E-mail: lilei@imnu.edu.cn
bInner Mongolia Key Laboratory for Physics and Chemistry of Functional Materials, Hohhot 010022, China
cDepartment of Physics, Xiamen University, Xiamen 361005, China. E-mail: yhwen@xmu.edu.cn
First published on 29th November 2023
Abstract
Development of low-cost and high-efficiency oxygen reduction reaction (ORR) catalysts is of significance for fuel cells and metal–air batteries. Here, by regulating the N environment, a series of dual-atom embedded N5-coordinated graphene catalysts, namely M1M2N5 (M1, M2 = Fe, Co, and Ni), were constructed and systematically investigated by DFT calculations. The results reveal that all M1M2N5 configurations are structurally and thermodynamically stable. The strong adsorption of *OH hinders the proceeding of ORR on the surface of M1M2N5, but M1M2N5(OH2) complexes are formed to improve their catalytic activity. In particular, FeNiN5(OH2) and CoNiN5(OH2) with the overpotentials of 0.33 and 0.41 V, respectively, possess superior ORR catalytic activity. This superiority should be attributed to the reduced occupation of d-orbitals of Fe and Co atoms in the Fermi level and the apparent shift of dyz and dz2 orbitals of Ni atoms towards the Fermi level after adsorbing *OH, thus regulating the active sites and exhibiting appropriate adsorption strength for reaction intermediates. This work provides significant insight into the ORR mechanism and theoretical guidance for the discovery and design of low-cost and high-efficiency graphene-based dual-atom ORR catalysts.
I. Introduction
The development of renewable energy conversion and storage devices, such as fuel cells and metal–air batteries, is of considerable importance for reducing the reliance on fossil fuels and resolving the environmental issues.1,2 However, the sluggish cathodic oxygen reduction reaction (ORR) heavily limits the overall performance of these devices and requires catalysts to accelerate the reaction process.3 At present, platinum (Pt)-based materials are still the most commonly used ORR catalysts, but the high cost and natural scarcity of Pt have severely restricted their widespread applications.4,5 Therefore, there is an urgent need to explore low-cost and high-performance alternatives to substitute Pt-based catalysts.Metal-embedded nitrogen-doped graphene (M–N–G) single-atom catalysts (SACs) have attracted increasing attention due to their abundant active sites, outstanding mechanical flexibility, and unique electronic properties.6–8 Unfortunately, their long-term stability and electrocatalytic performances are restrained by their simple geometric structure and single active site.9 Introducing a second metal atom into the single metal active site to form double-atom catalysts (DACs) not only inherits the advantages of SACs but also is beneficial to overcoming the restrictions above.10–13 Firstly, the chemical interactions between two metal atoms in DACs can prevent the agglomeration of individual species, which leads to higher metal loading and more stable active sites than SACs.10 Furthermore, DACs can provide adjacent coordinated reaction sites, modulate the adsorption strength and mode of reaction intermediates, and optimize the reaction pathway, thus leading to the remarkable improvement of the ORR catalytic activity.11,12 For example, Fe–Co and Co–Zn embedded N-doped graphene catalysts exhibit excellent ORR catalytic activity, operation durability, and structural stability, outperforming the commercial Pt/C.14,15 The incorporation of an Ir atom into the single Co active site can induce the re-arrangement of the d-electron of the Co atom toward intensified spin polarization and thus propel faster reaction kinetics.16 Recently, Fe–Se and Fe–Cu DACs were experimentally identified to possess better structural stability and ORR catalytic activity than Fe-based SACs.17,18
For N-doped graphene-based dual-metal–atom catalysts, the local coordination environment of dual metal atoms plays important roles in the modulation of their electronic structures and thus affecting their ORR catalytic activity. In particular, the N numbers and species have a significant effect on the ORR performance. FeCoN6 was identified as the best ORR catalyst among the FeCoNx (x = 1–6) DACs and the Fe atom was the main active center.19 Subsequently, the DFT calculations showed that FeCoN7 and FeCoN8 possess better catalytic performance as compared with FeCoN6, and the main active center was transformed from the Fe to Co atom.20 In addition, M1M2N8 (M1, M2 = Co, Pt, Fe, Ni) was confirmed to have better ORR catalytic activity than M1M2N6, and CoPtN8 has the smallest overpotential of 0.30 V.21 However, FeFeN6, FeCoN6, and CoZnN6 were found to exhibit the highest ORR catalytic activity by screening 63 M1–M2 (M1, M2 = Mn, Fe, Co, Ni, Cu, and Zn) embedded N8 and two kinds of N6 coordinated graphene.22 In our previous work, Ni–Ni dual-atom active sites displayed the highest ORR catalytic activity among all M1M2N6 (M1, M2 = Mn, Fe, Co, Ni, Cu, and Zn) catalysts through constructing a new N6 coordinated graphene support.23 Therefore, regulating the N coordination environment in DACs can be expected to further improve the ORR catalytic activity and enrich the structural possibilities of N-doped graphene-based dual-metal-atom catalysts.
In this work, by regulating the N numbers and sites, a N5-coordinated graphene support was designed to anchor the dual metal atoms. Considering that Fe, Co, and Ni are currently common participating metal atoms in ORR DACs with good catalytic performance, they were selected to construct M1M2N5 catalysts by embedding different dual metal combinations into N5-coordinated graphene. The structural stability, the electronic properties, the adsorption behaviors of intermediates, the ORR catalytic activity, and the reaction mechanism of these M1M2N5 catalysts were thoroughly investigated by DFT calculations. This work not only schemes out FeNiN5 and CoNiN5 DACs with excellent ORR catalytic activity, but provides a significant insight into the fundamental understanding of the ORR reaction mechanism at the atomic level.
II. Computational method
All the spin-polarized DFT calculations were performed using the Vienna ab initio simulation package (VASP).24,25 Electronic exchange–correlation interactions were treated with the Perdew–Burke–Ernzerhof (PBE) functional method within the generalized gradient approximation (GGA).26 The kinetic energy cutoff was chosen as 500 eV to sufficiently yield good energy convergence. The convergence criteria for the atomic force and the total energy were respectively set to 0.02 eV Å−1 and 10−5 eV. A sufficient vacuum of 15 Å in the c direction was used to eliminate the interactions between mirror images. The Brillouin zone was sampled with a 3 × 3 × 1 Monkhorst–Pack k-point grid for structural optimizations and a 11 × 11 × 1 grid for the electronic-structure calculations. The default setting of the ferromagnetic state for metal atoms in VASP was adopted and the initial magnetic moment for each metal atom was set to 3 μB. The DFT-D2 dispersion correction was adopted to describe the van der Waals interactions between catalysts and reaction intermediates.27,28 Ab initio molecular dynamics (AIMD) simulations were performed at the temperature of 300 K in the canonical ensemble with a time step of 2 fs and a total time of 10 ps.To evaluate the structural stability of M1M2N5, the formation energy (Ef) and the binding energy (Eb) between metal atoms and N5-coordinated graphene are calculated using the following equations
| Ef = EM1M2N5 − EM1 − EM2 − 5μN − 42μC, | (1) |
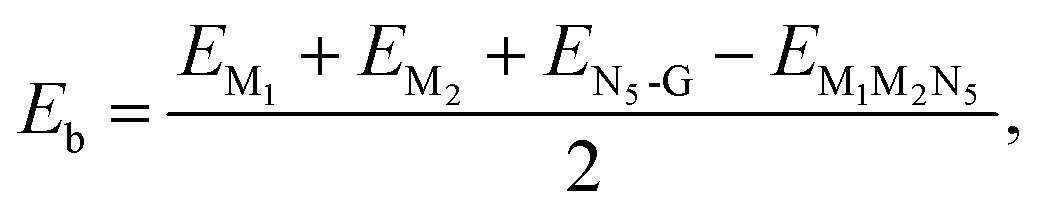 | (2) |
The adsorption energy (Eads) of the reaction intermediates on the catalysts is defined as
| Eads = Ecatalyst+X − Ecatalyst − EX, | (3) |
The free energy change (ΔG) of each reaction step is determined according to the following equation:
| ΔG = ΔE + ΔZPE − TΔS + ΔGU + ΔGpH, | (4) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 × pH, in which kB is the Boltzmann constant, and pH is set as 0.
10 × pH, in which kB is the Boltzmann constant, and pH is set as 0.
III. Results and discussion
The schematic diagram for constructing the M1M2N5 structure is illustrated in Fig. 1. Firstly, three connected C atoms were removed from a 5 × 5 pristine graphene supercell to form the tri-vacancy defect. Subsequently, five C atoms around the vacancy site were replaced by N atoms to form the N5-coordinated graphene. Finally, nine M1M2N5 catalysts (i.e., Fe2N5, Co2N5, Ni2N5, FeCoN5, CoFeN5, FeNiN5, NiFeN5, CoNiN5, and NiCoN5) were constructed by putting different diatomic combinations of Fe, Co, and Ni atoms into the vacancy site of N5-coordinated graphene. Due to the symmetry of N atoms in N5-coordinated graphene, CoFeN5, NiFeN5, and NiCoN5 respectively exhibited the same structure as FeCoN5, FeNiN5, and CoNiN5. Therefore, there are six possible structures in total, namely Fe2N5, Co2N5, Ni2N5, FeCoN5, FeNiN5, and CoNiN5. Among all the structures, the M1 atom was deviated from the plane of graphene, that is, these structures were divided into the raised side (RS) and the dented side (DS) (see step 3 in Fig. 1). To evaluate the structural stability of these catalysts, the formation energy (Ef) for M1M2N5, the binding energy (Eb) between metal atoms and N5-coordinated graphene, and the cohesive energy (Ecoh) for the bulk metal were calculated (see Table S1 for detailed results, ESI†). Evidently, the Ef values for all structures are negative. Moreover, the results of AIMD simulations show that the energy of all structures smoothly oscillates around a certain value, and there is no bond breakage, as indicated by Fig. S1 (ESI†). Furthermore, the larger Eb compared with Ecoh for all structures suggests that metal atoms tend to strongly combine with the N5-coordinated graphene rather than to aggregate together. Therefore, all the M1M2N5 structures mentioned above are thermodynamically stable and are considered in the following discussions.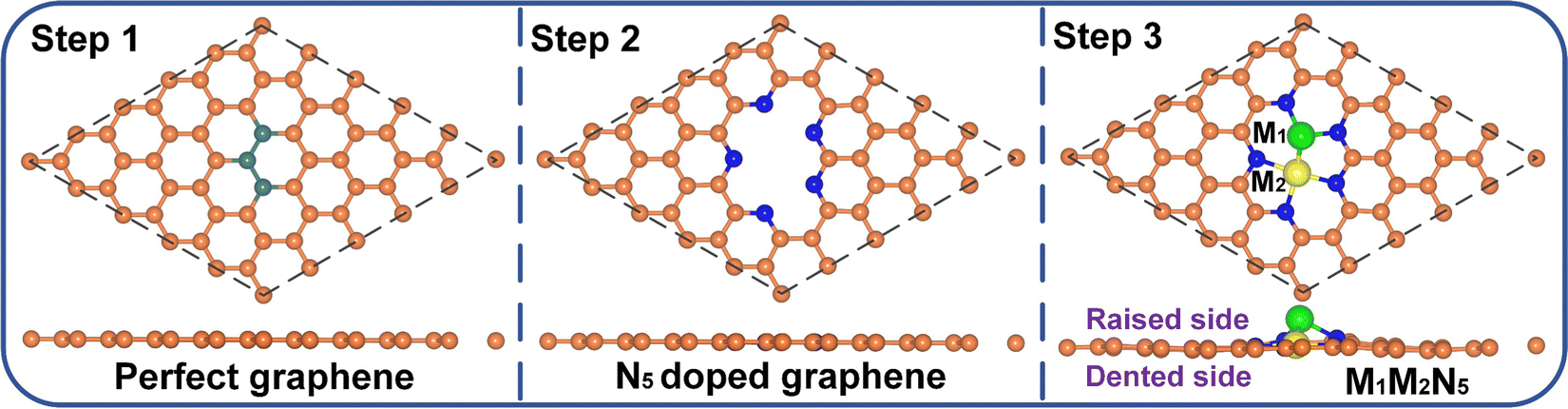 | ||
| Fig. 1 Schematic diagram for the formation of the M1M2N5 structure. The orange, olive, blue, green, and yellow balls denote C in perfect graphene, removed C, N, M1, and M2 atoms, respectively. | ||
In an acidic medium, the ORR process includes multiple reaction steps, and the adsorption of reaction intermediates plays a key role in the whole reaction process. Hence, the most stable adsorption structures of reaction intermediates on M1M2N5 were firstly explored by considering various possible adsorption configurations, and the calculated results are presented in Fig. 2(a). One can find that there is only one type of stable adsorption configuration for *O2 and *OH, while two possible stable configurations exist for *OOH (*OOH-I for Fe2N5, Co2N5, FeCoN5, and CoNiN5 as well as *OOH-II for Ni2N5 and FeNiN5) and *O (*O-I for FeNiN5 and CoNiN5 as well as *O-II for Fe2N5, Co2N5, Ni2N5, and FeCoN5). Among all stable adsorption structures, the M1 atom is the main active site to adsorb reaction intermediates. As is expected, the raised metal atom is more likely to capture the reaction intermediates. Meanwhile, the corresponding Eads and the M–O bond length (dM–O) are listed in Table 1 and Tables S2, S3 (ESI†).
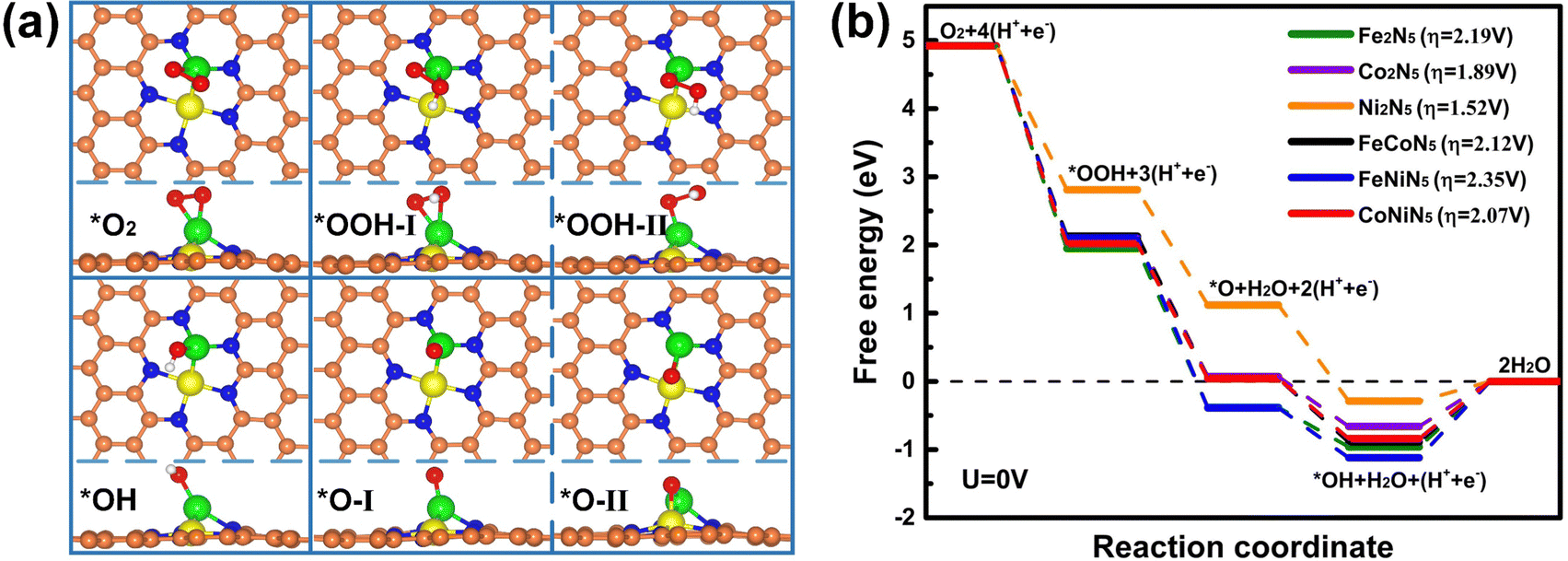 | ||
| Fig. 2 (a) Schematic illustration of the most stable adsorption structures of reaction intermediates on M1M2N5. (b) Free-energy diagrams of the ORR on M1M2N5. The orange, blue, green, yellow, red, and white balls denote C, N, M1, M2, O, and H atoms, respectively. | ||
| Model | E ads (eV) | Model | E ads (eV) | Model | E ads (eV) | ||
|---|---|---|---|---|---|---|---|
| RS | DS | RS | DS | ||||
| Fe2N5 | −3.06 | Fe2N5(OH) | −1.97 | −1.06 | Fe2N5(OH2) | −0.93 | −1.12 |
| Co2N5 | −2.84 | Co2N5(OH) | −2.11 | −1.85 | Co2N5(OH2) | −0.43 | −0.97 |
| Ni2N5 | −2.44 | Ni2N5(OH) | −1.02 | −0.40 | Ni2N5(OH2) | −0.23 | −0.34 |
| FeCoN5 | −2.89 | FeCoN5(OH) | −1.56 | −1.00 | FeCoN5(OH2) | −0.61 | −0.87 |
| FeNiN5 | −3.16 | FeNiN5(OH) | −1.22 | −0.38 | FeNiN5(OH2) | −0.28 | −0.45 |
| CoNiN5 | −2.91 | CoNiN5(OH) | −1.06 | −0.33 | CoNiN5(OH2) | −0.24 | −0.33 |
To further unveil the ORR process, the free-energy diagrams of the four-electron reaction pathway (*O2 → *OOH → *O + H2O → *OH + H2O → 2H2O) and the theoretical overpotential for all structures are presented in Fig. 2(b). It can be seen that only the free energy for the hydrogenation of *OH to H2O is uphill at zero electrode potential (U = 0 V), implying that this step is endothermic and thermodynamically unfavorable, and thus the ORR process is hindered by the formation of the second H2O molecule. Therefore, the rate-determining step (RDS) for the ORR on all M1M2N5 structures is the hydrogenation of *OH to H2O. This RDS arises from the strong adsorption of *OH on M1M2N5 (see Table S2, ESI†) and leads to the large overpotentials of 2.19, 1.89, 1.52, 2.12, 2.35, and 2.07 V, respectively. Meanwhile, the *OH can be regarded as a ligand to modify M1M2N5, thus forming M1M2N5(OH) complexes. Hence, the catalytic performances of M1M2N5(OH) complexes need to be further investigated.
Since the chemisorption of O2 is usually regarded as a prerequisite for triggering the ORR, the adsorption characteristics of O2 on the RS and DS of M1M2N5(OH) were firstly investigated. Through considering all the adsorption possibilities, the energetically favorable O2 adsorption configurations on the RS and DS of M1M2N5(OH) were obtained (see Fig. 3(a) and Fig. S2 for details, ESI†). It can be seen from Fig. 3(a) that O2 tends to be adsorbed on the RS of Fe2N5(OH), Co2N5(OH), and FeCoN5(OH) by bonding with dual metal atoms, but prefers to bond with the M1 atom of Ni2N5(OH), FeNiN5(OH), and CoNiN5(OH). This result is understandable since the spin charges are distributed on both M1 and M2 atoms in the former but only on the M1 atom in the latter, as indicated by Fig. 4. Different from the above results, O2 only bonded with the M2 atom on the DS of M1M2N5(OH) (see Fig. S2, ESI†). Furthermore, Eads of O2 on the RS and DS of M1M2N5(OH) were calculated to determine the most stable adsorption configurations. As shown in Table 1, the adsorption of O2 on the RS has higher Eads than that on the DS, suggesting that it is more likely to adsorb O2 on the RS of M1M2N5(OH). Therefore, the adsorptions of other reaction intermediates and the subsequent reaction steps of the ORR should occur in the RS of M1M2N5(OH). In addition, after forming the M1M2N5(OH) complexes, the Eads of O2 was remarkably decreased compared with M1M2N5 (see Table 1), and dM–O was stretched (see Table S3, ESI†). This is because the *OH obtained some charges from dual metal atoms (see Table S4, ESI†), thus reducing their adsorption capability. Besides O2, the most stable adsorption structures of other reaction intermediates (*OOH, *OH, and *O) on the RS of M1M2N5(OH), the corresponding Eads, and the dM–O are illustrated in Fig. 3(a) and Tables S2, S3 (ESI†). It can be seen that these intermediates are favorably bonding with a single M1 atom or dual M1 and M2 atoms. Moreover, the introduction of *OH also lowers the interactions between these reaction intermediates with M1M2N5, which may lead to the reduced overpotential.
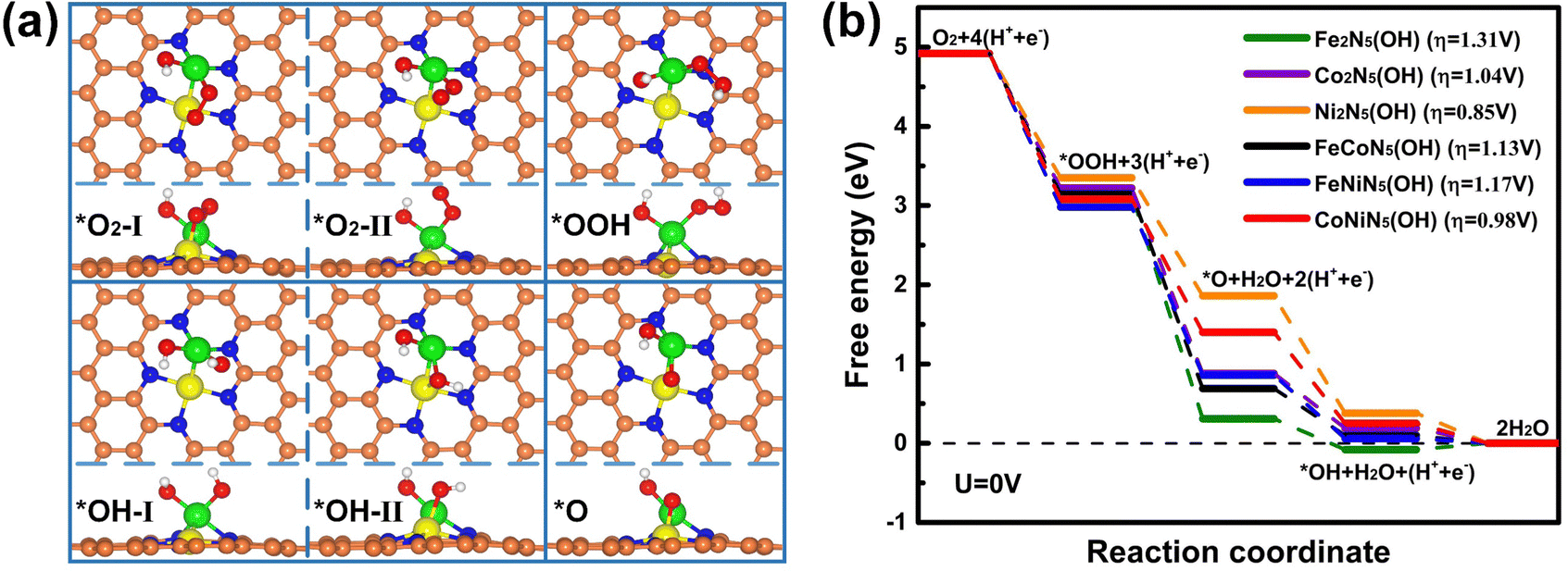 | ||
| Fig. 3 (a) Schematic illustration of the most stable adsorption structures of reaction intermediates on M1M2N5(OH). (b) Free-energy diagrams of the ORR on M1M2N5(OH). The orange, blue, green, yellow, red, and white balls denote C, N, M1, M2, O, and H atoms, respectively. | ||
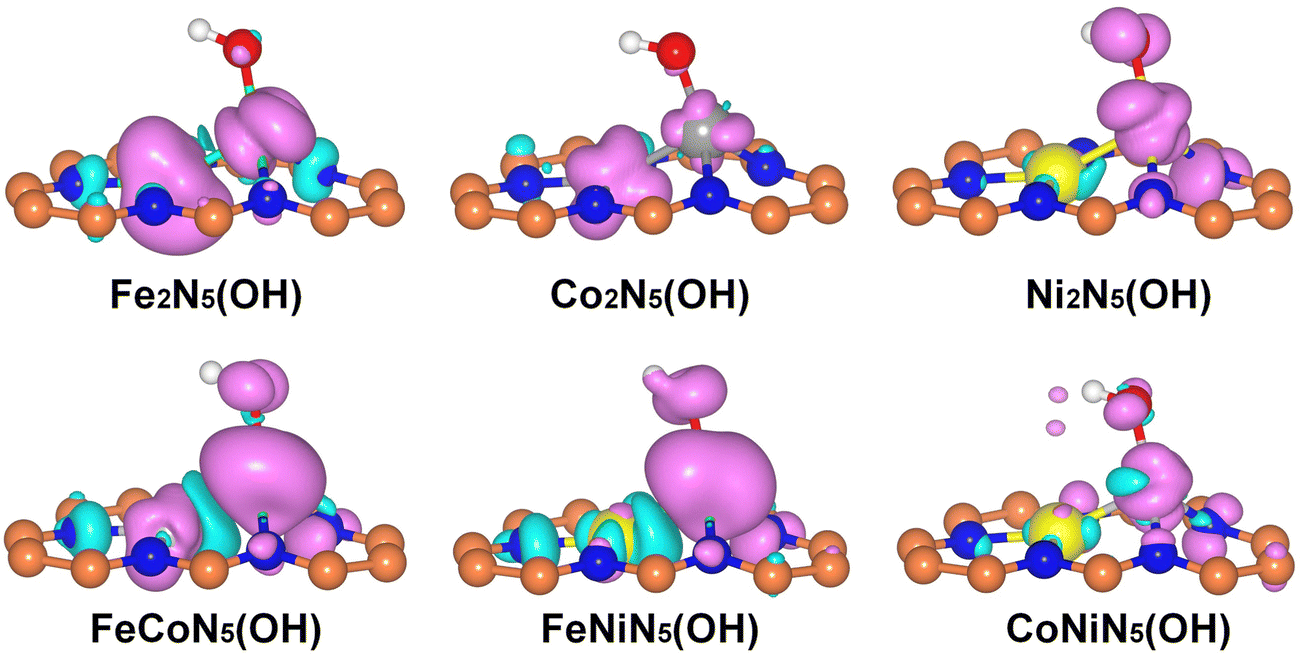 | ||
| Fig. 4 Spin-resolved charge density of M1M2N5(OH) with an isosurface value of 0.02 e Å−3. Purple and cyan bubbles represent positive and negative charges, respectively. The orange, blue, green, gray, yellow, red, and white balls denote C, N, Fe, Co, Ni, O, and H atoms, respectively. | ||
Based on the most stable adsorption structures of the reaction intermediates above, the free energy change and the overpotential for the ORR on M1M2N5(OH) were further investigated to unveil the catalytic activity. Different from M1M2N5, the free energy for all reaction steps of the ORR on M1M2N5(OH) is downhill at U = 0 V except for Fe2N5(OH) [see Fig. 3(b)], while the RDS is still the hydrogenation of *OH to H2O. Meanwhile, the overpotentials of ORR on Fe2N5(OH), Co2N5(OH), Ni2N5(OH), FeCoN5(OH), FeNiN5(OH), and CoNiN5(OH) are respectively reduced to 1.31, 1.04, 0.85, 1.13, 1.17, and 0.98 V, which could be attributed to the reduced binding strength between the reaction intermediates and these catalysts (see Table S2 for details, ESI†). However, these values are still significantly higher than those for Pt (111) (0.44 V)29 and Pt (100) (0.47 V).30 Fortunately, the *OH can be adsorbed on M1M2N5(OH) to form M1M2N5(OH2) complexes for further enhancing the ORR catalytic activity.
After incorporating *OH into M1M2N5(OH), its adsorption characteristics were significantly changed. The most stable adsorption configuration of O2 on M1M2N5(OH2), the corresponding Eads, and dM–O are presented in Table 1 and Fig. S2, Table S3 (ESI†). It can be found that the Eads of O2 on the RS of Ni2N5(OH2), FeNiN5(OH2), and CoNiN5(OH2), respectively, are only −0.23, −0.28, and −0.24 eV, implying the weak interactions between O2 and these catalysts. Moreover, the O–O bond lengths of O2 on these catalysts, respectively, are 1.250, 1.258, and 1.253 Å, which are very close to the O–O bond length of 1.232 Å in gas phase O2. These results indicate that an O2 molecule can be only physically adsorbed on the RS of Ni2N5(OH2), FeNiN5(OH2), and CoNiN5(OH2), and hence the ORR cannot proceed on the RS of these complexes. However, Fe2N5(OH2), Co2N5(OH2), and FeCoN5(OH2) can chemically adsorb O2 on their RS with Eads of −0.93, −0.43, and −0.61 eV, respectively. As compared with the RS of Fe2N5(OH2), Co2N5(OH2), Ni2N5(OH2), FeCoN5(OH2), FeNiN5(OH2), and CoNiN5(OH2), the DS of these structures show a stronger adsorption capacity for O2, and the Eads rises to −1.12, −0.97, −0.34, −0.87, −0.45, and −0.33 eV, respectively, indicating that the O2 prefers to be adsorbed on the DS of M1M2N5(OH2) to proceed the subsequent reactions of the ORR. Moreover, these values are smaller than that on M1M2N5(OH) because dual metal atoms lose some charges to *OH (see Table S4, ESI†). It is noteworthy that the values of −0.34, −0.45, and −0.33 eV fall in the range of −0.24 and −0.80 eV for some excellent dual-metal-atom embedded N-doped graphene ORR catalysts.19,31–33 Thus, Ni2N5(OH2), FeNiN5(OH2), and CoNiN5(OH2) may possess outstanding ORR catalytic activities.
Following the aforementioned O2 adsorption results, the free-energy diagrams of the whole reaction process on M1M2N5(OH2) and the corresponding overpotential are presented in Fig. 5(a) to identify their ORR catalytic activities. Compared with M1M2N5(OH), the RDS of the ORR on Ni2N5(OH2), FeNiN5(OH2), and CoNiN5(OH2) was transferred into the hydrogenation of O2 to *OOH. The overpotentials of the ORR on Fe2N5(OH2), Co2N5(OH2), Ni2N5(OH2), FeCoN5(OH2), FeNiN5(OH2), and CoNiN5(OH2) were reduced to 0.90, 0.73, 0.51, 0.81, 0.33, and 0.41 V, respectively. In particular, the overpotentials of 0.33 V for FeNiN5(OH2) and 0.41 V for CoNiN5(OH2) are smaller than or comparable to those for FeNiN6/8 (0.35–0.90 V),21–23,31,34–36 CoNiN6/8 (0.35–0.79 V),21–23,31,37 Pt (111) (0.44 V),29 and Pt (100) (0.47 V),30 indicating that the FeNiN5(OH2) and CoNiN5(OH2) complexes possess excellent ORR catalytic activity. The corresponding schematic illustration of the ORR process on FeNiN5(OH2) and CoNiN5(OH2) is given in Fig. 5(b). Considering that the FeNiN5(OH2) and CoNiN5(OH2) complexes are formed during the ORR process on FeNiN5 and CoNiN5 due to the strong adsorption of *OH, FeNiN5 and CoNiN5 should be promising alternatives to substitute for Pt catalysts.
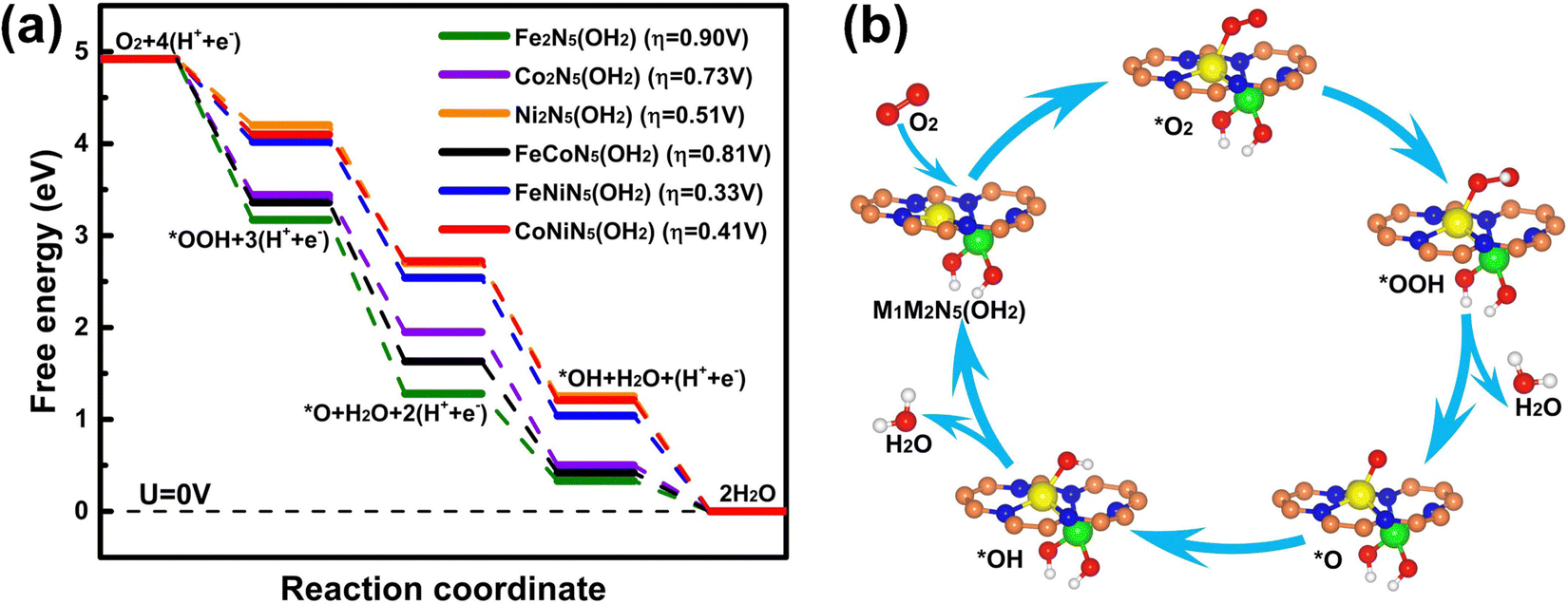 | ||
| Fig. 5 (a) Free-energy diagrams of the ORR on M1M2N5(OH2). (b) Schematic illustration of the ORR process on FeNiN5(OH2) and CoNiN5(OH2). The orange, blue, green, yellow, red, and white balls denote C, N, M1, M2, O, and H atoms, respectively. | ||
To concretely explore the origin of the outstanding catalytic activities of the FeNiN5(OH2) and CoNiN5(OH2), the projected density of states (PDOS) of FeNiN5, FeNiN5(OH), FeNiN5(OH2), CoNiN5, CoNiN5(OH), and CoNiN5(OH2) are presented in Fig. 6. It can be seen from this figure that dxy, dz2, and dx2−y2 orbitals of the Fe atom in FeNiN5 and dxy, dyz, and dz2 orbitals of Co atom in CoNiN5 are mainly distributed near the Fermi-level, but there are no d orbital electronic states of Ni atoms, indicating that Fe and Co atoms are the main active sites for the ORR. These results are consistent with the intermediate adsorption structures on FeNiN5 and CoNiN5. When incorporating *OH into FeNiN5 and CoNiN5, the *OH mainly obtained some electrons from Fe and Co atoms (see Table S4, ESI†), resulting in decreased d-orbitals of Fe and Co atoms near the Fermi-level. However, dyz and dz2 orbitals of Ni atoms in FeNiN5 and CoNiN5 only slightly moved towards the Fermi-level. Therefore, Fe and Co atoms are still the main active sites to adsorb reaction intermediates (as shown in Fig. 3), but their adsorption capabilities were weakened to reduce the overpotential. After forming FeNiN5(OH2) and CoNiN5(OH2) complexes, dyz and dz2 orbitals of Ni atoms continued to move towards the Fermi-level compared with those in FeNiN5(OH) and CoNiN5(OH). Meanwhile, the Fe and Co sites were occupied by the adsorbed *OH, and thus the active sites were transformed from Fe and Co atoms to Ni atoms, corresponding to moderate adsorption strength for reaction intermediates and improved ORR catalytic activity. For Fe2N5, Co2N5, Ni2N5, and FeCoN5, and the adsorption of *OH also moved the d-orbitals of M2 atoms towards the Fermi-level, as shown in Fig. S3 (ESI†). However, more d-orbital electronic states of these atoms distributed near the Fermi level lead to the stronger adsorption capability for intermediates, thus presenting lower ORR catalytic activity compared with FeNiN5 and CoNiN5. In addition, the above results also indicate that *OH ligand modification can be an effective strategy to modify the electronic structures and improve the ORR catalytic activity of graphene-based dual metal atom catalysts.
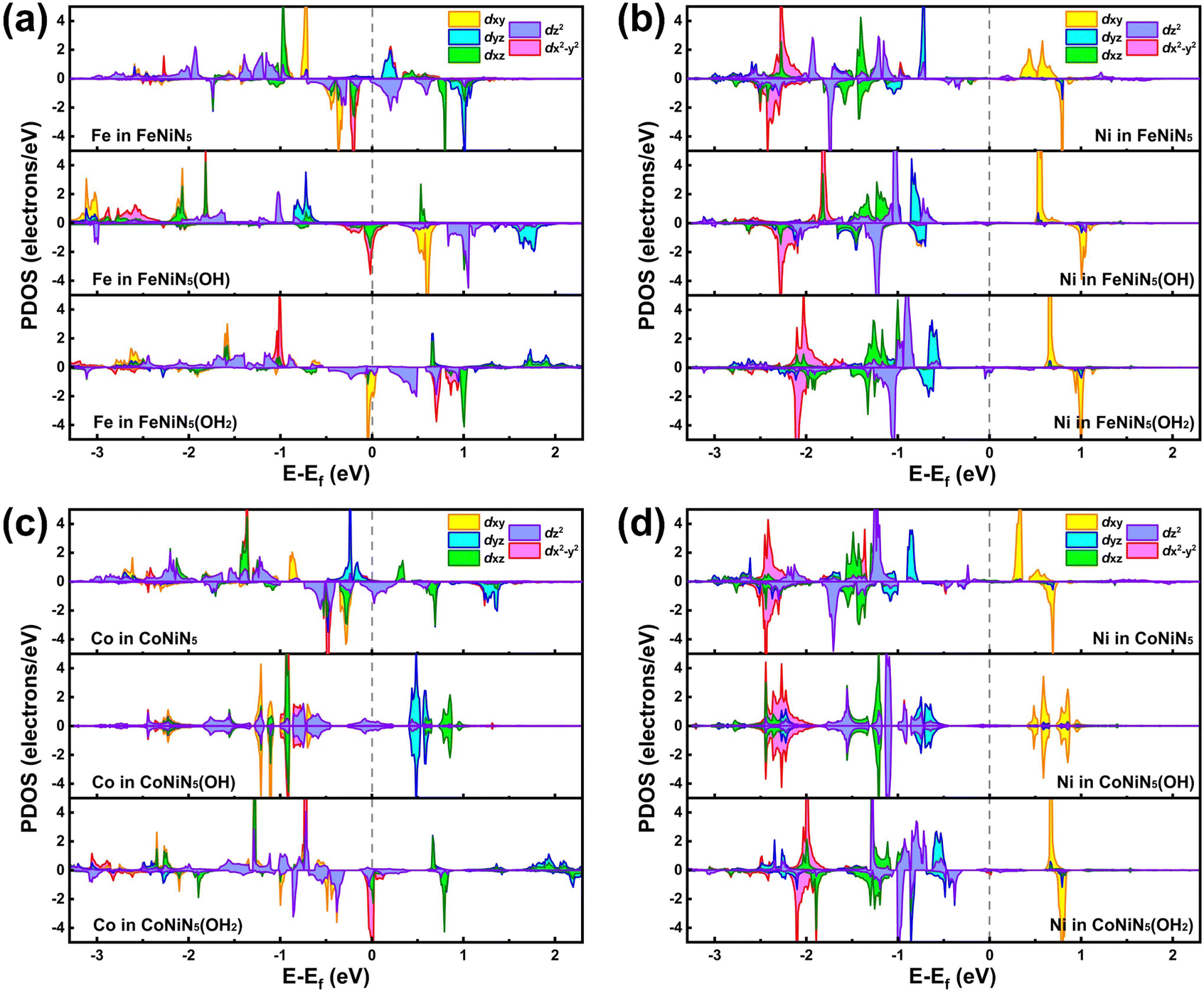 | ||
| Fig. 6 The PDOS of (a) Fe and (b) Ni atoms in FeNiN5, FeNiN5(OH) and FeNiN5(OH2) as well as (c) Co and (d) Ni atoms in CoNiN5, CoNiN5(OH) and CoNiN5(OH2). The dashed line indicates the Fermi level. | ||
IV. Conclusions
In summary, a series of dual-atom embedded N5-coordinated graphene catalysts M1M2N5 (M1, M2 = Fe, Co, and Ni) were constructed by regulating the N coordination environment. The geometric structure, electronic structure, ORR catalytic activity, and reaction mechanism of M1M2N5 catalysts were systematically investigated by DFT calculations. The results reveal that all M1M2N5 configurations are structurally and thermodynamically stable. The strong adsorption of *OH hinders the proceeding of ORR on the surface of M1M2N5 and M1M2N5(OH), but M1M2N5(OH2) complexes are formed in the reaction process. Among all M1M2N5(OH2), FeNiN5(OH2) and CoNiN5(OH2) respectively with the overpotentials of 0.33 and 0.41 V exhibit the best catalytic activity superior to Pt catalysts. The reason for this lies in the fact that the absorbed *OH lowers the occupation of the d-orbitals of Fe and Co atoms in the Fermi level, and shifts the dyz and dz2 orbitals of the Ni atom toward the Fermi level, thus transforming the active sites from Fe and Co atoms to Ni atoms and regulating the adsorption strength for reaction intermediates. The *OH ligand modification was identified to be an effective strategy for improving the ORR catalytic activity. This work provides a significant insight into the fundamental understanding of the ORR mechanism and greatly facilitates the computational screening and design of low-cost and high-efficiency graphene-based dual-atom ORR catalysts.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the Natural Science Foundation of Inner Mongolia Autonomous Region (Grant No. 2023QN01010), the Research Program of Science and Technology at Universities of Inner Mongolia Autonomous Region (Grant No. NJZZ23019, NJZZ23025), and the Fundamental Research Funds for the Inner Mongolia Normal University (Grant No. 2023JBYJ016).References
- Z. P. Cano, D. Banham, S. Y. Ye, A. Hintennach, J. Lu, M. Fowler and Z. W. Chen, Nat. Energy, 2018, 3, 279–289 Search PubMed.
- H. F. Wang and Q. Xu, Matter, 2019, 1, 565–595 CAS.
- X. L. Tian, X. F. Lu, B. Y. Xia and X. W. Lou, Joule, 2020, 9, 45–68 Search PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 CAS.
- L. Huang, S. Zaman, X. L. Tian, Z. T. Wang, W. S. Fang and B. Y. Xia, Acc. Chem. Res., 2021, 54, 311–322 CAS.
- L. Li, R. Huang, X. R. Cao and Y. H. Wen, J. Mater. Chem. A, 2020, 8, 19319–19327 CAS.
- M. B. Gawande, P. Fornasiero and R. Zbořil, ACS Catal., 2020, 10, 2231–2259 CAS.
- C. W. Ye and L. Xu, J. Mater. Chem. A, 2021, 9, 22218–22247 RSC.
- H. Y. Zhuo, X. Zhang, J. X. Liang, Q. Yu, H. Xiao and J. Li, Chem. Rev., 2020, 120, 12315–12341 CrossRef CAS PubMed.
- W. Y. Zhang, Y. G. Chao, W. S. Zhang, J. H. Zhou, F. Lv, K. Wang, F. X. Lin, H. Luo, J. Li, M. P. Tong, E. Wang and S. J. Guo, Adv. Mater., 2021, 33, 2102576 CrossRef CAS PubMed.
- R. Z. Li and D. S. Wang, Adv. Energy Mater., 2022, 12, 2103564 CrossRef CAS.
- H. S. Shang and D. Liu, Nano Res., 2023, 16, 6477–6506 CrossRef CAS.
- D. C. Zhong, Y. N. Gong, C. Zhang and T. B. Lu, Chem. Soc. Rev., 2023, 52, 3170–3214 RSC.
- J. Wang, Z. Q. Huang, W. Liu, C. R. Chang, H. L. Tang, Z. J. Li, W. X. Chen, C. J. Jia, T. Yao, S. Q. Wei, Y. E. Wu and Y. D. Lie, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed.
- Z. Y. Lu, B. Wang, Y. F. Hu, W. Liu, Y. F. Zhao, R. O. Yang, Z. P. Li, J. Luo, B. Chi, Z. Jiang, M. S. Li, S. C. Mu, S. J. Liao, J. J. Zhang and X. L. Sun, Angew. Chem., Int. Ed., 2019, 58, 2622–2626 CrossRef CAS PubMed.
- M. L. Xiao, J. B. Zhu, S. Li, G. R. Li, W. W. Liu, Y. P. Deng, Z. Y. Bai, L. Ma, M. Feng, T. P. Wu, D. Su, J. Lu, A. P. Yu and Z. W. Chen, ACS Catal., 2021, 11, 8837–8846 CrossRef CAS.
- Y. Wang, J. Wu, S. Tang, J. Yang, C. Ye, J. Chen, Y. Lei and D. Wang, Angew. Chem., Int. Ed., 2023, 62, e202219191 CrossRef CAS PubMed.
- Y. X. Zhang, S. Zhang, H. Huang, X. Liu, B. Li, Y. Lee, X. Wang, Y. Bai, M. Sun, Y. Wu, S. Gong, X. Liu, Z. Zhuang, T. Tan and Z. Niu, J. Am. Chem. Soc., 2023, 145, 4819–4827 CrossRef CAS PubMed.
- Y. L. Sun, J. Wang, Q. Liu, M. R. Xia, Y. F. Tang, F. M. Gao, Y. L. Hou, J. Tse and Y. F. Zhao, J. Mater. Chem. A, 2019, 7, 27175–27185 RSC.
- R. M. Hu, Y. C. Li, Q. W. Zeng and J. X. Shang, Appl. Surf. Sci., 2020, 525, 146588 CrossRef CAS.
- M. A. Hunter, J. M. T. A. Fischer, Q. H. Yuan, M. Hankel and D. J. Searles, ACS Catal., 2019, 9, 7660–7667 CrossRef CAS.
- F. T. Wang, W. B. Xie, L. J. Yang, D. Q. Xie and S. Lin, J. Catal., 2021, 396, 215–223 CrossRef CAS.
- L. Li, Y. M. Li, T. Zhao, R. Huang, X. R. Cao, T. E. Fan and Y. H. Wen, J. Electrochem. Soc., 2022, 169, 026524 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- V. Tripkovic, E. Skúlason, S. Siahrostami, J. K. Norskov and J. Rossmeisl, Electrochim. Acta, 2010, 55, 7975–7981 CrossRef CAS.
- Z. Y. Duan and G. F. Wang, J. Phys. Chem. C, 2013, 117, 6284–6292 CrossRef CAS.
- Z. Liang, M. M. Luo, M. W. Chen, C. Liu, S. G. Peera, X. P. Qi, J. Liu, U. P. Kumar and T. X. Liang, J. Colloid Interface Sci., 2020, 568, 54–62 CrossRef CAS PubMed.
- L. Li, Y. M. Li, R. Huang, X. R. Cao and Y. H. Wen, ChemCatChem, 2021, 13, 4645–4651 CrossRef CAS.
- K. W. Huang, W. H. Yang, L. Li, Y. M. Li, R. Huang and Y. H. Wen, Phys. Chem. Chem. Phys., 2023, 25, 18266–18274 RSC.
- Y. N. Meng, C. Yin, K. Li, H. Tang, Y. Wang and Z. J. Wu, ACS Sustainable Chem. Eng., 2019, 7, 17273–17281 CrossRef CAS.
- Y. D. Zhou, W. Yang, W. Utetiwabo, Y. M. Lian, X. Yin, L. Zhou, P. W. Yu, R. J. Chen and S. R. Sun, J. Phys. Chem. Lett., 2020, 11, 1404–1410 CrossRef CAS PubMed.
- X. F. Zhu, D. T. Zhang, C. J. Chen, Q. R. Zhang, R. S. Liu, Z. H. Xia, L. M. Dai, R. Amal and X. Y. Lu, Nano Energy, 2020, 71, 104597 CrossRef CAS.
- J. Y. Xu, A. Elangovan, J. Li and B. Liu, J. Phys. Chem. C, 2021, 125, 2334–2344 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04831d |
| This journal is © the Owner Societies 2024 |