Reactions of N2O and CO on neutral Rh10On clusters: a density functional study†
Received
11th October 2023
, Accepted 9th December 2023
First published on 19th December 2023
Abstract
Density functional theory calculations were performed to identify product, reactant and intermediate dissociative/associative structures for the oxygen abstraction and addition reactions: Rh10On + CO → Rh10On−1 + CO2, n = 1–5 and Rh10On + N2O → Rh10On+1 + N2, n = 0–4 reactions. In the case of the oxygen abstraction reactions, the energetics of the reaction path were very similar in energy regardless of the number of oxygen atoms on the Rh10On cluster, whereas for the addition of oxygen to the Rh10On cluster, the reaction was found to become significantly less exothermic with each successive addition of oxygen.
1 Introduction
Transition metal clusters have been of interest for several decades as both models of surfaces1 and entities in their own right, where their remarkable size-2 and structure-dependent3–5 properties make them highly attractive as potential heterogeneous catalysts.6–8 Late transition metal clusters, especially those of rhodium have maintained particular interest over that time with a multitude of studies employing a variety of spectroscopic and computational methods to probe their structure and behaviour.
Many of these studies have been motivated by the use of rhodium in automotive three-way catalytic converters9 and the reactions of N2O,10–12 NO,13–22 CO23,24 and CO225 have been extensively investigated on both pure and doped26–28 rhodium clusters. Given that spectroscopic methods such as infrared multiple photon dissociation spectroscopy (IRMPD)29 use mass spectroscopy to identify the species of interest, they generally provide information only about the products of reactions and reactivity studies therefore often include computational investigation,30,31 typically using density functional theory (DFT), but sometimes including higher level methods such as CCSD(T).32
Two particular reactions that are of interest for automotive catalytic converters are the reduction of NOx to N2 and the oxidation of CO to CO2. Both of these reactions have specifically been investigated on rhodium11,26 and platinum33,34 clusters. Aviles et al. undertook calculations to elucidate the N2O reduction mechanism on small Rhn clusters (n = 2–4), and determined that the N2–O bond was readily broken by the Rhn cluster, but the N2 bond was not.12 Yamamoto et al. used both IRMPD and DFT methods to determine reaction paths for the reduction of N2O by Pt6 and Pt7 clusters.,35 while Hernández and coworkers used DFT to elucidate the reaction of N2O on Pt8.36
Yet despite these successes, computational investigation remains challenging, especially on large and/or late transition metal clusters: Even employing frozen core/effective core potential approaches, clusters still possess a large number of electrons. A wide range of spin multiplicities need to be considered. And many clusters have multiple, very different geometries that all occur within a small energetic range,5,37,38 that is thermally accessible and/or within the error of computational methods. For some clusters, the manifold of low-energy geometries may be so dense and the ease of interconversion so facile, that the clear identification of a ground state is challenging. Rh10 has been identified as one such fluxional molecule.39 Such a dense manifold of structures, energies and spin multiplicities is additionally challenging when employing reaction path following methods such as Quasi-Synchronous transit and intrinsic reaction coordinate methods, as reaction paths may follow a comparatively broad path, where spin and cluster geometry may differ throughout a reaction.
In 2012, Yamada et al. undertook a mass-spectroscopic study on neutral rhodium clusters, Rhn (n = 10–28).40 By injecting both N2O and CO into their cluster source, they were able to demonstrate a full catalytic cycle and observed that the distribution of oxide products RhnOm was determined by an equilibrium between the oxygen being transferred to the RhnOmm = 0–4 cluster from N2O and oxygen being abstracted from the RhnOmm = 1–5, and that the rate constants for the oxygen abstraction reaction were 2–3 orders of magnitude higher on clusters where m ≥ 4 than those clusters with three or less oxygen atoms. They concluded that oxidized rhodium clusters were more effective as catalysts compared to bare clusters. Yamada's study used experimentally derived rate constants and did not include any calculated structures or reaction paths. Indeed the size of the rhodium clusters employed by Yamada et al., Rhn (n = 10–28) makes DFT challenging.
In this work, we revisit the catalytic cycle presented by Yamada et al. for the smallest rhodium cluster size, Rh10. As reaction path following on such flat potential energy surfaces (PES) is difficult, we employ a fully stochastic search procedure followed by DFT calculations to identify the reactants, products and the bond forming/breaking intermediate for both the oxygen transfer and oxygen abstraction reactions on the Rh10 cluster.
2 Computational method
The geometry of Rh10 was determined using the Kick3 stochastic structure generator supplied with 10 Rh atoms.41,42 Following optimization, the lowest energy Rh10 geometry was then employed as a fragment in further Kick3 runs with n oxygen atoms in order to determine the lowest energy Rh10On structures, for n = 1–5. A key point on each Rh10On → Rh10On±1 reaction path, is the species where the N2⋯O bond is being broken, or where the O⋯CO bond is being formed. To identify these intermediate species directly, N2O and OCO fragments with elongated NO and CO bonds respectively, were used as input to Kick3 to identify the Rh10On CO and Rh10On N2 intermediate species. 1000 stochastic geometries were generated for each stoichiometry.
The stochastically generated structures were optimized in a two-stage procedure: Firstly using the semi-empirical GFN1-xTB method,43 maintaining the geometry of all fragments employed in the stochastic structure generation, before a full optimization using BP86/DZP and then re-optimization using the TZ2P basis set. Up to the 4p electrons of rhodium were treated using a frozen core. Structures were initially optimized in the octet multiplicity before re-optimization with each multiplicity from doublet to 20-tet. 18- and 20-tet structures were found to lie very high in energy and were consequently not considered in analysis. All calculations were undertaken using AMS2022.1 using scalar relativivity and applying an electronic temperature of 373.15 K.44
To confirm that the BP8645,46 functional is sufficient, we also tested the TPSS47 functional with the D3(BJ)48,49 dispersion correction. The choice of functional made no significant difference to the relative ordering of different multiplicity Rh10On structures, as shown by difference-of-difference values in the ESI.† GGA functionals are expected to only give qualitative information about barrier heights, due to their self-interaction error50 and this is evident, with BP86 intermediate relative energies for oxygen abstraction reactions being consistently ≈0.4 eV higher in energy than the TPSS-calculated values and BP86 product energies being consistently ≈0.5 eV lower than the TPSS-calculated values. For the oxygen addition reaction paths, the TPSS relative energies for intermediate and product species are lowered w.r.t. the BP86 paths by ≈0.6 and 0.3 eV respectively.
After optimization of Rh10On, Rh10On CO and Rh10On N2 structures, Rh10On → Rh10On±1 reaction paths were determined by geometric RMSD. For each reaction, the lowest energy Rh10On reactant was chosen, and the nearest Rh10O±1 geometric match was determined (Rh10On+1 for Rh10On + N2O → Rh10On+1+ N2 and Rh10On−1 for Rh10On + CO → Rh10On−1 + CO2). The intermediate species was determined in the same manner, by determining the RMSD with the reactant and then confirming RMSD with the product. After the addition of the first two oxygen atoms to Rh10, multiple oxygen abstraction reaction paths are possible, corresponding to abstraction of different oxygen atoms, the path presented in this work is the one containing the most favourable (lowest energy) intermediate geometry.
It is not necessarily the case that the lowest energy Rh10On will lead to the lowest energy Rh10On±1 species, and because of the circular nature of the catalytic reaction, each Rh10On species appears as both a product and a reactant. Therefore, a second reaction path, beginning with the lowest energy reactant was also determined following the same geometric RMSD procedure. We denote the two reaction paths as lowest energy reactant (LER) and lowest energy product (LEP).
3 Results and discussion
Rh10
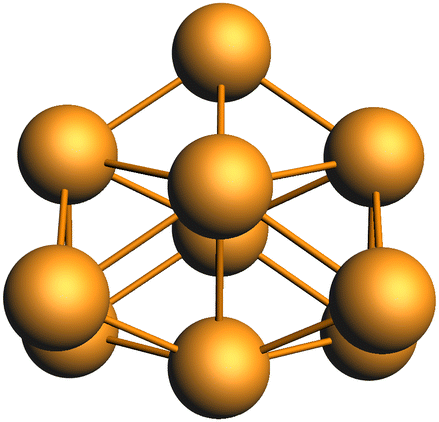
Several computational38,51 and experimental studies have investigated the structure of the Rh10 cluster in neutral, cationic and anionic charge states, identifying a dense manifold of low-energy structures. Knickelbein et al. measured the static electric dipole polarizabilities of Rhnn = 5–28 clusters and suggested that Rh10 is a fluxional molecule.39 The lowest energy Rh10 octet species identified in this work is a structure with C2v symmetry and a binding energy of −1.7686006 a.u. and is shown in Fig. 1. Re-optimization of each identified Rh10 isomer at other multiplicities (doublet to 20-tet) revealed the lowest energy isomer overall, is the same structure, but with 16-tet multiplicity and a binding energy of −1.7909319 a.u. This structure corresponds to the sc3 structure identified for Rh10+ by Harding et al.51
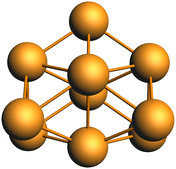 |
| | Fig. 1 Lowest energy geometry of Rh10. | |
Rh10On + CO → Rh10On−1 + CO2 reactions
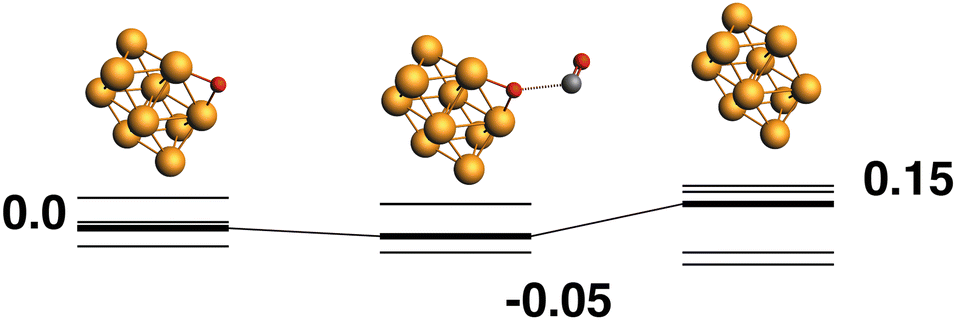
Rh10O + CO → Rh10 + CO2.
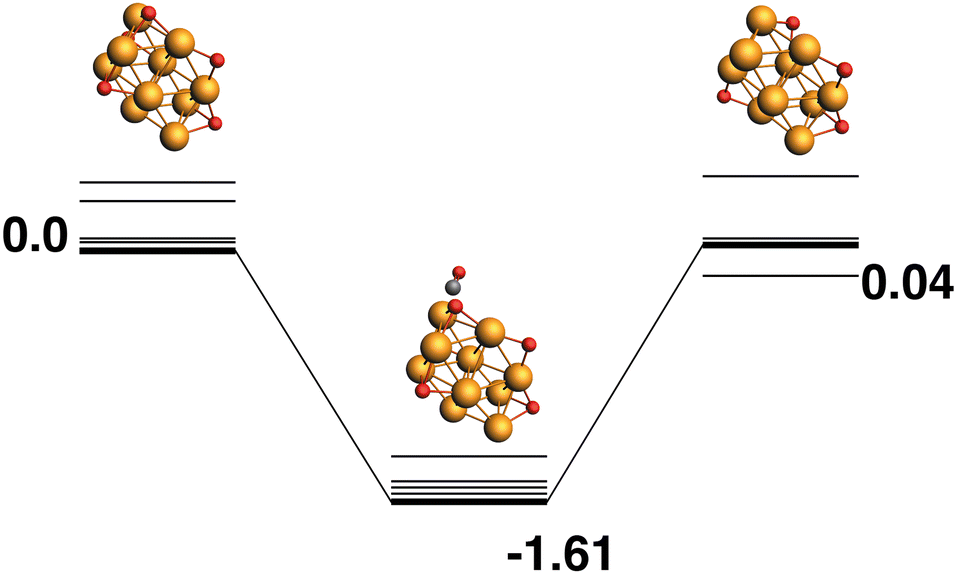
Only the predetermined minimum Rh10 structure is considered in this work, therefore there is only one Rh10O + CO → Rh10 + CO2 reaction path, defined by the lowest energy Rh10O species, which has a μ2-bound oxygen. The reaction path from this species is shown in Fig. 2. The Rh10O⋯C distance in the intermediate state is 2.72 Å. Overall, the reaction path is very flat with the separated products (Rh10 + CO2) lying only 0.15 eV above the reactants and the intermediate Rh10O⋯CO structure being effectively isoenergetic with the Rh10O + CO reactants. For the reactant and intermediate structures, all multiplicities (doublet to 16-tet) were found within a range of 0.35 eV. Different multiplicities of the Rh10 product span an energetic range of 0.71 eV.
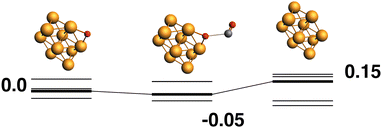 |
| | Fig. 2 Rh10O + CO → Rh10 + CO2 reaction path for the lowest energy Rh10O species. Relative energies are shown for the octet surface in eV. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
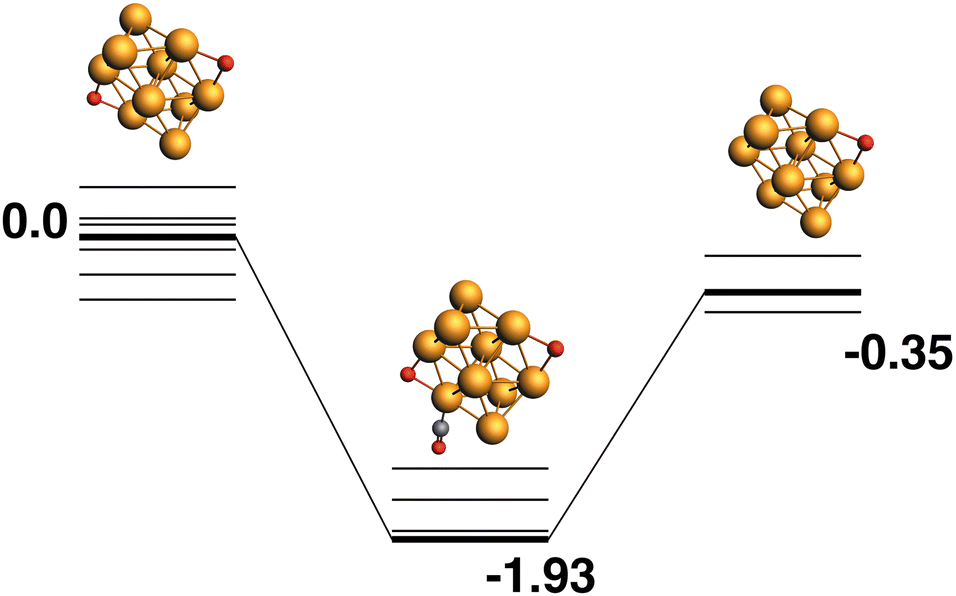
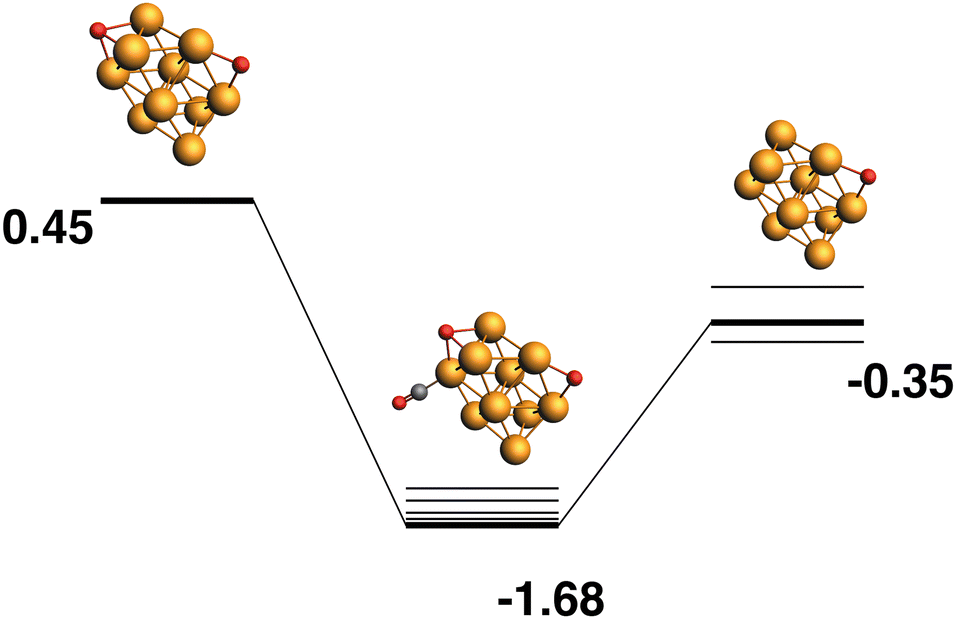
Rh10O2 + CO → Rh10O + CO2.
Both the LER (Fig. 3) and LEP (Fig. 4) reaction paths arrive at the same μ2 Rh10O product – i.e. the lowest energy Rh10O2 can follow a reaction path that results in the lowest energy Rh10O. The lowest energy Rh10O2 structure has two μ2-bound oxygen atoms in symmetry-equivalent positions. The intermediate CO-bound species, removing either of these oxygen atoms is −1.93 eV lower in energy than the separated reactants and has a Rh10O⋯C distance of 3.19 Å. Beginning with the lowest energy μ2 Rh10O product and searching for a reaction path towards any Rh10O2 reactant, yields a μ3μ2 reactant 0.45 eV higher in energy than the lowest energy Rh10O2 and a corresponding Rh10OO·CO intermediate. The intermediate structure removes the μ3-bound oxygen atom and is at −1.68 eV, 0.25 eV higher in energy than the intermediate in the LER pathway.
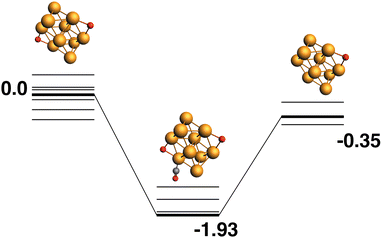 |
| | Fig. 3 Rh10O2 + CO → Rh10O + CO2 reaction path for the lowest energy Rh10O2 species. Relative energies are shown for the octet surface in eV. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
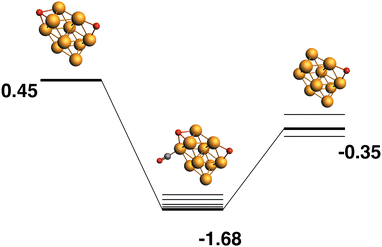 |
| | Fig. 4 Rh10O2 + CO → Rh10O + CO2 reaction path for the lowest energy product (Rh10O) species. Relative energies are shown for the octet surface in eV. Zero energy is the lowest energy Rh10O2 reactant, as shown in Fig. 3. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
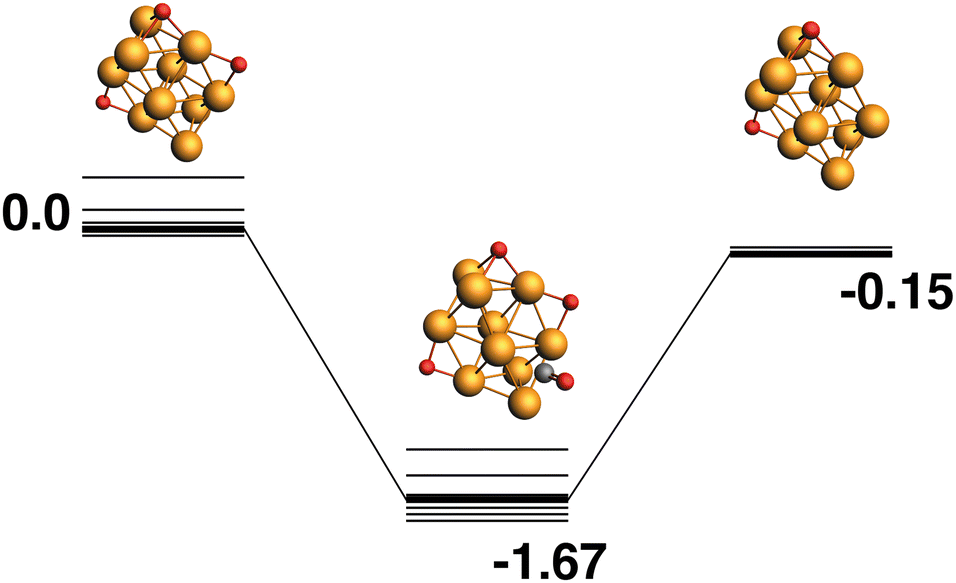
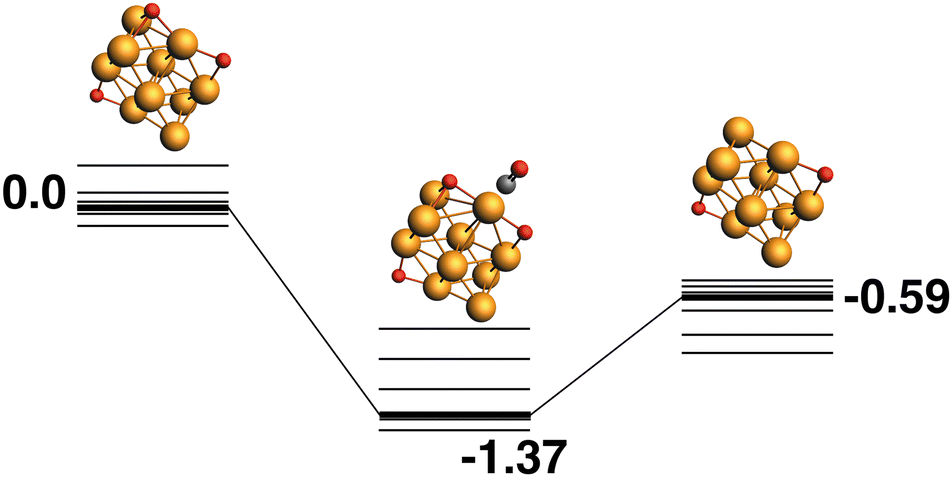
Rh10O3 + CO → Rh10O2 + CO2.
The reaction path from the lowest energy Rh10O3 structure is shown in Fig. 5. The lowest energy Rh10O3 structure has the same two μ2-bound oxygen atoms as in Rh10O2 plus a μ3-bound oxygen atom that shares a Rh atom with one of the μ2-bound oxygen atoms. In the Rh10O2O·CO intermediate structure, the incoming CO approaches the side of the cluster with both the μ3 and μ2 oxygen atoms, but abstracts the μ2-bound oxygen. The intermediate is at −1.67 eV and the Rh10O⋯C distance is 3.39 Å. The Rh10O2 product is at −0.15 eV. Removal of the μ3-bound oxygen instead of the μ2-bound oxygen yields the lowest energy Rh10O2 structure at −0.59 eV, and this pathway is shown in Fig. 6. The intermediate in this pathway is at −1.37 eV, 0.30 eV higher in energy than the LER path intermediate and the Rh10O⋯C distance is 2.91 Å.
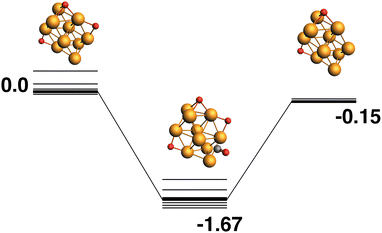 |
| | Fig. 5 Rh10O3 + CO → Rh10O2 + CO2 reaction path for the lowest energy Rh10O3 species. Relative energies are shown for the octet surface in eV. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
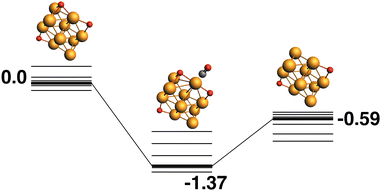 |
| | Fig. 6 Rh10O3 + CO → Rh10O2 + CO2 reaction path for the lowest energy product (Rh10O2) species. Relative energies are shown for the octet surface in eV. Zero energy is the lowest energy Rh10O3 reactant, as shown in Fig. 5. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
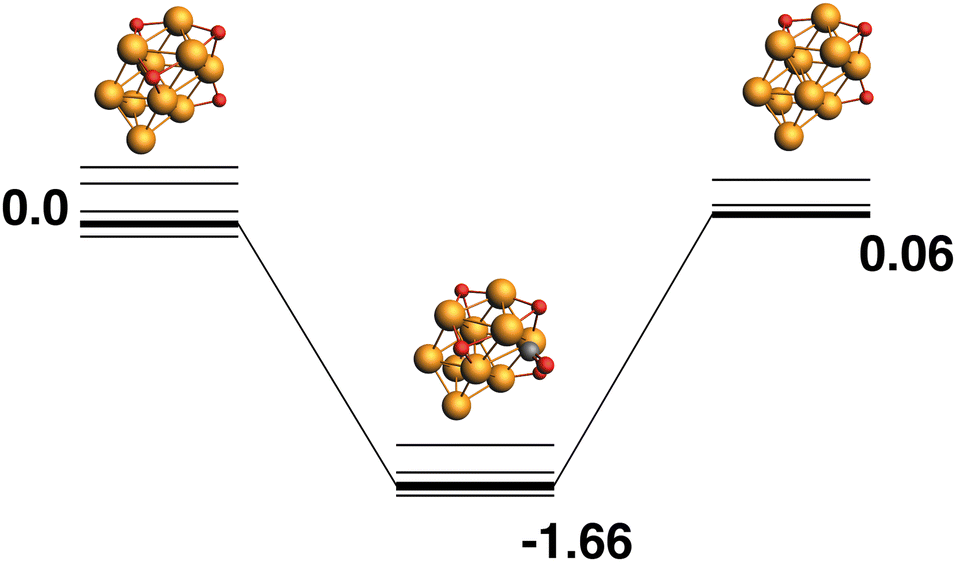
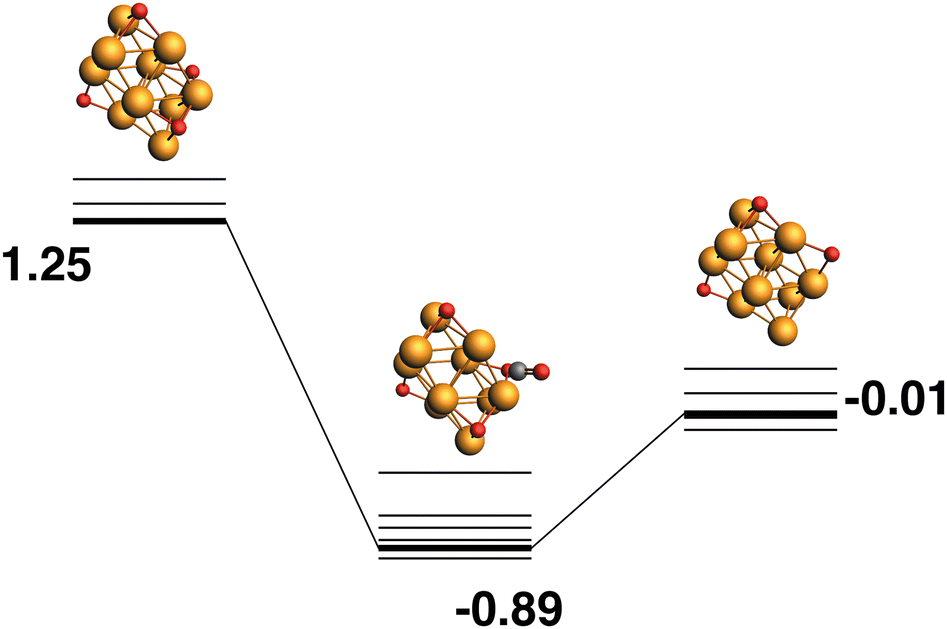
Rh10O4 + CO → Rh10O3 + CO2.
The lowest energy Rh10O4 structure has three μ3 -bound oxygen atoms and a μ2-bound oxygen. The reaction path from this species is shown in Fig. 7 and removes one of the μ3 oxygen atoms. The products (Rh10O3 + CO2) are nearly isoenergetic with the reactants. As the oxygen atoms in the Rh10O4 cluster are all bound (in part) to the same four Rh atoms, the approaching CO is likely to interact with more than one oxygen atom. In the lowest energy intermediate structure identified, the CO could possibly abstract one of two a μ3-bound oxygen atoms, with Rh10O⋯C distances of 3.08 and 3.22 Å respectively.
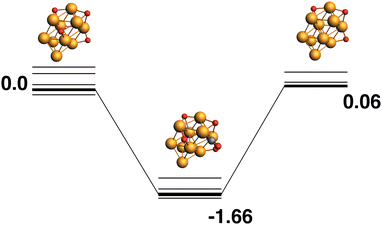 |
| | Fig. 7 Rh10O4 + CO → Rh10O3 + CO2 reaction path for the lowest energy Rh10O4 species. Relative energies are shown for the octet surface in eV. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
The lowest energy Rh10O3, as above, possesses two μ2-bound oxygen atoms and one μ3-bound oxygen, so it is not a possible product (without rearrangement) from the lowest energy Rh10O4 structure. The path ending with the lowest energy Rh10O3 structure beings with a high energy Rh10O4 structure (+1.25 eV), which has two μ3-bound oxygen atoms and two μ2-bound oxygen atoms (Fig. 8). The close Rh10O⋯C distances in the intermediate structure, are 3.16 Å for the μ3-bound oxygen that is abstracted by the CO and 3.39 Å for the μ2-bound oxygen that remains.
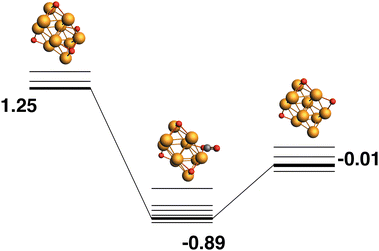 |
| | Fig. 8 Rh10O4 + CO → Rh10O3 + CO2 reaction path for the lowest energy product (Rh10O3) species. Relative energies are shown for the octet surface in eV. Zero energy is the lowest energy Rh10O4 reactant, as shown in Fig. 7. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
Rh10O5 + CO → Rh10O4 + CO2.
The lowest energy Rh10O5 structure has four μ3-bound oxygen atoms and a μ2-bound oxygen. The reaction path from this species is shown in Fig. 9 and removes one of the μ3 oxygen atoms. Similarly to the Rh10O4 to Rh10O3 LER reaction path, the products (Rh10O4 + CO2) are nearly isoenergetic with the reactants, the relative energy of the Rh10O5·CO intermediate species is also similar to that of the Rh10O4·CO intermediate (Fig. 7), at −1.61 eV. The approaching CO abstracts a μ3 -bound oxygen atom from 3.04 Å.
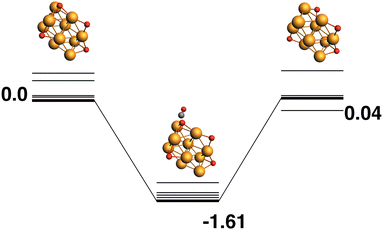 |
| | Fig. 9 Rh10O5 + CO → Rh10O4 + CO2 reaction path for the lowest energy Rh10O5 species. Relative energies are shown for the octet surface in eV. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
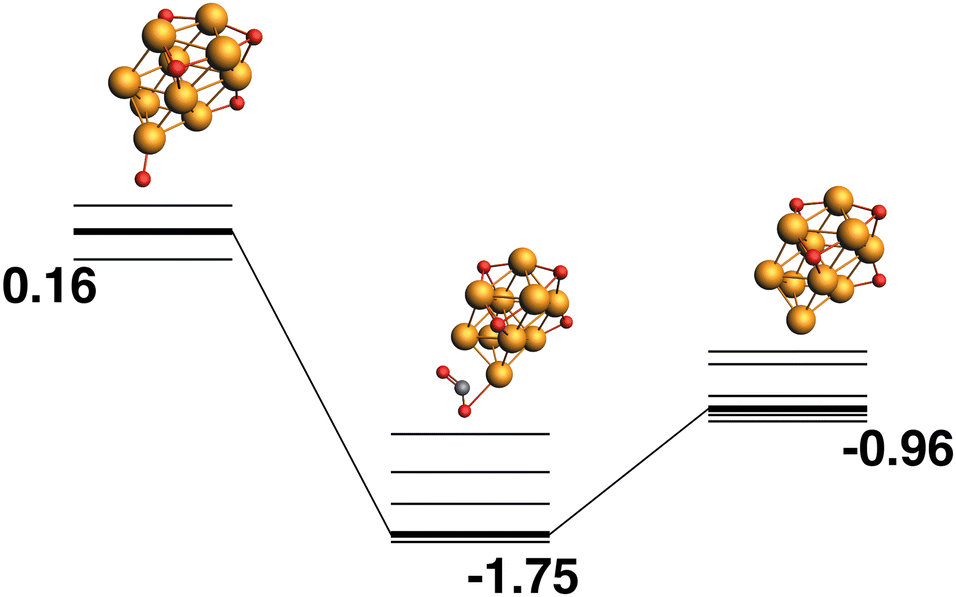
The LEP reaction path is shown in Fig. 10. The Rh10O4 product has a relative energy of −0.96 eV and the lowest energy reaction path removes a μ1-bound oxygen atom that is bound to a previously bare Rh atom.
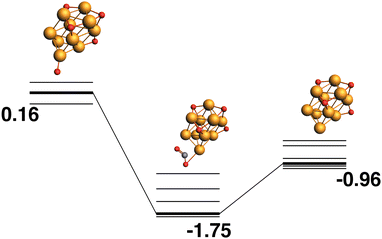 |
| | Fig. 10 Rh10O5 + CO → Rh10O4 + CO2 reaction path for the lowest energy product (Rh10O4) species. Relative energies are shown for the octet surface in eV. Zero energy is the lowest energy Rh10O5 reactant, as shown in Fig. 9. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
Rh10On + N2O → Rh10On+1 + N2 reactions
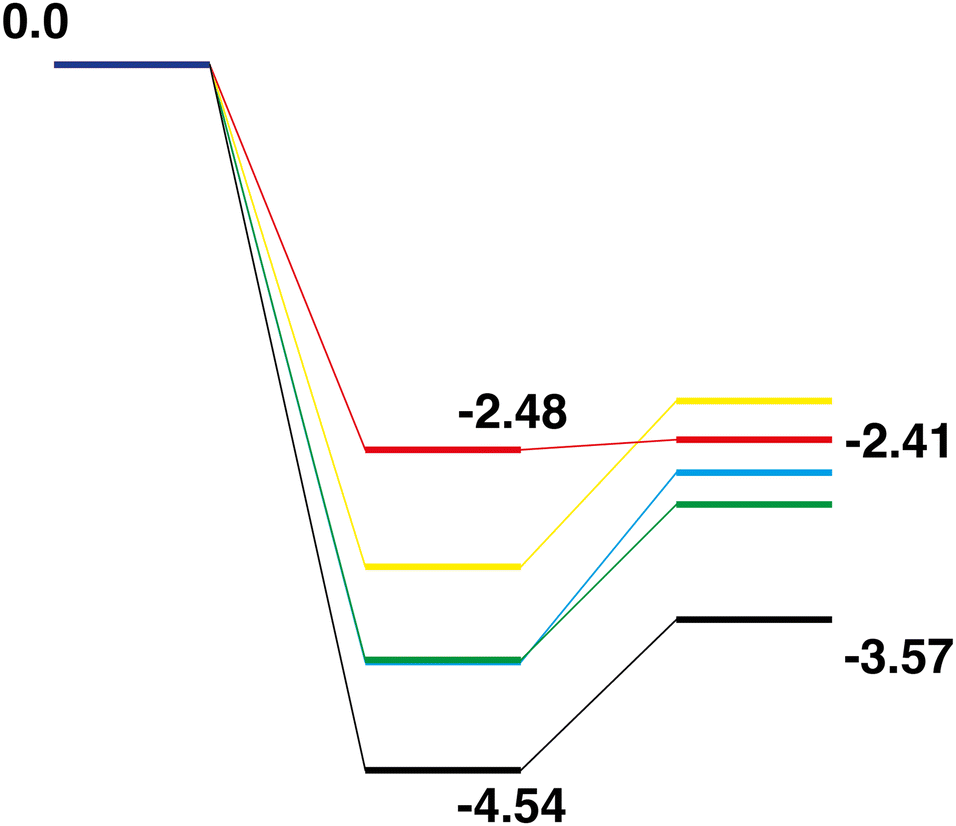
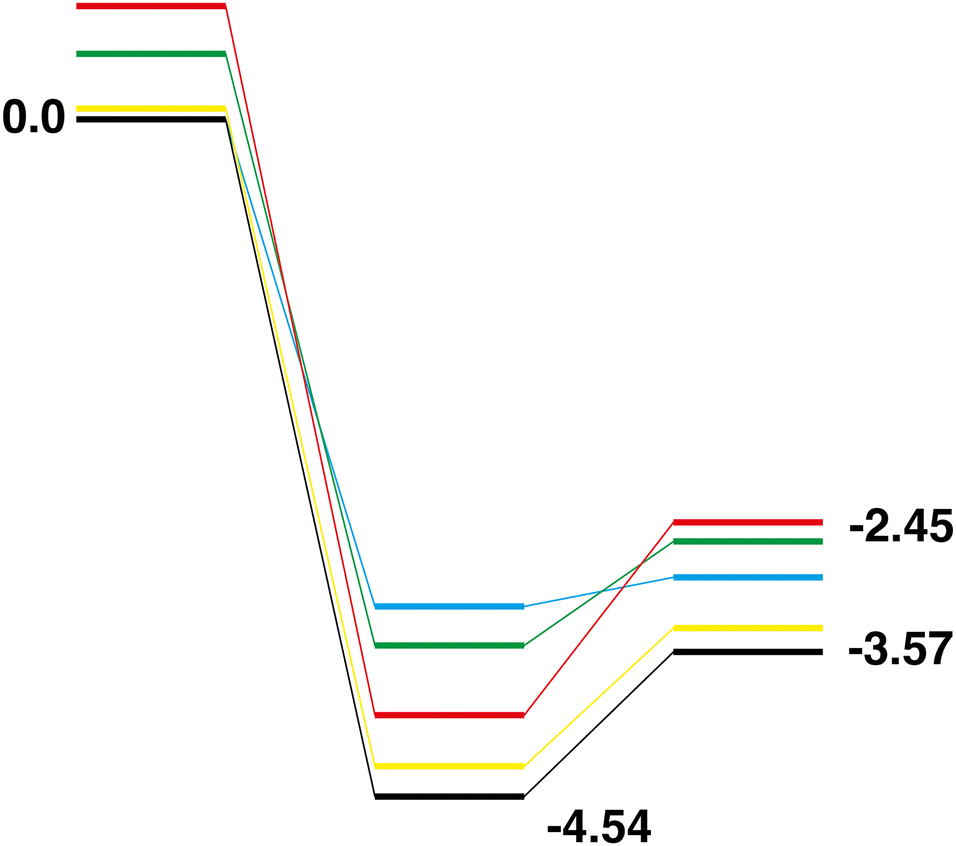
Rh10 + N2O → Rh10O + N2.
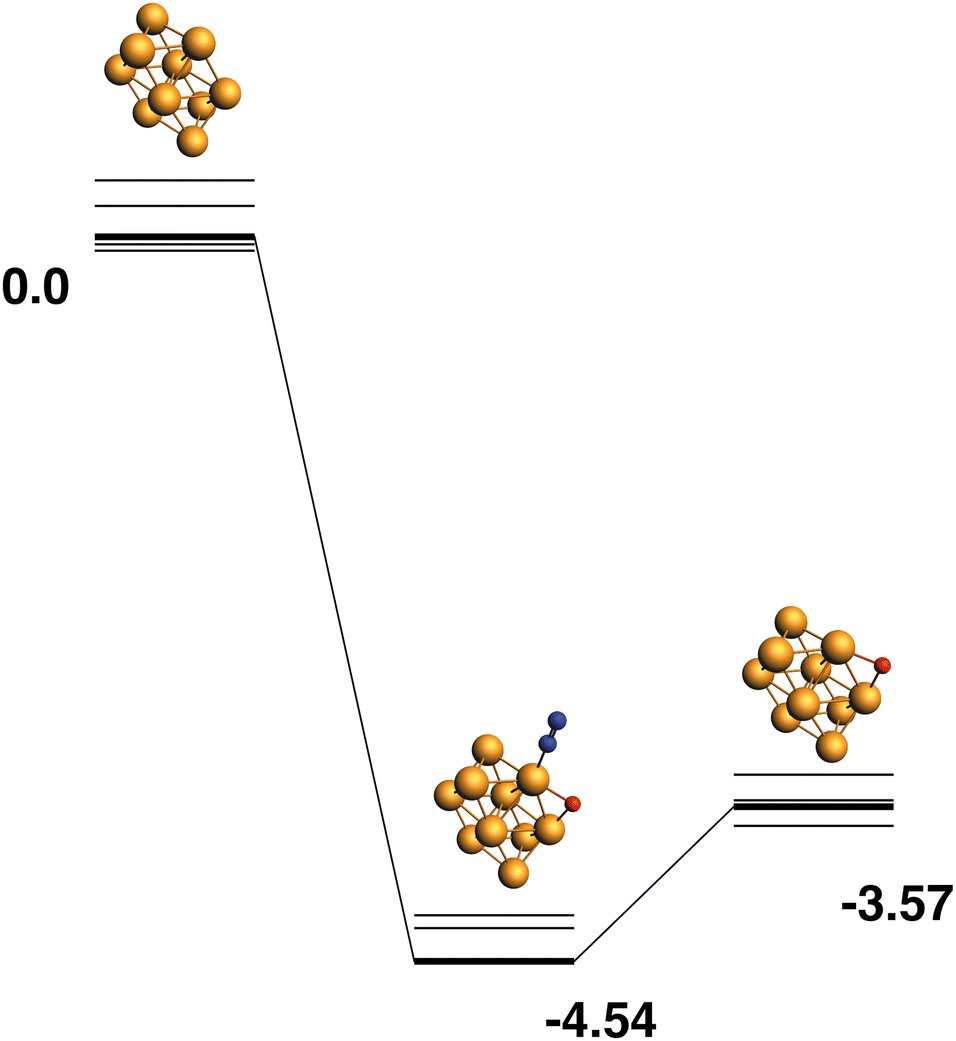
Only the predetermined minimum Rh10 structure is considered in this work, therefore there is only one Rh10 + N2 → Rh10O + N2 reaction path, defined by the lowest energy Rh10O species, which has a μ2-bound oxygen. The reaction path from this species is shown in Fig. 11. The energetic profile of the reaction path is quite different from the reduction reactions, with the Rh10O.N2 intermediate structure being −4.54 eV below zero energy and the Rh10O + N2 products being found at −3.57 eV. The Rh10O·N2 separation is 3.07 Å.
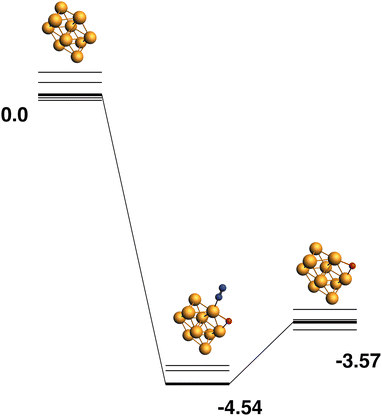 |
| | Fig. 11 Rh10 + N2O → Rh10O + N2 reaction path for the lowest energy product (Rh10O) species. Relative energies are shown for the octet surface in eV. Zero energy is the defined Rh10 reactant, as shown in Fig. 1. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
Rh10O + N2O → Rh10O2 + N2.
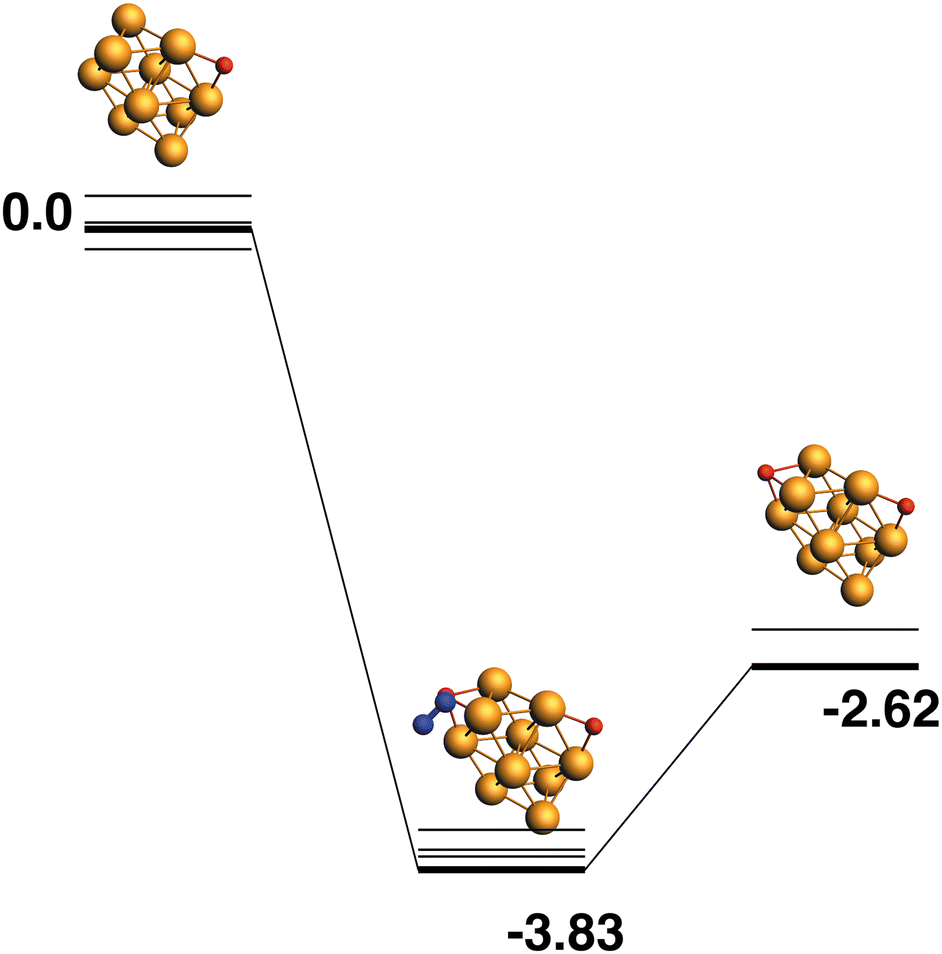
The lowest energy Rh10O structure, as previously seen, has a μ2-bound oxygen. The reaction path from this species is shown in Fig. 12 and deposits a μ3 oxygen atom such that no Rh atoms bind both oxygen atoms. The intermediate structure has a relative energy of −3.83 eV and the Rh10O·N2 distance is 3.96 Å. The products are found at −2.62 eV.
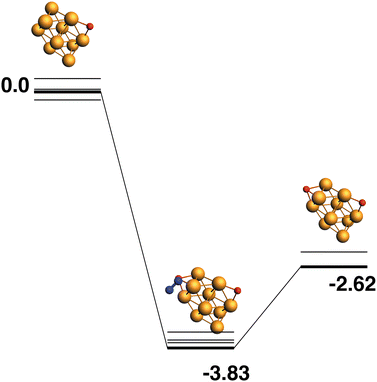 |
| | Fig. 12 Rh10O + N2O → Rh10O2 + N2 reaction path for the lowest energy Rh10O species. Relative energies are shown for the octet surface in eV. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
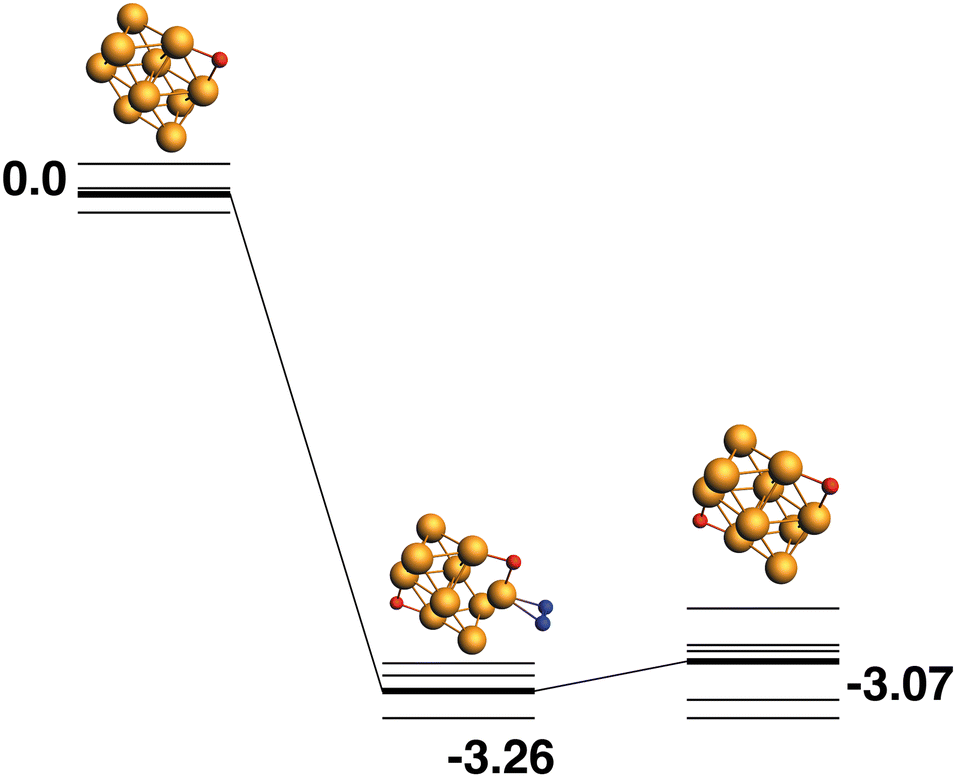
The LEP reaction path is shown in Fig. 13. The Rh10O2 product has a relative energy of −3.07 eV and deposits the second of the two symmetry-equivalent μ2-bound oxygen atoms.
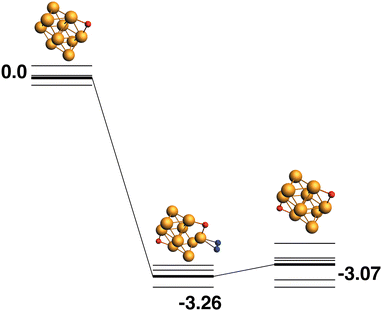 |
| | Fig. 13 Rh10O + N2O → Rh10O2 + N2 reaction path for the lowest energy product (Rh10O2) species. Relative energies are shown for the octet surface in eV. Zero energy is the lowest energy Rh10O reactant, as shown in Fig. 12. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
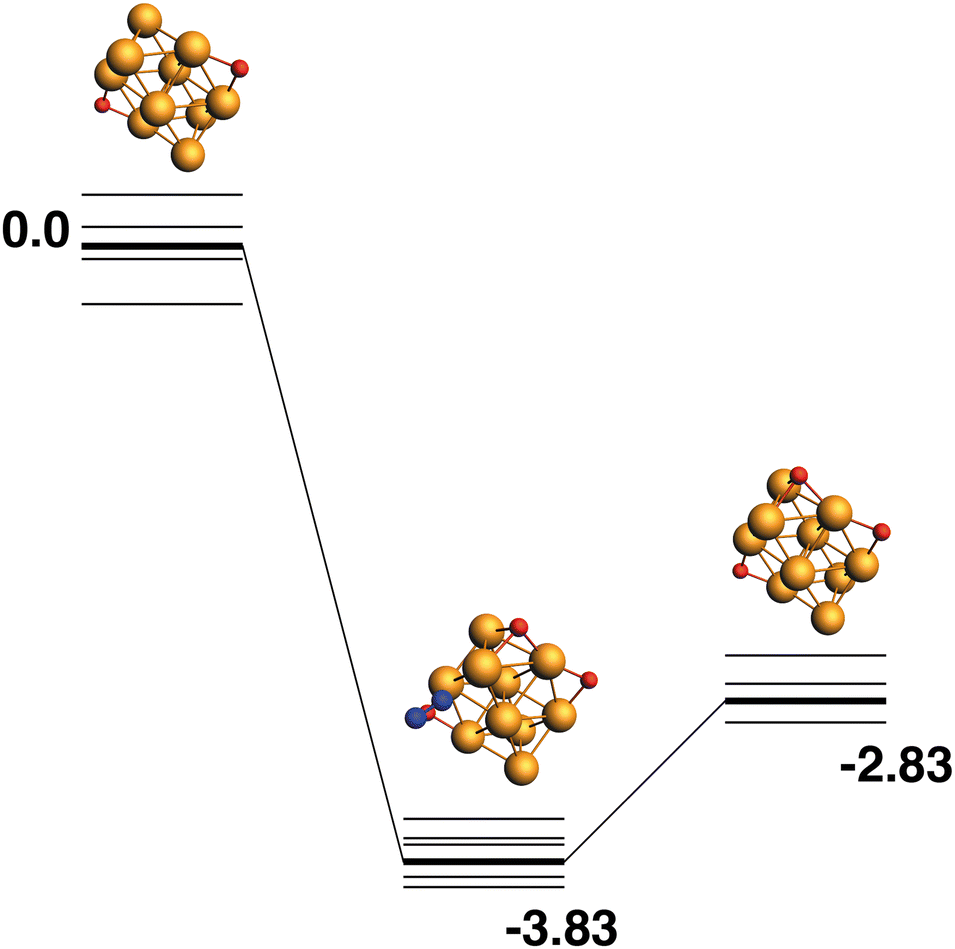
Rh10O2 + N2O → Rh10O3 + N2.
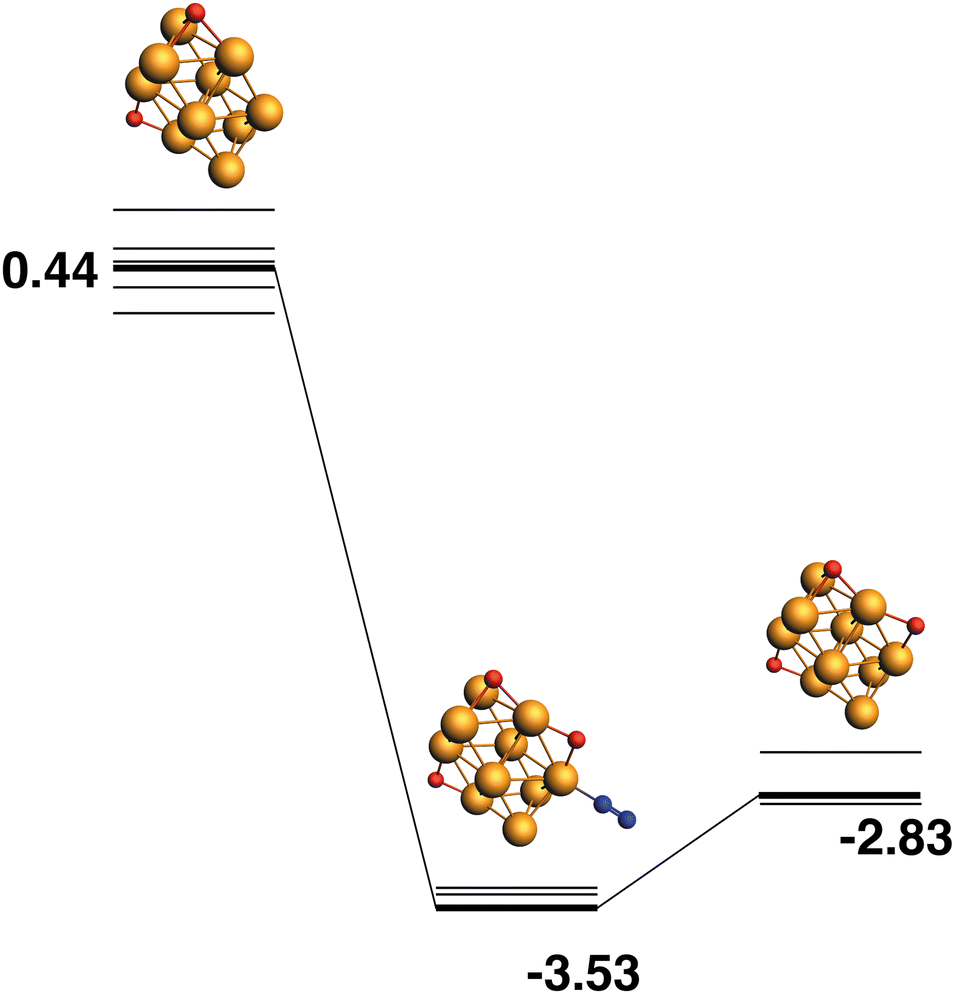
Both the LER (14) and LEP (15) reaction paths form the lowest energy Rh10O3 product, which has two μ2 and one μ3-bound oxygen atom. This product (Rh10O2 + N2) has an energy of −2.83 eV relative to the lowest energy Rh10O2 + N2O. The intermediate structure connecting these two lowest energy Rh10O2 and Rh10O3 structures, must therefore deposit the μ3 -bound oxygen atom. This intermediate is found at −3.83 eV and the Rh10O·N2 distance is 3.96 Å. If instead, of depositing the μ3 -bound oxygen atom, the intermediate is chosen to deposit one of the two μ2-oxygen atoms, the reaction path in Fig. 15 is observed. The Rh10O2 reactant and Rh10O3·N2 intermediate structures are found at +0.44 and −3.83 eV respectively - i.e. the intermediate structure is 0.3 eV higher in energy than the LER intermediate and has a Rh10O·N2 distance of 2.99 Å.
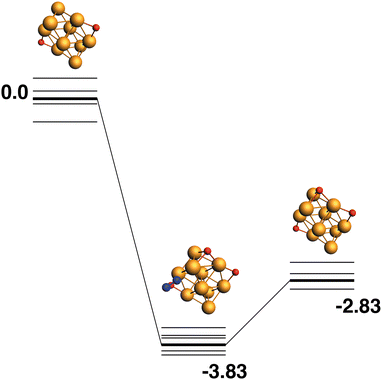 |
| | Fig. 14 Rh10O2 + N2O → Rh10O3 + N2 reaction path for the lowest energy Rh10O2 species. Relative energies are shown for the octet surface in eV. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
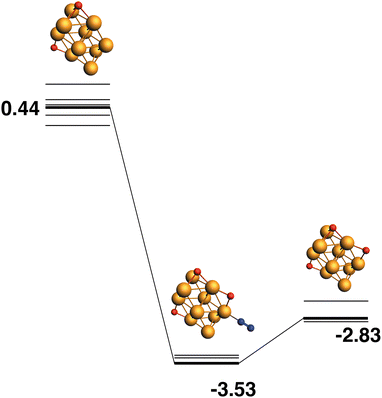 |
| | Fig. 15 Rh10O2 + N2O → Rh10O3 + N2 reaction path for the lowest energy product (Rh10O3) species. Relative energies are shown for the octet surface in eV. Zero energy is the lowest energy Rh10O2 reactant, as shown in Fig. 14. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
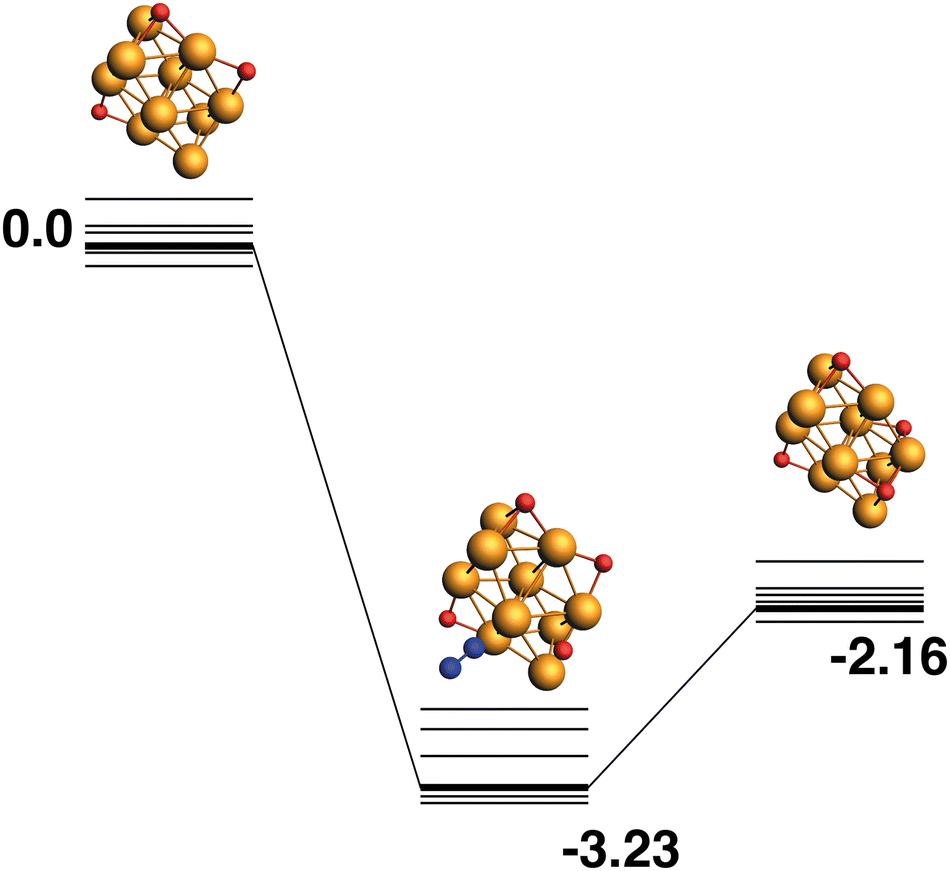
Rh10O3 + N2O → Rh10O4 + N2.
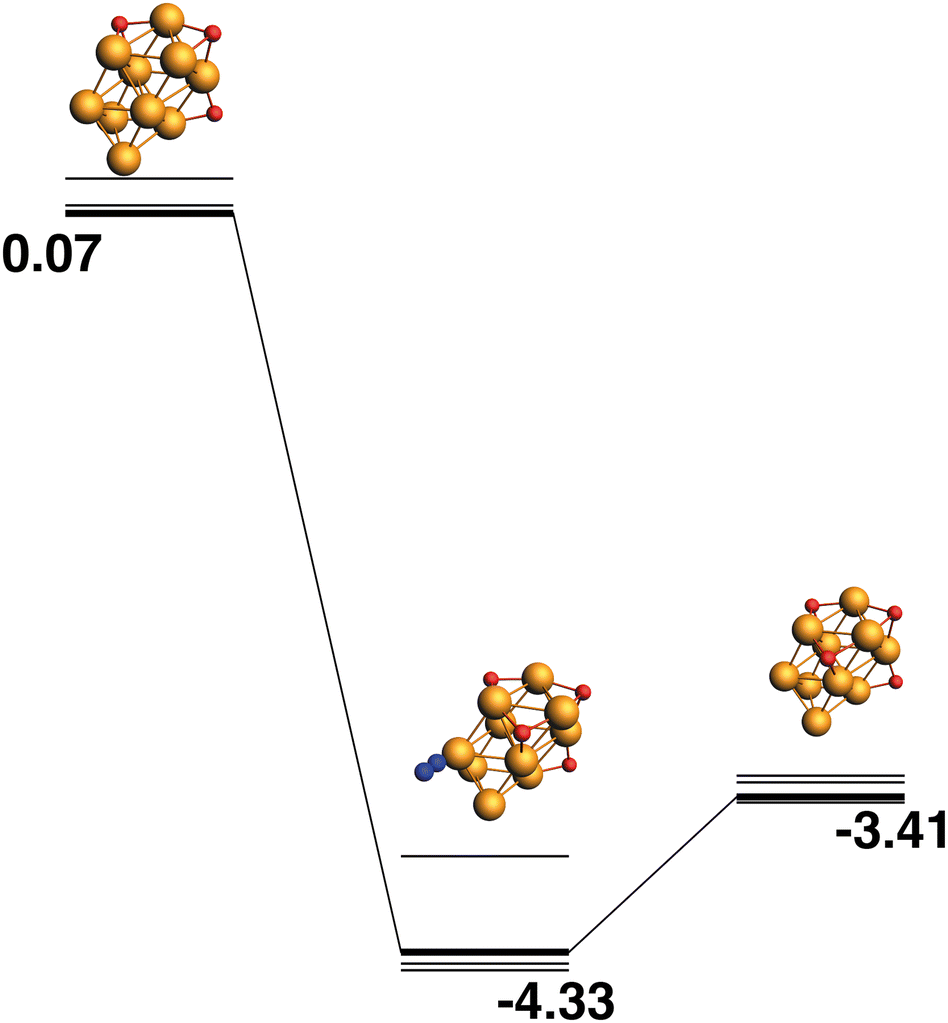
The LER reaction path for the Rh10O3 + N2O → Rh10O4 + N2 reaction is shown in Fig. 16. The Rh10O4 product at −2.16 eV has two μ2 and two μ3-bound oxygen atoms and the corresponding intermediate structure at −3.23 eV has a Rh10O⋅N2 distance of 3.20 Å, depositing the second μ3 oxygen atom. The path leading to the lowest energy Rh10O4 product is shown in Fig. 17. The Rh10O4 product has three μ3 and one μ2-bound oxygen atom, and the lowest energy intermediate structure deposits the third μ3-bound oxygen atom. The geometrically matching Rh10O3 reactant with two μ3 and one μ2-bound oxygen, is very close in energy to the lowest energy Rh10O3 structure (two × μ3 and 1 × μ2), with a relative energy of only 0.07 eV. The Rh10O·N2 distance in the intermediate structure is 3.56 Å.
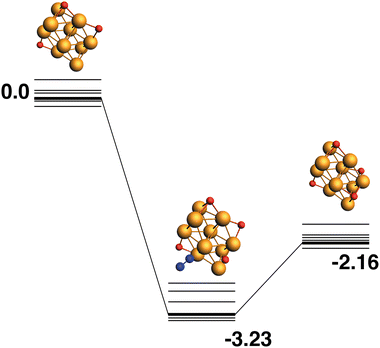 |
| | Fig. 16 Rh10O3 + N2O → Rh10O4 + N2 reaction path for the lowest energy Rh10O3 species. Relative energies are shown for the octet surface in eV. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
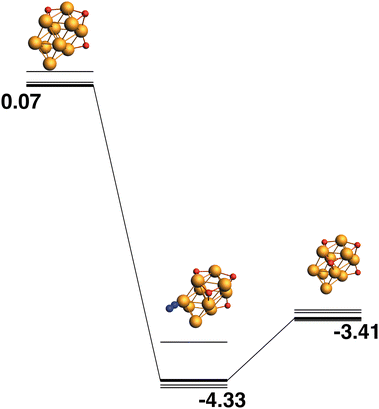 |
| | Fig. 17 Rh10O3 + N2O → Rh10O4 + N2 reaction path for the lowest energy product (Rh10O4) species. Relative energies are shown for the octet surface in eV. Zero energy is the lowest energy Rh10O3 reactant, as shown in Fig. 16. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
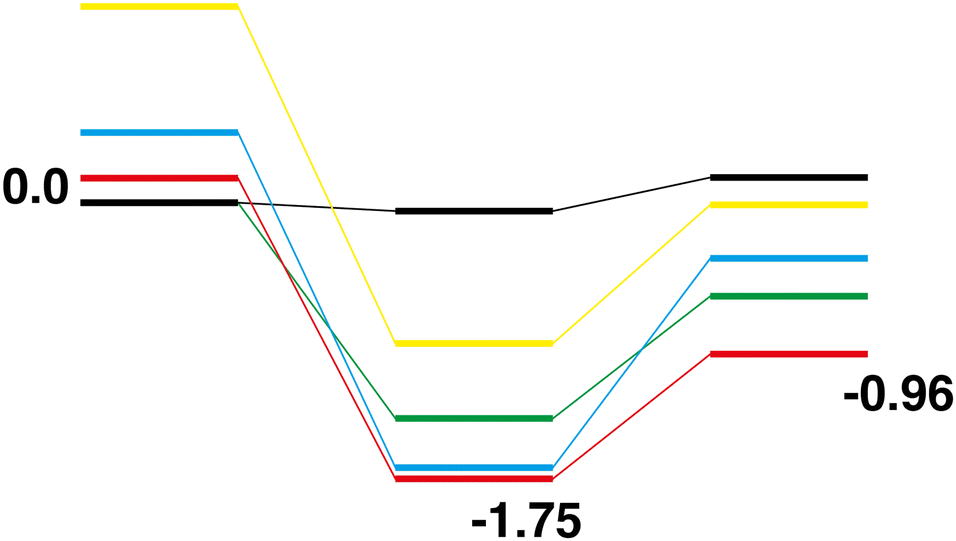
Rh10O4 + N2O → Rh10O5 + N2.
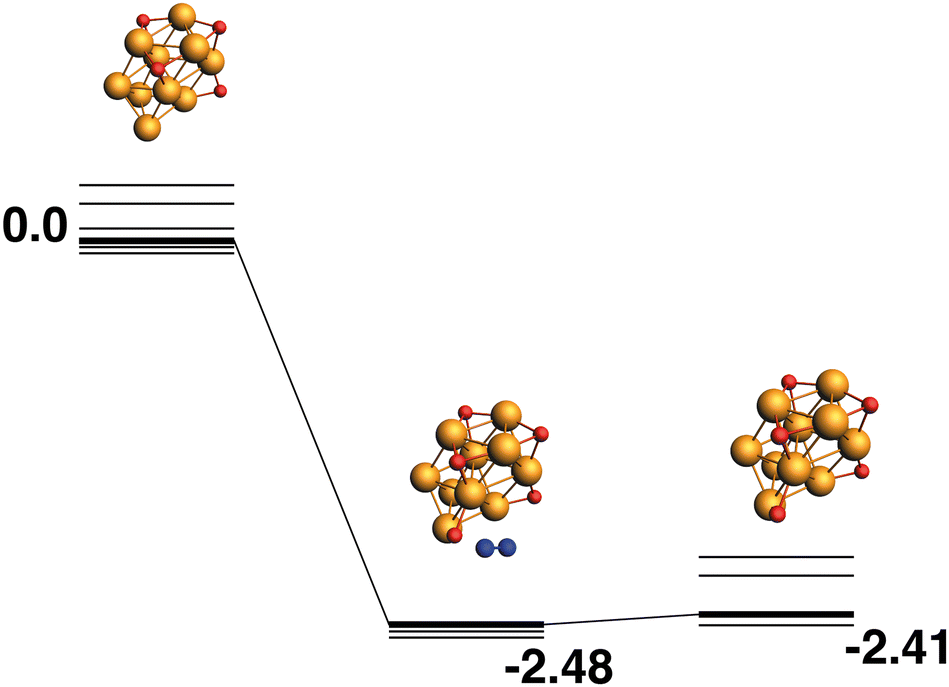
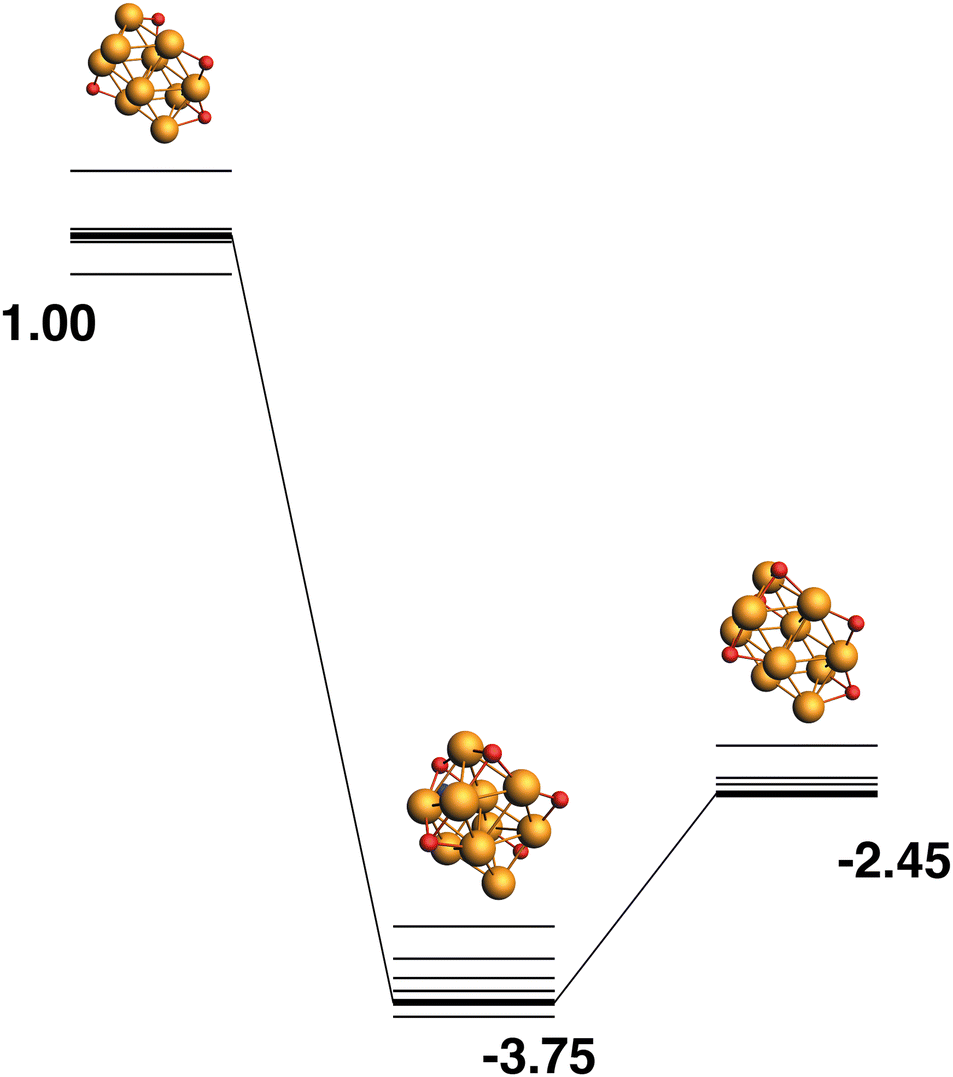
The reaction path from the lowest energy Rh10O4 structure is shown in Fig. 18. The Rh10O5 product lies at −2.41 eV has two μ2 and three μ3-bound oxygen atoms and the corresponding intermediate structure, depositing the second μ2 oxygen atom, lies very close in energy at −2.48 eV and has a Rh10O·N2 distance of 3.38 Å. The lowest energy Rh10O5 structure has one μ2 and four μ3-bound oxygen atoms and the path leading to this product is shown in Fig. 19. The lowest energy matching intermediate structure at −3.75 eV corresponds to depositing the fourth μ3-bound oxygen atom, but the corresponding Rh10O4 reactant is high in energy, at +1.00 eV. The Rh10O·N2 distance in the intermediate structure is 3.94 Å.
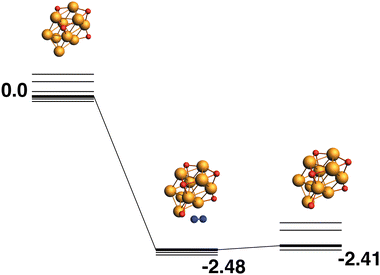 |
| | Fig. 18 Rh10O4 + N2O → Rh10O5 + N2 reaction path for the lowest energy Rh10O4 species. Relative energies are shown for the octet surface in eV. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
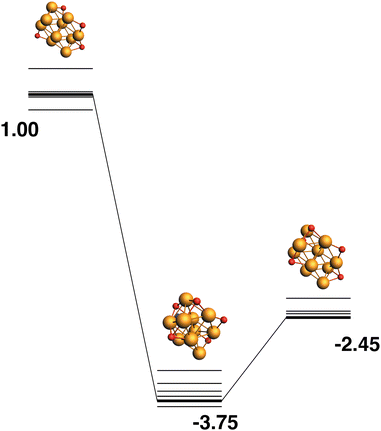 |
| | Fig. 19 Rh10O4 + N2O → Rh10O5 + N2 reaction path for the lowest energy product (Rh10O5) species. Relative energies are shown for the octet surface in eV. Zero energy is the lowest energy Rh10O4 reactant, as shown in Fig. 18. The octet surface is bolded, all other multiplicities are shown with thin lines. | |
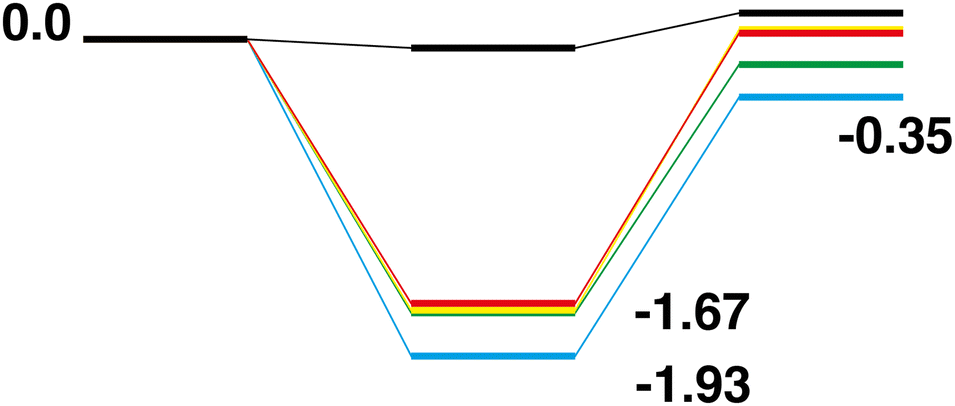
Comparison of the Rh10On + CO → Rh10On−1 + CO2 reaction paths for n = 1–5 reveals a largely similar energetic landscape regardless of n, as shown in Fig. 20 and 21 for the LER and LEP reaction paths respectively. For the pathways beginning with the lowest energy reactant, with the exception of the last abstraction of oxygen, resulting in a bare rhodium cluster (i.e. Rh10O + CO → Rh10O + CO2, Fig. 2), where the intermediate structure lies very close to the energy of both the products and reactants, the four reaction paths have almost the same energetics, with the Rh10On·CO intermediate structure being found in a narrow range between −1.67 and −1.93 eV relative to the reactants. The Rh10On−1 products are found in a similarly narrow energy range, from −0.35 eV for the Rh10O2 reaction to +0.06 eV for the Rh10O4 reaction. Considering the reaction paths arriving at the lowest energy products, shows similar behaviour, with intermediate structures consistently 2 eV lower in energy than the corresponding reactants. The ΔE (products-reactants) shows some deviation with values of 0.80, 0.59, 1.26 and 1.17 eV for n = 2–5 respectively, indicating that the Rh10On−1 products are more stable for the Rh10O4 and Rh10O5 reactants. This is consistent with the higher rates observed for the RhmOn + CO → RhmOn− 1 + CO2 reactions for n = 4,5 by Yamada et al.40
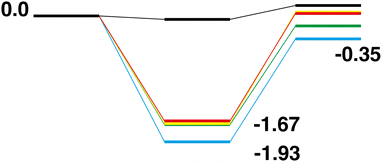 |
| | Fig. 20 Rh10On + CO → Rh10On−1 + CO2, n = 1–5 LER reaction paths overlaid. The Rh10O +CO2 path is black, the Rh10On paths are coloured blue, green, yellow and red, for n = 2–5 respectively. | |
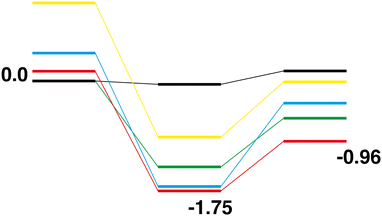 |
| | Fig. 21 Rh10On + CO → Rh10On−1 + CO2, n = 1–5 LEP reaction paths overlaid. Zero energy is the lowest energy Rh10O reactant, as shown in Fig. 2. The Rh10O + CO2 path is black, the Rh10On paths are coloured blue, green, yellow and red, for n = 2–5 respectively. | |
Comparison of the Rh10On + N2O → Rh10On+1 + N2 reaction paths for n = 0–4, shows a very different pattern to the abstraction of oxygen reactions. Considering first the LER paths, shown overlaid in Fig. 22. As the Rh10On reactant is increasingly oxidized (i.e. as n increases), the relative energy of both the intermediate and product increases. With increasing oxygen content, the energy of the intermediate species rises by approximately 0.7 eV. The relative energy of the Rh10On+1 product also rises, but less linearly, with the Rh10O4 and Rh10O5 products switching order. A change in behaviour is clearly seen for the Rh10O4 + N2O → Rh10O5 + N2 reaction, where the intermediate and product species are almost isoenergetic, compared to the n = 0–3 reactions, where the products are less stable than the intermediate by approximately 1 eV. The LEP reaction paths, shown overlaid in Fig. 23 show broadly similar energy profiles. The most notable feature is the difference in the relative energies of the intermediate and product species, which increases from 0.195 eV for the Rh10O + N2 reaction, to 0.71, 0.93 and 1.29 eV for the n = 2–4 paths respectively.
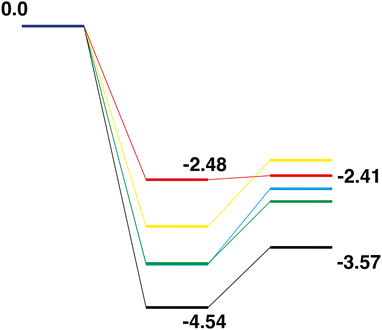 |
| | Fig. 22 Rh10On + N2O → Rh10On+1 + N2, n = 0–4 LER reaction paths overlaid. The Rh10 + N2O path is black, the Rh10On paths are coloured blue, green, yellow and red, for n = 1–4 respectively. | |
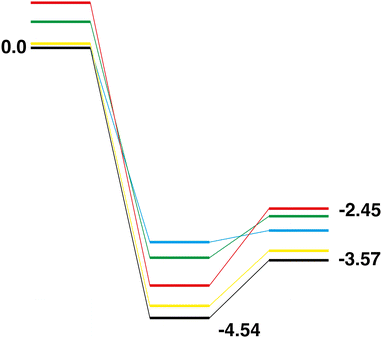 |
| | Fig. 23 Rh10On + N2O → Rh10On+1 + N2, n = 0–4 LEP reaction paths overlaid. Zero energy is the lowest energy Rh10 reactant, as shown in Fig. 11. The Rh10 + N2O path is black, the Rh10On paths are coloured blue, green, yellow and red, for n = 1–4 respectively. | |
4 Conclusion
We have determined the key points on the reaction paths of Rh10On + CO → Rh10On−1 + CO2 and Rh10On + N2O → Rh10On+1 + N2 reactions at multiplicities ranging from doublet up to 16-tet. The vast majority of structures were found to lie within a relatively small energetic range, indicating that most multiplicities would be accessible in reaction conditions.
For the Rh10On + CO → Rh10On−1 + CO2 reactions, the relative energy of the O⋯CO intermediate. Structure was found to be remarkably consistent as the oxidation of the Rh10 cluster increased, lying between −1.93 and −1.67 eV below the energy of the reactants. Similarly, the relative energy of the Rh10On−1 product was found to lie in a narrow range of only 0.35 eV.
The oxygen addition reactions, Rh10On + N2O → Rh10On+1 + N2, showed significantly different behaviour, with the energy of the O⋯N2 intermediate rising by approximately 0.7 eV with each addition oxygen on the Rh10On cluster. The relative energy of the Rh10On+1 product also rose significantly, by ≈0.3 eV, though a linear relationship was not seen for these species.
These reaction paths combined suggest that the rate of oxygen addition to the cluster surface is governed by both thermodynamic and kinetic effects, whereas, the rate of oxygen abstraction reaction is largely governed by access to oxygen on the cluster surface. Paths defined by the lowest energy reactant and lowest energy product show similar energetic profiles, suggesting that the experimentally observed reaction rates are unlikely to be significantly affected by the presence of a mixture of Rh10On isomers.
Author contributions
ATD, VM and OA: investigation MAA: conceptualization of this study, methodology, writing – review and editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
MAA is grateful to the Engineering and Physical Sciences Research Council for funding through grant no. EP/S015868/1 and HPC time via the UK Materials and Molecular Modelling Hub on Young, via grant no. (EP/T022213).
References
- M. Bowker, Automotive catalysis studied by surface science, Chem. Soc. Rev., 2008, 37, 2204–2211 RSC.
- Y. Jia, L. Geng, H. Zhang and Z. Luo, Dramatic Size-dependence of Rhn+ Clusters in Reacting with Small Hydrocarbons: Rh3+ Cluster Catalysis for Dehydrogenation, ChemistrySelect, 2022, 7, e202203632 CrossRef CAS.
- F. Mafune, K. Koyama, T. Nagata, S. Kudoh, T. Yasuike, K. Miyajima, D. M. M. Huitema, V. Chernyy and J. M. Bakker, Structures of Rhodium Oxide Cluster Cations Rh7Om+ (m = 4–7, 12, 14) Revealed by Infrared Multiple Photon Dissociation Spectroscopy, J. Phys. Chem. C, 2019, 123, 5964–5971 CrossRef CAS.
- E. M. Cunningham, A. S. Gentleman, P. W. Beardsmore and S. R. Mackenzie, Structural isomers and low-lying electronic states of gas-phase M+ (N2O)n (M = Co, Rh, Ir) ion-molecule complexes, Phys. Chem. Chem. Phys., 2019, 21, 13959–13967 RSC.
- D. Harding, M. S. Ford, T. R. Walsh and S. R. Mackenzie, Dramatic size effects and evidence of structural isomers in the reactions of rhodium clusters, Rhn±, with nitrous oxide, Phys. Chem. Chem. Phys., 2007, 9, 2130–2136 RSC.
- J. A. Alonso, Electronic and Atomic Structure, and Magnetism of Transition-Metal Clusters, Chem. Rev., 2000, 100, 637–678 CrossRef CAS PubMed.
- M. B. Knickelbein, Reactions of Transition Metal Clusters with Small Molecules, Annu. Rev. Phys. Chem., 1999, 50, 79–115 CrossRef CAS PubMed.
- P. Armentrout, Reactions and Thermochemistry of Small Transiton Metal Cluster Ions, Ann. Rev. Phys. Chem., 2001, 52, 423–461 CrossRef CAS PubMed.
- P. Granger and G. Leclercq, Reduction of N2O by CO over Ceria-Modified Three-Way Pt-Rh Catalysts: Kinetic Aspects, J. Phys. Chem. C, 2007, 111, 9905–9913 CrossRef CAS.
- S. M. Hamilton, W. S. Hopkins, D. J. Harding, T. R. Walsh, M. Haertelt, C. Kerpal, P. Gruene, G. Meijer, A. Fielicke and S. R. Mackenzie, Infrared-Induced Reactivity of N2O on Small Gas-Phase Rhodium Clusters, J. Phys. Chem. A, 2011, 115, 2489–2497 CrossRef CAS PubMed.
- P. Rodrguez-Kessler and A. Rodrguez-Domnguez, N2O dissociation on small Rh clusters: A density functional study, Comput. Mater. Sci., 2015, 97, 32–35 CrossRef.
- R. Aviles, E. Poulain, O. Olvera-Neria and V. Bertin, The spin significance in the capture and activation of N2O by small Rh nanoparticles, J. Mol. Catal. A: Chem., 2013, 376, 22–33 CrossRef CAS.
- F. Mafune, J. M. Bakker and S. Kudoh, Dissociative adsorption of NO introduces flexibility in gas phase Rh6+clusters leading to a rich isomeric distribution, Chem. Phys. Lett., 2021, 780, 138937 CrossRef CAS.
- J.-J. Chen, Q.-Y. Liu, S.-D. Wang, X.-N. Li and S.-G. He, Catalytic NO Reduction by NO Pre-Adsorbed RhCeO2NO− Clusters, Chem. Phys. Chem., 2023, 24, e202200743 CrossRef CAS.
- T. Nagata, S. Kudoh, K. Miyajima, J. M. Bakker and F. Mafune, Adsorption of Multiple NO Molecules on Rhn+ (n = 6, 7) Investigated by Infrared Multiple Photon Dissociation Spectroscopy, J. Phys. Chem. C, 2018, 122, 22884–22891 CrossRef CAS.
- T. Nagata, K. Koyama, S. Kudoh, K. Miyajima, J. M. Bakker and F. Mafune, Adsorption Forms of NO on Rhn+ (n = 6-16) Revealed by Infrared Multiple Photon Dissociation Spectroscopy, J. Phys. Chem. C, 2017, 121, 27417–27426 CrossRef CAS.
- M. J. Piotrowski, P. Piquini, Z. Zeng and J. L. F. Da Silva, Adsorption of NO on the Rh13, Pd13, Ir13, and Pt13 Clusters: A Density Functional Theory Investigation, J. Phys. Chem. C, 2012, 116, 20540–20549 CrossRef CAS.
- H. Xie, M. Ren, Q. Lei, W. Fang and F. Ying, Explore the Catalytic Reaction Mechanism in the Reduction of NO by CO on the Rh7+ Cluster: A Quantum Chemical
Study, J. Phys. Chem. C, 2012, 116, 7776–7781 CrossRef CAS.
- J.-D. Lee, W.-P. Fang, C.-S. Li and C.-H. Cheng, Catalytic reduction of nitrous oxide by carbon monoxide in the presence of rhodium carbonyl and hydroxide. Evidence for an electron-transfer and an oxygen-transfer mechanism, J. Chem. Soc., Dalton Trans., 1991, 1923–1927 RSC.
- S. L. Romo-Ávila and R. A. Guirado-López, Adsorption of Nitric Oxide on Small Rhn± Clusters: Role of the Local Atomic Environment on the Dissociation of the N-O Bond, J. Phys. Chem. A, 2012, 116, 1059–1068 CrossRef.
- J. M. Bakker and F. Mafuné, Zooming in on the initial steps of catalytic NO reduction using metal clusters, Phys. Chem. Chem. Phys., 2022, 24, 7595–7610 RSC.
- P. Ghosh, R. Pushpa, S. de Gironcoli and S. Narasimhan, Interplay between bonding and magnetism in the binding of NO to Rh clusters, J. Chem. Phys., 2008, 128, 194708 CrossRef PubMed.
- H. Yoshida, K. Koizumi, M. Boero, M. Ehara, S. Misumi, A. Matsumoto, Y. Kuzuhara, T. Sato, J. Ohyama and M. Machida, High Turnover Frequency CO–NO Reactions over Rh Overlayer Catalysts: A Comparative Study Using Rh Nanoparticles, J. Phys. Chem. C, 2019, 123, 6080–6089 CrossRef CAS.
- A. Dutta and P. Mondal, Density Functional Study on Structure and Bonding Nature of CO Adsorbed Rhn± (n = 2–8) Clusters, J. Cluster Sci., 2017, 28, 2601–2622 CrossRef CAS.
- J.-J. Chen, X.-N. Li, L.-H. Mou, Q.-Y. Liu and S.-G. He, Catalytic conversion of NO and CO into N2 and CO2 by rhodium-aluminum oxides in the gas phase, J. Mater. Chem. A, 2022, 10, 6031–6037 RSC.
- A. C. Hermes, S. M. Hamilton, W. S. Hopkins, D. J. Harding, C. Kerpal, G. Meijer, A. Fielicke and S. R. Mackenzie, Effects of Coadsorbed Oxygen on the Infrared Driven Decomposition of N2O on Isolated Rh5+ Clusters, J. Phys. Chem. Lett., 2011, 2, 3053–3057 CrossRef CAS.
- Y. Zhang, M. Yamaguchi, K. Kawada, S. Kudoh, O. Lushchikova, V. J. M. Bakker and F. Mafune, Adsorption Forms of NO on Iridium-Doped Rhodium Clusters in the Gas Phase Revealed by Infrared Multiple Photon Dissociation Spectroscopy, J. Phys. Chem. A, 2022, 126, 36–43 CrossRef CAS PubMed.
- L. Zhao, J. Du and G. Jiang, Reduction Reaction of Nitric Oxide on the Rh5V+ Cluster: A Density Functional Theory Mechanistic Study, J. Phys. Chem. C, 2019, 123, 24495–24500 CrossRef CAS.
- K. Koyama, T. Nagata, S. Kudoh, K. Miyajima, D. M. M. Huitema, V. Chernyy, J. M. Bakker and F. Mafune, Geometrical Structures of Partially Oxidized Rhodium Cluster Cations, Rh6Om+ (m = 4, 5, 6), Revealed by Infrared Multiple Photon Dissociation Spectroscopy, J. Phys. Chem. A, 2016, 120, 8599–8605 CrossRef CAS PubMed.
- M. A. Addicoat, M. A. Buntine, B. Yates and G. F. Metha, Associative versus dissociative binding of CO to 4d transition metal trimers: A density functional study, J. Comput. Chem., 2008, 29, 1497–1506 CrossRef CAS PubMed.
- M. A. Addicoat, K. F. Lim and G. F. Metha, Reactions of Nb2 and Nb3 with CO, D2, N2, and O2: Reconciling experimental kinetics with density functional theory-calculated reaction profiles, J. Chem. Phys., 2012, 137, 034301 CrossRef PubMed.
- J. Song, Y. Quan, Y. Zhao and G. Wang, Infrared spectroscopic and density functional theoretical study on the binary rhodium-oxygen Rh2O9+ cation, Chem. Phys. Lett., 2021, 780, 138926 CrossRef CAS.
- Y. Shi and K. M. Ervin, Catalytic oxidation of carbon monoxide by platinum cluster anions, J. Chem. Phys., 1998, 108, 1757–1760 CrossRef CAS.
- F. Zaera and C. S. Gopinath, Evidence for an N2O intermediate in the catalytic reduction of NO to N2 on rhodium surfaces, Chem. Phys. Lett., 2000, 332, 209–214 CrossRef CAS.
- H. Yamamoto, K. Miyajima, T. Yasuike and F. Mafuné, Reactions of Neutral Platinum Clusters with N2O and CO, J. Phys. Chem. A, 2013, 117, 12175–12183 CrossRef CAS PubMed.
- E. Hernandez, V. Bertin, J. Soto, A. Miralrio and M. Castro, Catalytic Reduction of Nitrous Oxide by the Low-Symmetry Pt8 Cluster, J. Phys. Chem. A, 2018, 122, 2209–2220 CrossRef CAS PubMed.
- C. Adlhart and E. Uggerud, C–H activation of alkanes on Rhn+.(n = 1–30) clusters: Size effects on dehydrogenation, J. Chem. Phys., 2005, 123, 214709 CrossRef PubMed.
- J. L. F. Da Silva, M. J. Piotrowski and F. Aguilera-Granja, Hybrid density functional study of small Rhn (n = 2–15) clusters, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 125430 CrossRef.
- M. K. Beyer and M. B. Knickelbein, Electric deflection studies of rhodium clusters, J. Chem. Phys., 2007, 126, 104301 CrossRef PubMed.
- A. Yamada, K. Miyajima and F. Mafune, Catalytic reactions on neutral Rh oxide clusters more efficient than on neutral Rh clusters, Phys. Chem. Chem. Phys., 2012, 14, 4188–4195 RSC.
- M. Addicoat and G. Metha, Kick: Constraining a stochastic search procedure with molecular fragments, J. Comput. Chem., 2009, 30, 57–64 CrossRef CAS PubMed.
- M. A. Addicoat, S. Fukuoka, A. J. Page and S. Irle, Stochastic structure determination for conformationally flexible heterogenous molecular clusters: Application to ionic liquids, J. Comput. Chem., 2013, 34, 2591–2600 CrossRef CAS PubMed.
- S. Grimme, C. Bannwarth and P. Shushkov, A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All spd-Block Elements (Z = 1–86), J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef CAS PubMed.
-
R. Rüger, M. Franchini, T. Trnka, A. Yakovlev, E. van Lenthe, P. Philipsen, T. van Vuren, B. Klumpers and T. Soini, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, AMS 2022.1, 2023, https://www.scm.com.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Climbing the Density Functional Ladder: Nonempirical Meta-Generalized Gradient Approximation Designed for Molecules and Solids, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- P. Mori-Sánchez, A. J. Cohen and W. Yang, Many-electron self-interaction error in approximate density functionals, J. Chem. Phys., 2006, 125, 201102 CrossRef PubMed.
- D. J. Harding, P. Gruene, M. Haertelt, G. Meijer, A. Fielicke, S. M. Hamilton, W. S. Hopkins, S. R. Mackenzie, S. P. Neville and T. R. Walsh, Probing the structures of gas-phase rhodium cluster cations by far-infrared spectroscopy, J. Chem. Phys., 2010, 133, 214304 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spreadsheet of reaction path energies, zipfile of reaction path structures. Structures are additionally available at https://zenodo.org/record/8428605. See DOI: https://doi.org/10.1039/d3cp04929a |
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a