
The counteracting influence of 2-hydroxypropyl substitution and the presence of a guest molecule on the shape and size of the β-cyclodextrin cavity†
Avilasha A.
Sandilya
and
M. Hamsa
Priya
 *
*
Department of Biotechnology, Bhupat and Jyoti Mehta School of Biosciences, Indian Institute of Technology Madras, Chennai-600036, India. E-mail: hamsa@iitm.ac.in; Tel: +91 044 2257 4132
First published on 19th January 2024
Abstract
The aqueous solubility of β-cyclodextrin (β-CD), a cyclic carbohydrate comprising seven α-D-glucose molecules, is enhanced by 2-hydroxypropyl (2-HP) substitution of the hydroxyl groups at the CD rims. Our thorough analysis of the structural and solvation properties with different degrees of 2-hydroxypropyl substitution on β-CD using molecular dynamics simulations reveals that the solubility is enhanced at the cost of the structural distortion of the CD cyclic structure. Substitution at the secondary rim predominantly enhances the favourable interactions between CD and water by decreasing CD–CD hydrogen bonding and promoting CD–water hydrogen bonding. However, the effect of substitution at the primary rim on the CD–water interactions is minimal; the hydrogen bonds between water and the primary hydroxyl group in native CD merely get replaced by those between water and 2-HP, since the substitution makes the primary hydroxyl oxygen (O6 atom) inaccessible to water. In contrast, substitution at the primary rim maintains the structural integrity of CD, while substitution at the secondary rim results in structural distortion due to the disruption of the intramolecular hydrogen bond belt, even leading to cavity closure. Certain strategic substitutions of the primary hydroxyl groups can help in the reduction of structural distortion, depending upon the degree of substitution at the secondary hydroxyl rim. A detailed inspection of the simulation trajectory revealed that the tilting of glucose units with the primary hydroxyl oxygen (O6) pointing inward is the primary driver for cavity closure. Even though the dynamics of glucose tilting can influence the kinetics of host–guest complex formation, once the guest is well incorporated into the cavity, glucose tilting is inhibited and the cavity opens up as in native β-CD.
1 Introduction
Cyclodextrin (CD) is a cyclic oligosaccharide of α-D-glucose molecules connected by 1–4 glycosidic bonds. The native forms of cyclodextrin are referred as α-, β- and γ-CDs depending on the number of glucose units, i.e., 6, 7 and 8 units, respectively.1–3 The primary and secondary hydroxyl groups of the glucose units form the narrower and wider rims of the CD structure, respectively. The cyclic linkage of the glucose units leads to the formation of a central cavity that can comfortably house small organic molecules. Although the outer CD surface resembles a truncated cone, the inner cavity assumes the shape of a conical hourglass because of the inward protrusion of glycosidic oxygen (O1 atoms).4 The cavity is always occupied by a guest molecule and/or by water molecules.1–3Since its discovery, CD has been widely used industrially for separating enantiomers, entrapping volatile compounds, enhancing the solubility of hydrophobic molecules, and protecting chemical compounds against external factors such as heat, light, and so on.5–7 CDs are also popular as drug carriers and excipients in drug formulation owing to their biodegradability, biocompatibility, and their capacity to increase the bioavailability of drug molecules.8–11 Among the native CDs, β-CD is the most widely used drug carrier because of its high stability, low cost of production and its perfect cavity size to host many drug molecules.1–3,8–11
The stability of β-CD is imparted by the flip-flop hydrogen bond between the O2 and O3 atoms of the hydroxyl groups in the secondary rim,1,4,12,13 however, its aqueous solubility is only ∼18.5 mg mL−1 at 25 °C, i.e., 10 times lower than for α- or γ-CDs.2,3 Substitution at the hydroxyl groups in the rims is the common strategy to enhance the aqueous solubility of β-CDs. About a 20–60 fold increase in aqueous solubility has been reported upon substitution with methyl (M), ethyl (E), 2-hydroxypropyl (2-HP), sulfobutylether (SB) groups, and so on.3 Some substitutions, like that with 2-HP, also help in reducing the toxicity of the native CD.14
The reaction of propylene oxide with β-CD under alkaline conditions results in 2-HP substituted β-CD (HP-β-CD). The degree and position of substitution on the hydroxyl groups could be modulated by varying the amount of NaOH and propylene oxide in solution. When the alkalinity of the solution is low, substitutions tend to occur at O2 atoms of the secondary rim, as the hydrogen (H2) associated with the hydroxyl group (O2H2) bonded to C2 is the most acidic amongst all the glucose hydroxyl groups.2,15–19 With an increase in the alkalinity, the substitution tends to occur at the O6 atom in the primary rim. The relative ratio of reactivity of O6 and O2 is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 at low alkali concentration, whereas it is 7:1 at high alkali concentration.19 The extent of substitution is usually expressed in terms of the degree of substitution (DS). It is influenced by factors such as the ratio of the reactants, the time of reaction and reaction temperature.2,15 Experimentally, DS is determined using GC-MS, FTIR, TG-DTA and NMR techniques.20,21
:5 at low alkali concentration, whereas it is 7:1 at high alkali concentration.19 The extent of substitution is usually expressed in terms of the degree of substitution (DS). It is influenced by factors such as the ratio of the reactants, the time of reaction and reaction temperature.2,15 Experimentally, DS is determined using GC-MS, FTIR, TG-DTA and NMR techniques.20,21
HP-β-CDs with DS values of 2.8–10.5 are preferably used for pharmaceutical applications.15 The solubility of HP-β-CD is ∼1200 mg mL−1; it is a versatile excipient in pharmaceutical formulations delivered by various modes including ocular,22 oral,23 rectal,24 parenternal25 and dermal.26 Unlike native β-CD, there have been only limited experimental studies investigating the structure of HP-β-CDs. These only probed HP-β-CDs with degree of substitution values of 1–2, since it is hard to obtain good crystals of HP-β-CDs14 because the gradual increase in the degree of substitution results only in amorphous precipitation. Interestingly, the HP-β-CDs with DS values of 12–14 exist in semi-crystalline form.2,27 The properties of HP-β-CDs are highly influenced by the degree and site of substitution. For example, HP-β-CD with mono-substitution (DS = 1) at O6 is found to have a 13% larger cavity volume compared with the mono-substitution at O2.28 The crystal structure of the mono-substituted HP-β-CD revealed that 2-HP of HP-β-CD gets inserted into the cavity of an adjacent HP-β-CD molecule.28,29 On the other hand, despite the unavailability of crystal structures of substituted HP-β-CDs, there are reports on the powder X-ray diffraction of some HP-β-CD–guest inclusion complexes.30–33 Yet, it is difficult to obtain single crystals of HP-β-CD–guest inclusion complexes, while there are several studies on single crystal X-ray diffractometry of native β-CD–guest inclusion complexes.34–38
Commercially available samples of HP-β-CDs show high variability in their properties15 because of the difference in the site of substitution and poly-dispersity in the DS of the samples. Two samples of HP-β-CDs with DS values of 4.2 and 4.3 showed a contrasting effect in the treatment of Niemann-Pick disease type C in mice; the former delayed the neurological symptoms while the latter did not.15 It is, therefore, necessary to characterise HP-β-CDs based on the site of substitution, not just by the DS. A unified method in reporting the degree of substitution is essential as many physicochemical properties of CD including its ability to complex are greatly influenced by the nature, degree and position of substitution.39 No systematic study has explored and mapped the variation in the structural properties with the DS and the site of substitution. The crystal diffraction investigation is inhibited because of the unavailability of good crystals for the whole range of DS values. Spectroscopic techniques like NMR21 and IR20 can be used to probe the structure, but one-on-one mapping of the DS and structural properties is not possible because of the poly-dispersity in the HP-β-CD samples and the inability to exactly quantify the DS. Computational techniques like molecular dynamics simulation serve as an apt tool for the one-on-one mapping.
In this work, we examine the structural properties of twenty HP-β-CDs with DS values ranging from 4–14 using molecular dynamics simulation. Some HP-β-CDs with the same DS but different positions of substitution are also investigated. We determine that the substitution at the secondary rim imparts conformational flexibility to the glucose units. The conformational flexibility promotes sugar puckering, i.e., transition of the 4C1 chair conformation to the inverted chair 1C4 and other intermediate boat and skew forms. Substitution at the secondary rim leads to a breach in the O2–O3 hydrogen bond belt, leading to the closure of the CD cavity. Encapsulation of a guest molecule in the CD cavity, however, stabilises the structure of the CD cavity, though the constituent glucose units undergo a conformational transition. The primary cause for cavity closure has been identified as the tilting of glucose units about the glycosidic plane with the primary hydroxyl group pointing inward. The presence of a guest molecule inside the cavity restores the cavity shape and structure by inhibiting the tilting of the glucose units. The substitution of 2-HP at the secondary rim predominantly aids in enhancing the favourable interactions between CD and water required for increasing the aqueous solubility. We found that the extent of the CD–water interactions is strongly correlated to the number of CD–water hydrogen bonds, which remains unaffected in the presence of a guest molecule inside the CD cavity. We have systematically mapped the effect of CD ring flexibility on the observed changes in the structural parameters.
2 Methods
2.1 Simulation details
Table 1 lists twenty HP-β-CDs investigated in this work with varying degree of substitution values in the range of 4–14; multiple possible variations in the position of substitution for a given degree of substitution are also considered. The initial structures of the substituted CDs were generated by computationally substituting the structure of native cyclodextrin (3cgt.pdb) with the 2-hydroxypropyl moiety at the desired positions using the Avogadro molecule builder.40 The partial charges and other parameters for the 2-hydroxypropyl moiety are taken from Khuntawee et al.41Fig. 1 presents the partial charges used for a glucose unit and 2-hydroxypropyl unit. Carbohydrates are modelled using the Gromos56A6Carbo_R force field, a united atom force field developed for hexopyranose.42,43 It is one of the three force fields recommended for simulating cyclodextrin.12 Moreover, it is the only force field that captures the difference in the hydrogen bonding capability of the two hydroxyl groups (O2H2 and O3H3) at the secondary rim.4 The initial structure and topology for the guest molecules – p-nitrophenol, neral and phenolphthalein – were obtained from the Automated Topology Builder (ATB)44 based on the Gromos 54A7 force field. Although, the Gromos56A6Carbo_R force field was originally based on the Gromos 53A6 force field, we verified that the involved parameters are unchanged in the Gromos 54A7 force field. Thus, the parameters for the guest molecule and CD are compatible. The initial structures for the CD–guest simulations were obtained by placing the guest molecule at the centre of the energy-minimised structure of the HP-β-CDs.| Molecule | DS | g O2 | g O6 |
|---|---|---|---|
| The subscripts i and j in hij imply the number of 2-hydroxypropyl substitutions at the secondary hydroxyl rim (O2) and at the primary hydroxyl rim (O6) of β-CD, respectively; DS – degree of substitution; gO2 and gO6 are the list of the indices of glucose units substituted at the O2 and O6 positions, respectively. | |||
| β-CD | 0 | None | None |
| h 04 | 4 | None | 1,3,5,7 |
| h 22 | 4 | 1,5 | 3,7 |
| h 40 | 4 | 1,3,5,7 | None |
| h 05 | 5 | None | 1,3,5,6,7 |
| h 23 | 5 | 3,6 | 1,4,7 |
| h 32 | 5 | 1,4,7 | 3,6 |
| h 50 | 5 | 1,3,5,6,7 | None |
| h 33 | 6 | 1,4,7 | 1,4,7 |
| h 33′ | 6 | 1,3,5 | 2,4,6 |
| h 07 | 7 | None | All |
| h 34 | 7 | 2,4,6 | 1,3,5,7 |
| h 43 | 7 | 1,3,5,7 | 2,4,6 |
| h 70 | 7 | All | None |
| h 44 | 8 | 1,3,5,7 | 1,3,5,7 |
| h 72 | 9 | All | 1,4 |
| h 73 | 10 | All | 1,3,5 |
| h 74 | 11 | All | 1,3,5,7 |
| h 75 | 12 | All | 1,3,5,6,7 |
| h 76 | 13 | All | 1,2,3,4,5,6 |
| h 77 | 14 | All | All |
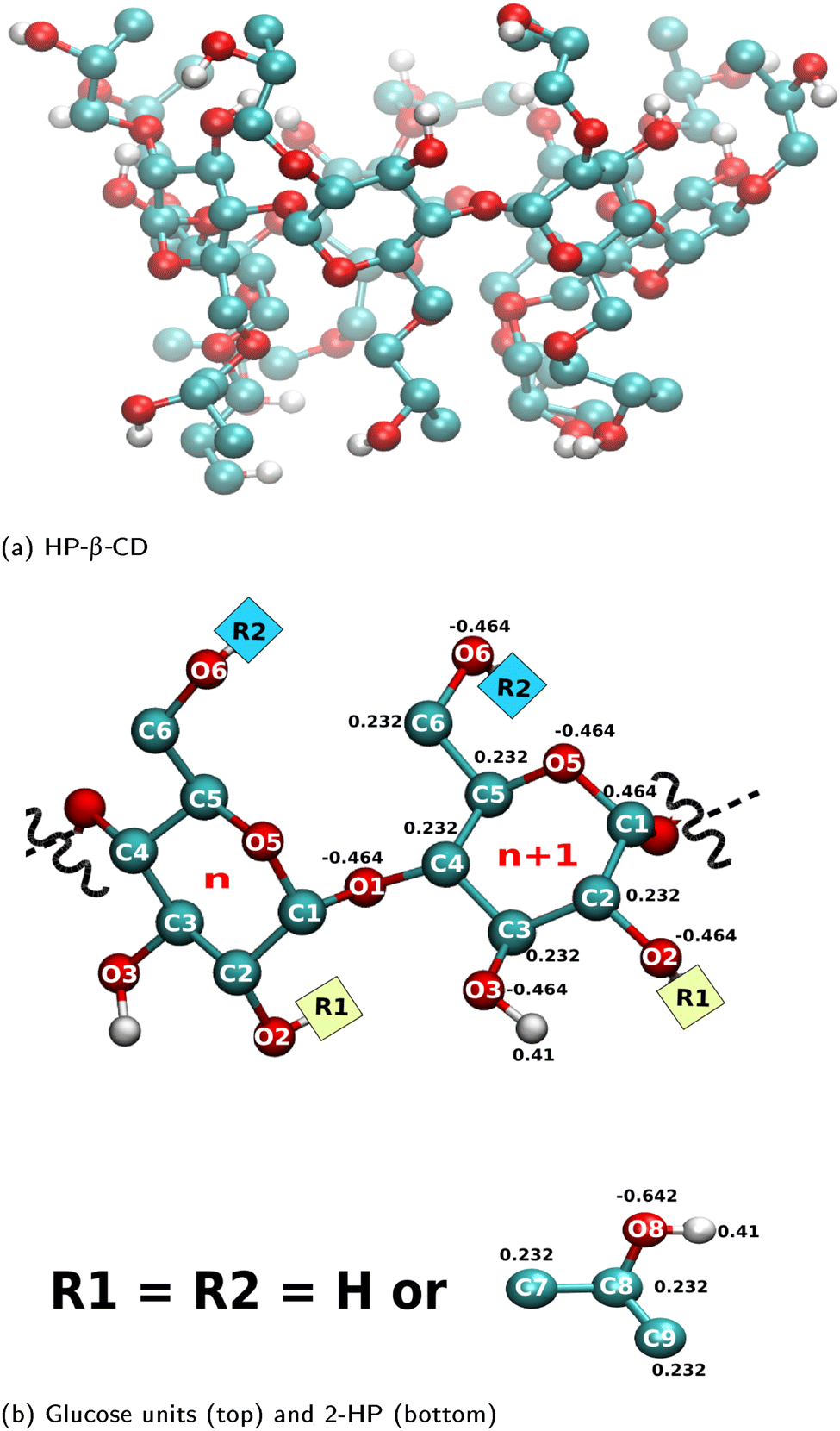 | ||
| Fig. 1 (a) Representative molecular structure of a HP-β-CD; (b) atom names and the partial charges of glucose units (top) and 2-hydroxypropyl, i.e., 2-HP group (bottom) used in the simulation. Colour code: cyan – carbon; red – oxygen; white – hydrogen. | ||
All the simulations were carried out with the Gromacs 2016.4 package. Each CD molecule and CD–guest complex was solvated in a cubic SPC/E water box with 1.2 nm of water padding on all sides. Energy minimisation was carried out for 500 steps through the steepest descent algorithm, then the system was equilibrated for 100 ps under NVT conditions at 298 K using a Berendsen thermostat.45 An NPT run of 1 ns was then done at 298 K and 1 bar using a Nose–Hoover Thermostat46,47 and a Parrinello–Rahman barostat,48 respectively. Following this, the simulation was carried out under NVT conditions for 1 ns allowing the system to equilibrate further. All bonds were constrained using the LINCS algorithm. Dispersion interactions were truncated at 1 nm. The particle mesh Ewald (PME)49 method was used for the electrostatic interactions with a real-space cut-off radius of 1 nm. An integration time step of 2 fs was used in all MD runs. Finally, an MD run of 100 ns under NVT conditions at 298 K was carried out and used for further analysis.
2.2 Analysis
| εCD–water = εsystem − εCD–CD − εwater–water |
3 Results & discussion
3.1 Substitution distorts & a guest restores the cavity structure
Fig. 2(a)–(g) show the surface plot of the average structure (averaged over the 100 ns simulation trajectory) of native β-CD and some HP-β-CDs as viewed from the secondary hydroxyl rim. The structure of β-CD (Fig. 2a) is symmetric and its cavity is wide-open, although the complete view of the cavity is not possible because of the conical hour-glass structure of the CD cavity with the constriction at the glycosidic rim.4 Among the HP-β-CDs, only the molecule h07 (Fig. 2c) where only the primary rim is completely substituted with 2-HP resembles the structure of β-CD; all other CDs appear to be deformed and the cavities seem to be closed. The most deformation is seen in h44 (Fig. 2e) when both the rims are partially and equally substituted with di-substitution of the glucose units. In the case of substitution at only one rim, the HP-β-CDs with substitution at only the primary hydroxyl rim (Fig. 2b and c) are less deformed than the HP-β-CDs with the same degree of substitution in the secondary hydroxyl rim (Fig. 2d and f).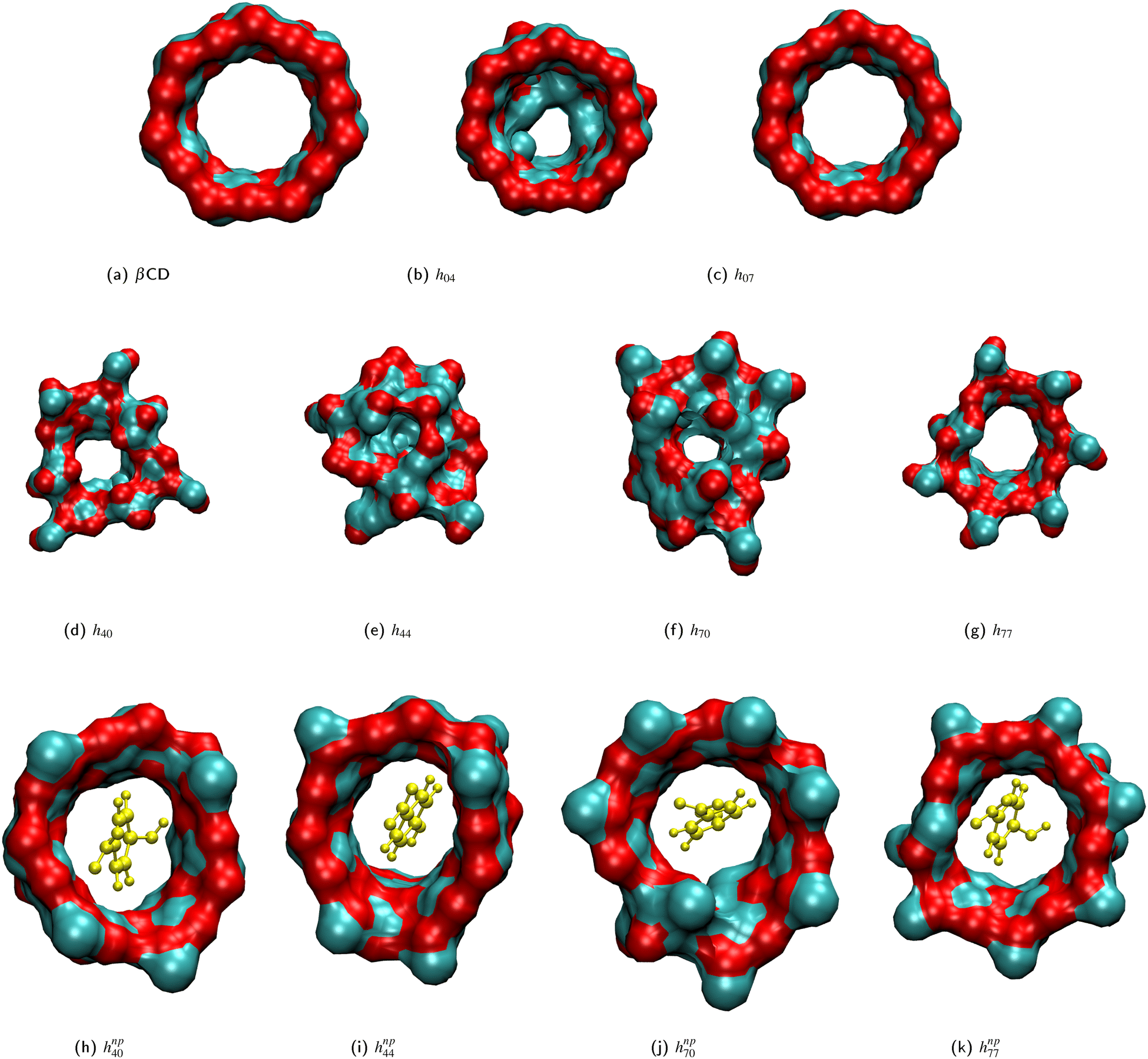 | ||
| Fig. 2 Surface plots of the average structure depicting the closure of the central cavity (b)–(g) in HP-β-CDs, hxy, and cavity opening (h)–(k) in the presence, hnpxy, of the guest p-nitrophenol (yellow CPK representation): (a) native β-CD; (b) h04; (c) h07; (d) h40; (e) h44; (f) h70; (g) h77; (h) hnp40; (i) hnp44; (j) hnp70; (k) hnp77. 2-HP groups are not shown for uniformity and visual clarity. | ||
In order to quantify the shape and size of the cavity of the HP-β-CDs, we computed the circularity of the glycosidic rim and the volume of the cavity. The native β-CD has a circularity value of 0.89 indicating that the glycosidic rim is nearly circular, but the circularity values of the HP-β-CDs (red curve in the top panel of Fig. 3) are low, only in the range of 0.5–0.74, considerably smaller than the native β-CD. The volume of the cavity is determined by the grid technique (see Methods); we obtained a value of 300 Å3 for the β-CD cavity, which is in reasonable agreement with the reported value of 262 Å3.3 The red curve in the bottom panel of Fig. 3 presents the difference in the cavity volume of HP-β-CDs and β-CD. We have defined the CD cavity based only on the position of the O2 and O6 atoms to facilitate a uniform and fair comparison between different degrees of substitution of HP-β-CDs and β-CD. We can see that in all the HP-β-CDs, the cavity volume decreases; about a 50% reduction in cavity volume (∼150 Å3) is observed for 6–7 HP-β-CDs.
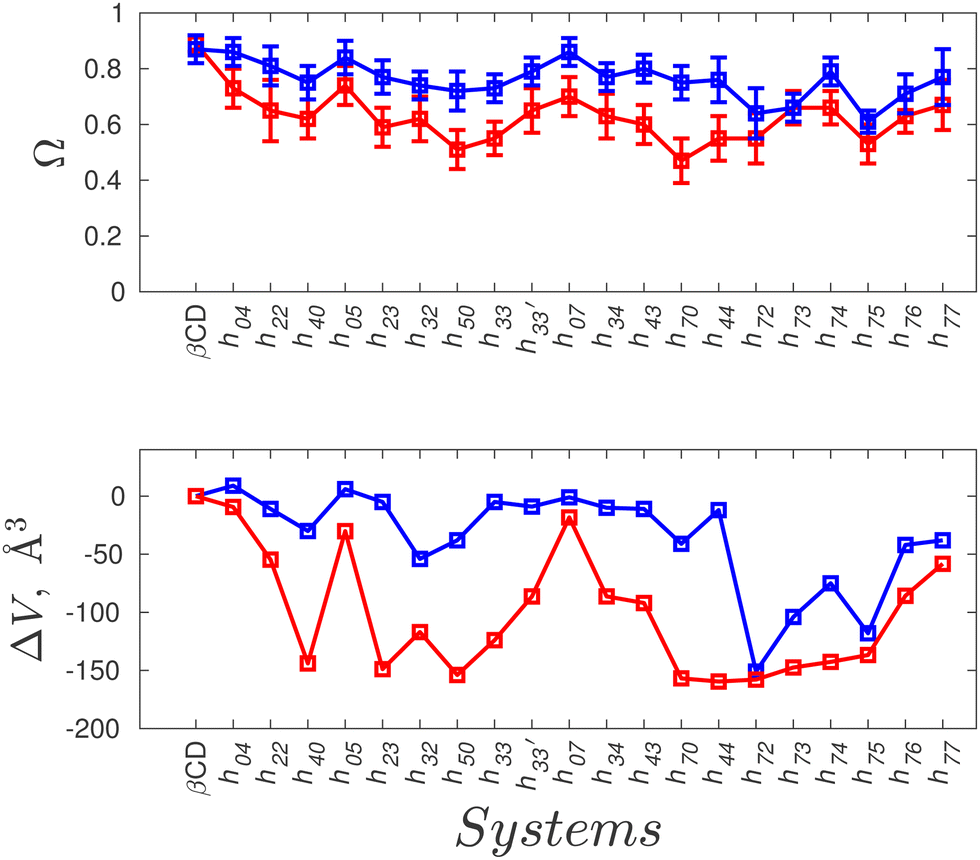 | ||
| Fig. 3 Quantitative measurements of the cavity shape and size of HP-β-CDs in the absence (red curve) and presence of the guest p-nitrophenol (blue curve). Top panel: Circularity of the glycosidic rim, Ω, defined as the ratio of the smallest to the largest distance of glycosidic oxygen atoms (O1) from the CD centre of mass; bottom panel: the difference in the cavity volume of HP-β-CDs and native β-CD, computed by the grid approach. | ||
The general trend is that the cavity volume decreases with an increase in the substitution at the secondary rim: Vh40 > Vh50 > Vh70. For the same degree of substitution at only one of the rims, the volume of the HP-β-CD with substitution at the primary rim is larger than that for the HP-β-CD with substitution at the secondary hydroxyl rim – Vh04 > Vh40; Vh05 > Vh50; Vh07 > Vh70. Even the crystallographic studies on mono-substituted HP-β-CD have also reported that mono-substituted HP-β-CD with substitution at O6 had 13% greater volume than when substituted at the O2 site.28 We can see that the cavity opens up when the substitution at the primary hydroxyl rim is increased, especially when the secondary hydroxyl rim is completely substituted: Vh70 < Vh72 < Vh73 < Vh74 < Vh75 < Vh76 < Vh77. Interestingly, the cavity volume of the HP-β-CD with the alternate glucose units substituted in either of the rims is higher than that for HP-β-CDs with the same glucose unit substituted in both rims – 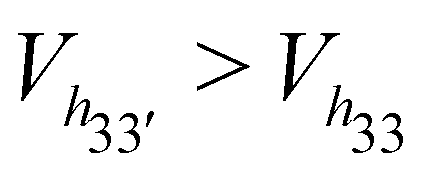 – although the degree of substitution is the same. Also, the HP-β-CDs – h33′, h34 and h43 – with alternating glucose units substituted at the primary and secondary rims, have almost the same cavity volume, which is intermediate to that of the h0x and hx0 systems.
– although the degree of substitution is the same. Also, the HP-β-CDs – h33′, h34 and h43 – with alternating glucose units substituted at the primary and secondary rims, have almost the same cavity volume, which is intermediate to that of the h0x and hx0 systems.
We believe that the drastic cavity closure can result in CD agglomeration and therefore it is difficult to obtain good crystals of HP-β-CDs for structural investigation. The X-ray structures of HP-β-CDs have only been reported for mono-substituted HP-β-CD.28,29 However, there are several structural reports on HP-β-CD host–guest complexes with guest molecules like sulfanilamide,30 fluconazole,31 nifepidine32 and flavonoids.33 So, we carried out the simulation of HP-β-CDs with p-nitrophenol inserted into the cavity of the initial energy-minimized structure of HP-β-CDs. We choose p-nitrophenol because it is the most common guest compound studied in the literature.52–56Fig. 2(h)–(k) are the surface plots of HP-β-CDs with p-nitrophenol encapsulated into the cavity. In contrast to the structures shown in Fig. 2(d)–(g), we noticed that in the presence of the guest molecule, the cavity of the HP-β-CDs is open. Fig. 3 confirms that both the circularity of the glycosidic rim and cavity volume improves for most of the HP-β-CDs upon insertion of the guest molecule. The exceptions being h72, h73, h74 and h75 systems, since the guest molecule didn't remain bound in the cavity during the simulation; instead 2-HP groups were preferably found to be pointed inward into the cavity.
3.2 Deformed HP-β-CDs exhibit multi-modal distribution of structural parameters
To understand the origin of the deformation of the structure of HP-β-CDs, we determined the key structural parameters characterising the molecular arrangement of adjacent α-glucose units (Table 2). The glycosidic dihedral angles ϕ and ψ are the primary parameters that represent the orientation of one glucose unit with respect to its neighbour. In native β-CD, two units are aligned such that the average ϕ and ψ values are 117° and 116°, respectively. HP-β-CDs exhibit a huge variation in ϕ and ψ values ranging from 66–117° and 50–116°, respectively. We can observe that when the cavity volume decreases, ϕ decreases and ψ increases. Only for HP-β-CDs with substitution at the primary hydroxyl rim (hCD0x) are the ϕ and ψ values comparable to native β-CD as the reduction in their cavity volume is also minimal. For the systems whose cavity volumes are enhanced after inserting the guest molecule inside the cavity, the ϕ and ψ values approached the values of native β-CD. Interestingly, the angle between three consecutive glycosidic oxygens, δ, does not show much variation among HP-β-CDs; a maximum variation of only about 4° is observed from that of native β-CD. The value of 128° in the native β-CD corresponds to the angle between the sides of a regular heptagon, emphasising the symmetric heptagonal arrangement of the constituent glucose units.4 Despite the decrease in circularity in HP-β-CDs (Fig. 3), the change in the average δ angle is small; it is mainly attributable to the requirement of maintaining the covalently bonded cyclic structure.| Molecule | ϕ, deg | ϕ np, deg | ψ, deg | ψ np, deg | δ, deg | δ np, deg |
|---|---|---|---|---|---|---|
| ϕ and ψ are the glycosidic dihedral angles, defined as O5(n)–C1(n)–O1(n)–C4(n − 1) and C1(n)–O1(n)–C4(n − 1)–C3(n − 1), respectively; δ is the angle between three consecutive O1 atoms (O1(n − 1)–O1(n)–O1(n + 1)); the superscript np denotes the structural properties in the presence of guest p-nitrophenol in the cavity. The reported average and standard deviation represent the mean and the variation in the structural properties, respectively, observed throughout the simulation trajectory. | ||||||
| β-CD | 117.2 ± 11.5 | 117.5 ± 10.9 | 116.6 ± 11.1 | 116.3 ± 9.9 | 128.1 ± 6.5 | 127.9 ± 6.5 |
| h 04 | 119.9 ± 10.1 | 121.1 ± 10.1 | 114.2 ± 13.5 | 113.5 ± 10.6 | 128.1 ± 6.7 | 128.0 ± 6.5 |
| h 22 | 104.6 ± 19.6 | 112.3 ± 14.1 | 122.2 ± 24.3 | 117.6 ± 14.6 | 126.5 ± 17.1 | 127.5 ± 10.2 |
| h 40 | 92.5 ± 22.7 | 106.2 ± 15.1 | 132.3 ± 32.4 | 116.7 ± 16.8 | 126.7 ± 22.4 | 127.5 ± 14.2 |
| h 05 | 114.9 ± 13.5 | 117.0 ± 11.5 | 117.5 ± 18.4 | 114.0 ± 12.1 | 127.9 ± 6.7 | 127.9 ± 8.0 |
| h 23 | 94.5 ± 23.7 | 110.7 ± 14.6 | 126.9 ± 30.2 | 113.2 ± 13.3 | 125.6 ± 18.6 | 126.3 ± 11.4 |
| h 32 | 87.2 ± 22.8 | 90.7 ± 18.2 | 121.5 ± 31.1 | 114.9 ± 24.3 | 126.7 ± 20.7 | 127.4 ± 16.4 |
| h 50 | 94.7 ± 18.3 | 104.1 ± 14.5 | 122.3 ± 31.7 | 117.7 ± 19.7 | 124.6 ± 23.5 | 127.3 ± 15.2 |
| h 33 | 100.2 ± 22.5 | 102.5 ± 16.2 | 125.5 ± 25.2 | 120.8 ± 20.6 | 124.0 ± 19.9 | 125.7 ± 16.9 |
| h 33′ | 106.3 ± 17.6 | 104.3 ± 11.7 | 114.6 ± 22.3 | 108.2 ± 13.3 | 127.5 ± 14.9 | 128.0 ± 12.1 |
| h 07 | 114.5 ± 13.5 | 116.5 ± 11.6 | 117.2 ± 15.4 | 116.3 ± 12.2 | 127.8 ± 8.6 | 127.9 ± 7.4 |
| h 34 | 104.6 ± 16.2 | 106.3 ± 11.1 | 117.3 ± 24.2 | 109.0 ± 13.4 | 127.0 ± 17.0 | 127.9 ± 12.7 |
| h 43 | 99.1 ± 18.5 | 103.9 ± 12.4 | 119.4 ± 24.8 | 110.3 ± 15.4 | 126.3 ± 20.7 | 127.9 ± 11.8 |
| h 70 | 93.8 ± 20.2 | 99.2 ± 13.2 | 119.2 ± 31.8 | 116.9 ± 20.2 | 125.9 ± 23.5 | 127.6 ± 15.2 |
| h 44 | 88.5 ± 22.5 | 110.3 ± 12.2 | 129.2 ± 28.8 | 114.5 ± 12.9 | 123.6 ± 23.9 | 127.9 ± 12.5 |
| h 72 | 69.7 ± 20.8 | 69.3 ± 19.8 | 142.4 ± 40.0 | 140.2 ± 52.8 | 124.9 ± 21.6 | 124.9 ± 19.1 |
| h 73 | 80.7 ± 19.5 | 85.5 ± 16.2 | 122.8 ± 37.3 | 117.3 ± 33.3 | 126.4 ± 20.1 | 126.4 ± 18.8 |
| h 74 | 81.5 ± 24.2 | 90.5 ± 13.8 | 126.4 ± 38.0 | 116.2 ± 33.7 | 124.5 ± 15.7 | 125.8 ± 12.6 |
| h 75 | 94.3 ± 21.5 | 95.0 ± 21.4 | 124.2 ± 28.2 | 124.5 ± 27.0 | 125.4 ± 21.5 | 125.3 ± 21.2 |
| h 76 | 104.4 ± 17.6 | 107.4 ± 15.4 | 125.0 ± 20.7 | 122.2 ± 18.1 | 126.3 ± 17.2 | 127.1 ± 15.4 |
| h 77 | 106.7 ± 16.9 | 110.6 ± 14.1 | 122.7 ± 18.2 | 121.2 ± 14.7 | 127.0 ± 14.0 | 127.4 ± 12.5 |
The probability distribution of the δ angle in HP-β-CDs shown in Fig. 4 clearly reveals the actual variation. In the case of native β-CD and HP-β-CDs with substitution only at the primary rim, the distribution is unimodal. In all other systems, we can observe trimodal distribution – the central peak corresponds to native-like distribution, while the other two peaks at 90° and 150° result from the glucose units that close the cavity by pointing inward. The population of the glucose units with low and high δ values are almost identical contributing to the cancellation of the deviation from the native region, hence eventually leading to minimal variation in the average δ values. Upon inclusion of the guest inside the cavity of HP-β-CDs, the distribution narrows down and become unimodal, except for h72, h73, h74 and h75.
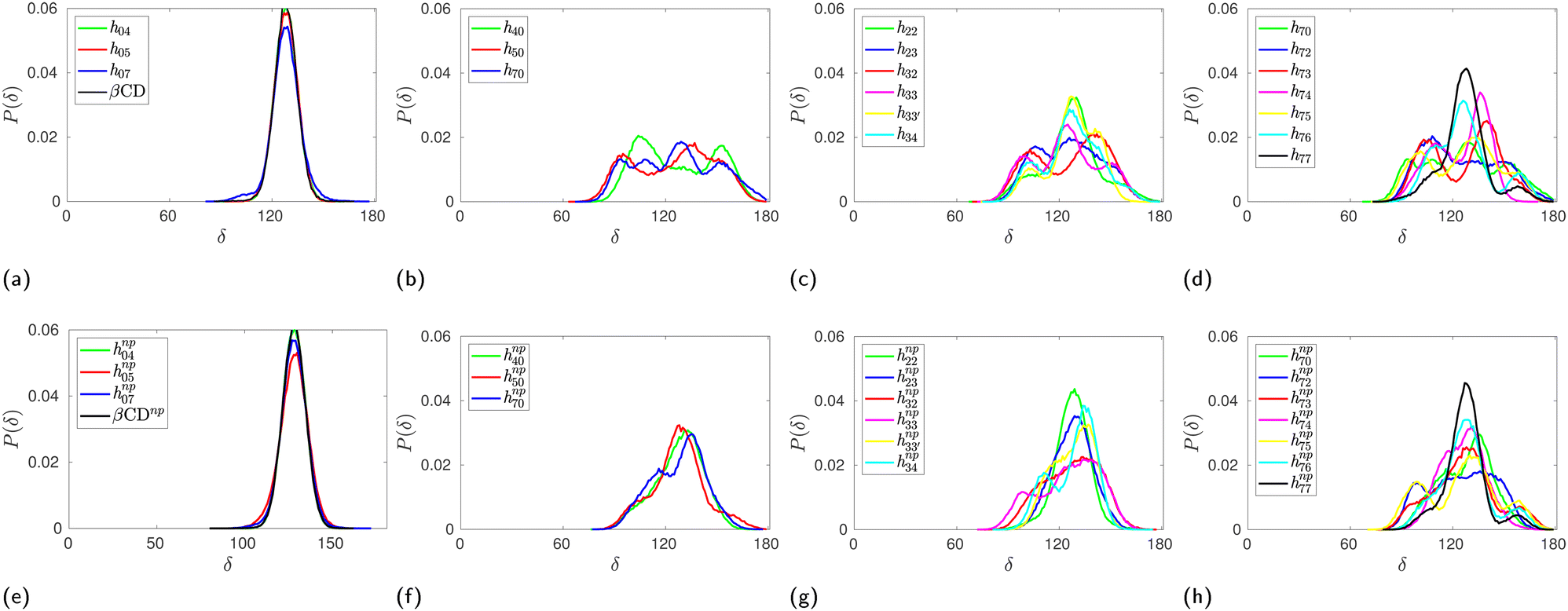 | ||
| Fig. 4 Probability distribution of the angle between three consecutive glycosidic oxygen atoms, δ angle, in HP-β-CDs in the absence (top panel) and the presence (bottom panel) of the guest p-nitrophenol inside the cavity: (a) and (e) substitution only in the primary hydroxyl rim; (b) and (f) substitution only in the secondary hydroxyl rim; (c) and (g) glucose units are substituted either at the primary or the secondary hydroxyl rim but not both; (d) and (h) increase in substitution in the primary hydroxyl rim when all the glucose units in the secondary hydroxyl rim are substituted. The distributions for native β-CD in the absence and the presence of the guest are shown in (a) & (e), respectively, for comparison. | ||
Similarly, we computed the joint probability distribution for ϕ and ψ, i.e., P(ϕ,ψ), and determined the free-energy difference of observing a CD at a given ϕ and ψ against the most dominant conformation using the expression: 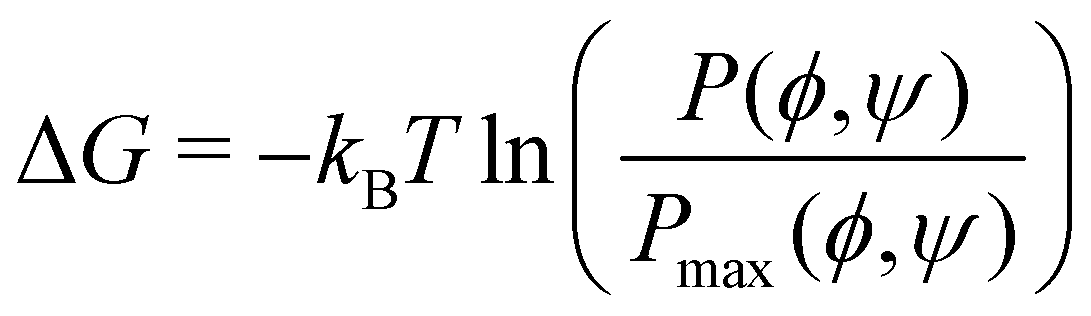 . From Fig. 5, we observe that the native β-CD, h04, and h07 predominantly have the same single energy basin about the mean (ϕ,ψ) values reported in Table 2, however, the spread of the basin is a little wider in h07. On the other hand, other HP-β-CDs exhibit three free-energy basins, as depicted for h40, h44 and h70. The central basin of the free-energy surface plot lies in the range 100 ≤ ϕ/ψ ≤ 130, whereas two other basins are seen with the shift in ϕ and ψ values, respectively. The population of the central native-like region is only 8.6%, 8.7% and 14% in h40, h44 and h70, respectively; the rest lies in the non-native region. The trimodal distribution, however, reduces to a single basin in the presence of guest p-nitrophenol inside the cavity, as shown in Fig. 5(g)–(i).
. From Fig. 5, we observe that the native β-CD, h04, and h07 predominantly have the same single energy basin about the mean (ϕ,ψ) values reported in Table 2, however, the spread of the basin is a little wider in h07. On the other hand, other HP-β-CDs exhibit three free-energy basins, as depicted for h40, h44 and h70. The central basin of the free-energy surface plot lies in the range 100 ≤ ϕ/ψ ≤ 130, whereas two other basins are seen with the shift in ϕ and ψ values, respectively. The population of the central native-like region is only 8.6%, 8.7% and 14% in h40, h44 and h70, respectively; the rest lies in the non-native region. The trimodal distribution, however, reduces to a single basin in the presence of guest p-nitrophenol inside the cavity, as shown in Fig. 5(g)–(i).
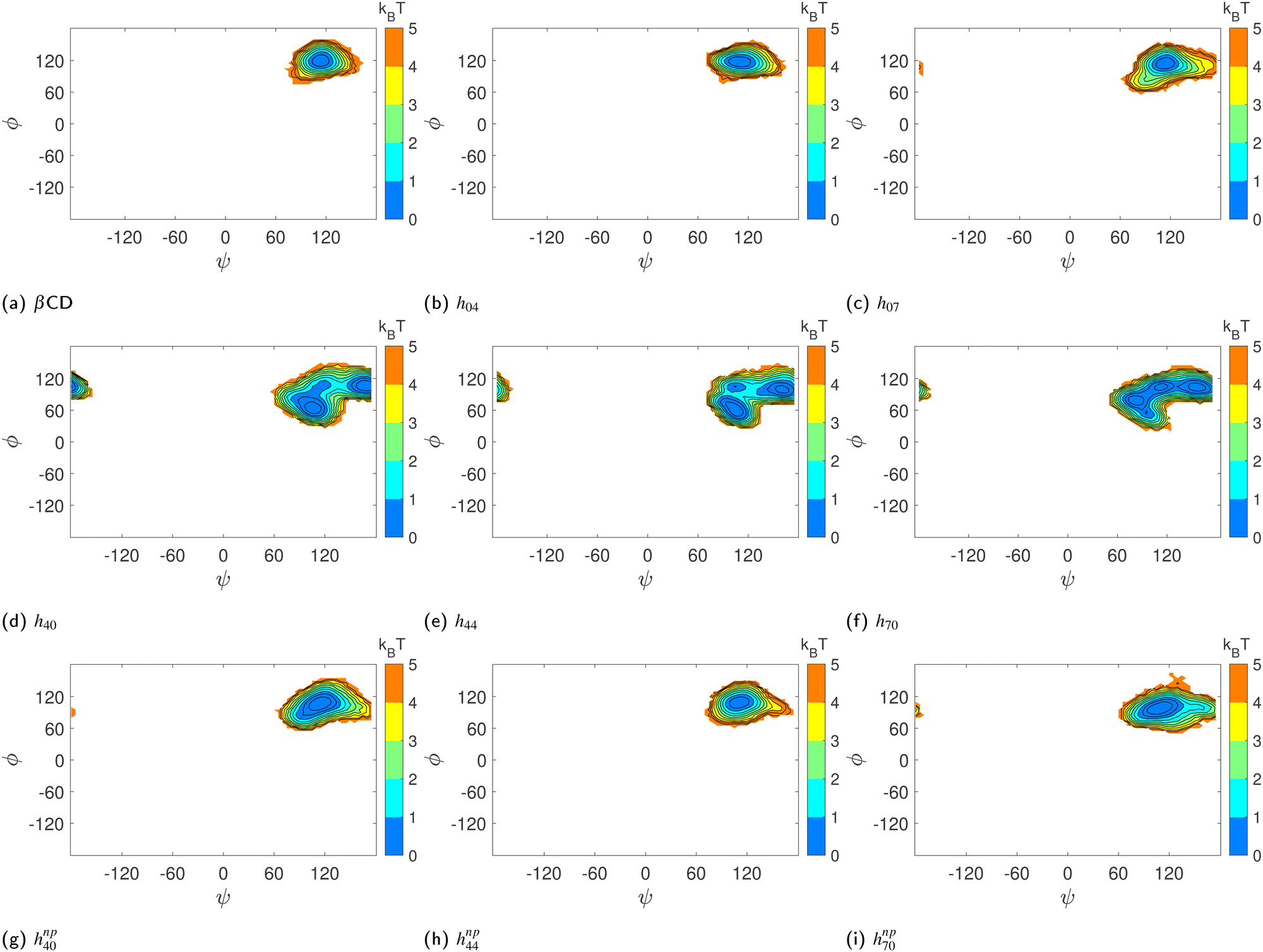 | ||
| Fig. 5 Contour plots of the potential of the mean force in the glycosidic dihedral angle space (ϕ,ψ) of the HP-β-CDs in the absence (a)–(f) and the presence (g)–(i) of guest p-nitrophenol inside the cavity: (a) native β-CD; (b) h04; (c) h07; (d) h40; (e) h44; (f) h70; (g) hnp40; (h) hnp44; (i) hnp70. The colour bar shows the energy values in kBT and the contour lines are drawn for every 1 kJ mol−1. | ||
3.3 Conformational transition cannot explain the cavity closure
The monomer glucose predominantly assumes the 4C1 chair conformation in solution.3,43,50,51,57 About 90% of the glucose units in native β-CD also assume the 4C1 conformation. In the X-ray crystallographic structure of trimethyl β-CD,58 one glucose unit has been found in the inverted chair 1C4 conformation; the δ value about the puckered glucose unit has been reported to be ∼180° and the corresponding adjacent δ angle exhibited a value of 90°. This prompted us to identify the glucose conformations in HP-β-CDs using Pickett–Strauss parameters50 that help in identifying 14 glucose conformations. We mainly observed the six conformations shown in Fig. S1 (ESI†) and the corresponding Pickett–Strauss parameters for these conformations are tabulated in Table S1 (ESI†). Those conformations that couldn't be identified as one of the 14 conformations listed in Table S1 (ESI†) are denoted as an undefined (U) conformation. The probabilities of observing a glucose unit in HP-β-CDs in these conformations, both in the absence and presence of the guest in the CD cavity, are shown in Fig. S2 (ESI†).We observe a small fraction of inverted chair (I) conformations in HP-β-CDs except for the h04, h05, h07 and h33′ systems. The population of I is the highest in h72, the most distorted HP-β-CD, but the population of U (undefined conformation) is also higher than the N (native 4C1) population. In systems like h23, h40, h34, h43 and h44, in the absence of a guest molecule, the population of the inverted chair conformation was seen to be about 5–25%, whereas in the presence of the guest p-nitrophenol in the cavity, glucose inversion didn't happen. But in the h32, h33, h50 and h7x systems, the change in the inverted chair population in the absence and presence of the guest is minimal, but we have observed an increase in the circularity and the cavity volume in the presence of a guest molecule in these systems (Fig. 3). In the case of h33′, although we didn't observe any inverted chair conformations in the absence of a guest inside the cavity, there was an ∼80 Å3 increase in cavity volume in the presence of the guest. When we simulated h70 with three different guests – neral, p-nitrophenol and phenolphthalein – varying in size and chemistry, we observed only about a 5–10% change in the glucose conformational population in the absence and the presence of these three guests (Fig. 6a). Still, we had observed >150 Å3 increase in cavity volume in the presence of these guests compared to that in the absence of the guest. It has to be noted that the population of the undefined conformations in all the cases is >20%. This suggests that glucose conformational changes do not necessarily explain the cavity closure and opening in the absence and the presence of a guest molecule, respectively.
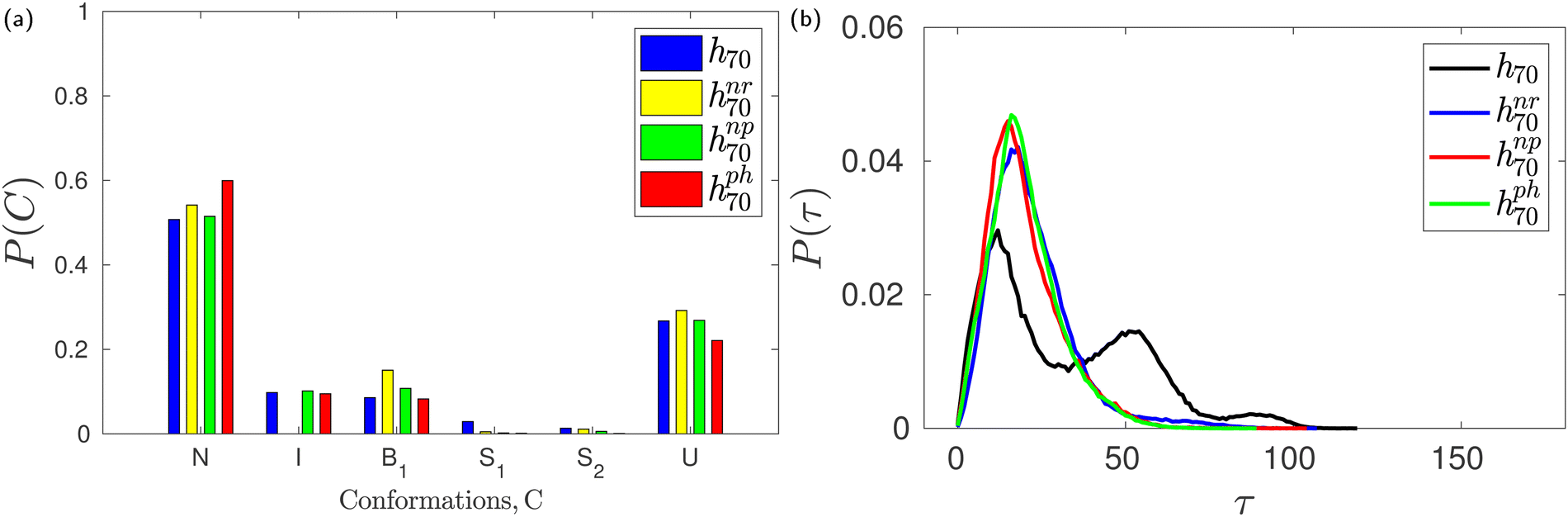 | ||
| Fig. 6 (a) The fractional population of glucose units in different conformations and (b) the distribution of tilt angle in the absence (h70) and presence of guests neral (hnr70), p-nitrophenol (hnp70) and phenolphthalein (hph70) inside a HP-β-CD. | ||
3.4 Structural changes upon substitution are attributed to glucose tilting
During careful inspection of the simulation trajectory, we observed an inclination of the glucose units with their O6 atoms pointing towards the interior of the CD cavity. Interestingly, such inward inclination of the O6 unit and subsequent encapsulation in the cavity is observed in one of the glucose units in the crystal structure of mono-substituted HP-β-CD.29 The inclination of the glucose units with respect to the CD axis is quantified in terms of the tilt angle (τ) as defined in the Methods section. The tilt angle of the glucose units in native β-CD lies in the range of 0–50° with an average value of 13.8°. Fig. 6b shows the distribution of tilt angle for h70 in the absence and presence of guest molecules like neral, p-nitrophenol and phenolphthalein. We can clearly see that in the absence of the guest, the distribution is trimodal with peaks at 15°, 50° and 90°. Contrastingly, in the presence of either of the guests, the distribution becomes unimodal with 99% of the glucose units exhibiting a tilt angle of <50°.Furthermore, Fig. 7 confirms that all the HP-β-CDs that demonstrated cavity structural distortion in the absence of a guest exhibit multi-modal tilt angle distribution. In the presence of the guest p-nitrophenol, the tilt angle distribution (bottom panel of Fig. 7) becomes unimodal with the distribution ranging mostly within 50°. The systems like h72, h73, and h75, however, exhibited large tilt angles of 90–150° in Fig. 7f as the p-nitrophenol moved out of the cavity during the simulation. This clearly reveals that in the absence of a guest, the glucose units are highly flexible and are tilted more, while the presence of a guest inside the CD cavity arrests the tilting motion, thereby restoring the open cavity structure.
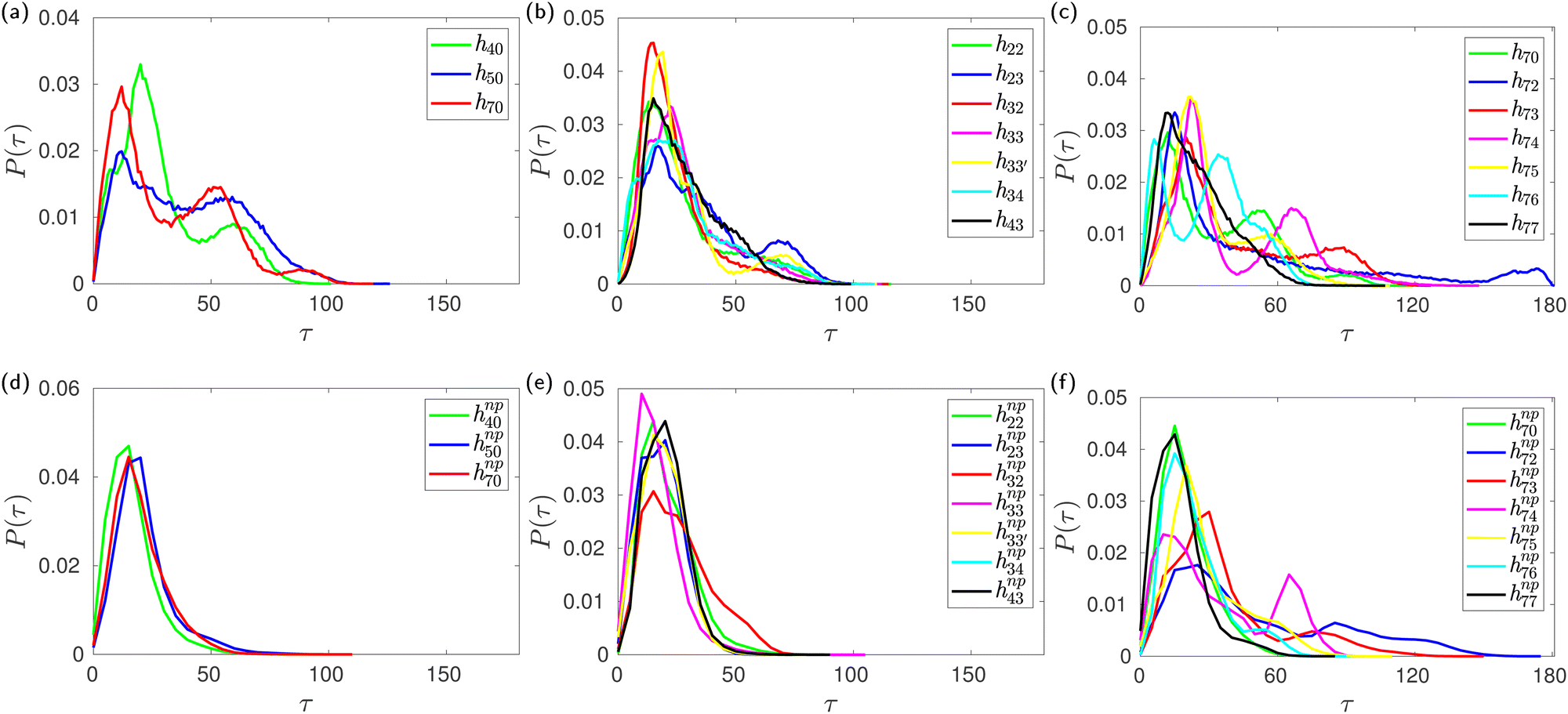 | ||
| Fig. 7 Probability distribution of the tilt angle of glucose units in HP-β-CDs in the absence (top panel) and the presence of the guest p-nitrophenol (bottom panel) inside the CD cavity: (a) and (d) substitution only in the secondary hydroxyl rim; (b) and (e) glucose units are substituted either at the primary or the secondary hydroxyl rim, but not both; (c) and (f) increase in the substitution in the primary hydroxyl rim when all the glucose units in the secondary hydroxyl rim are substituted. | ||
To verify whether glucose tilting explains the multiple basins we have observed in the (ϕ,ψ) free-energy surfaces (Fig. 5d–f) of HP-β-CDs without guests, we computed the conditional joint probability of the glycosidic dihedral angles when the adjacent glucose units are untilted as in native β-CD (N–N), when only the first unit is tilted (T–N), and when only the second unit is tilted (N–T). The glucose units with τ > 50° are characterized as tilted (T) units. We have very rarely observed two adjacent glucose units in HP-β-CDs as tilted units. When a native-like glucose unit is followed by a tilted unit (N–T), both the ϕ and ψ angles are shifted to lower values, thereby occupying the basin with values lower than 90° for both the dihedral angles. When a tilted unit is followed by a untilted glucose unit (T–N), a shift in the ψ value closer to 180° is observed, whereas the ϕ remains almost unchanged with respect to the native β-CD (Fig. 8).
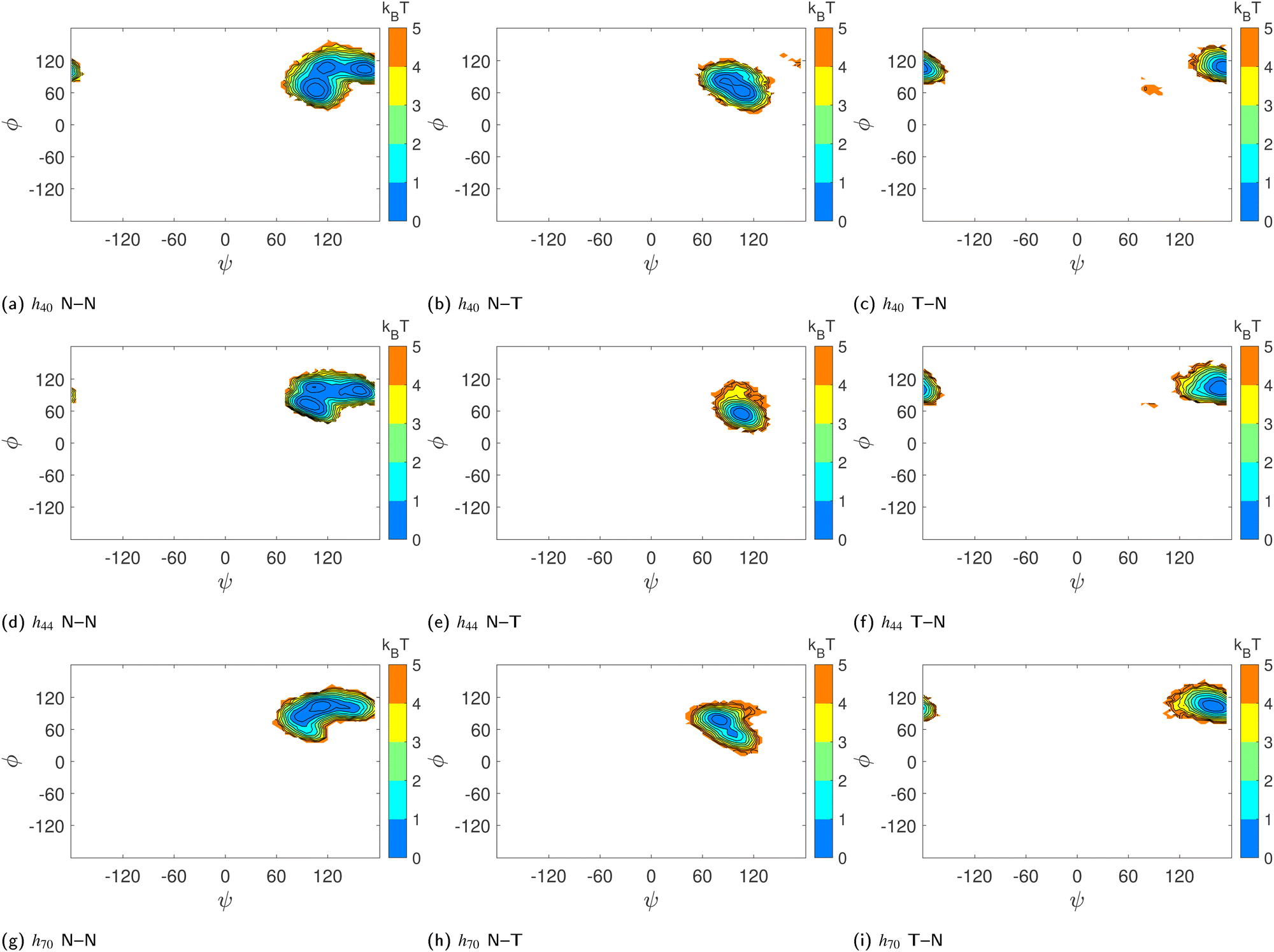 | ||
| Fig. 8 Effect of glucose tilting on the (ϕ,ψ) population of (a)–(c) h40, (d)–(f) h44 and (g)–(i) h70 systems: (a), (d) and (g) – two adjacent glucose units are native-like or untilted (N–N); (b), (e) and (h) – an untilted glucose unit followed by a tilted glucose unit (N–T); (c), (f) and (i) – tilted glucose unit followed by an untilted glucose unit (T–N). The colour bar shows the energy values in kBT and the contour lines are drawn for every 1 kJ mol−1. | ||
However, the population of two adjacent normal untilted glucose units (N–N) shows three basins; this could be attributed to the presence of other tilted units in the vicinity of those normal units. Any N–N groups directly connected to a tilted unit on either side shows a basin in the same region as the N–T population. However, the impact of other tilted units not directly connected to the N–N neighbours could not be ascertained definitively. Since the tilted units alter the shape of the CD ring, the structural parameters of the N–N units somehow have to adjust as they are covalently bonded to form a cyclic structure. Furthermore, we investigated the influence of glucose tilting on the angle between three consecutive glycosidic oxygen atoms, i.e., the δ angle. Fig. 9 shows the distribution of the δ angle for the N–N, N–T and T–N populations of h40, h44 and h70 in the absence of a guest. As in the ϕ–ψ distribution, we see that the N–T and T–N populations exhibit distinct distributions with a mean value of 90° and 150°, respectively. Once again, however, the distribution for the N–N population samples the whole range, possibly influenced by the neighboring tilted glucose units and to maintain the covalently bonded cyclic CD structure.
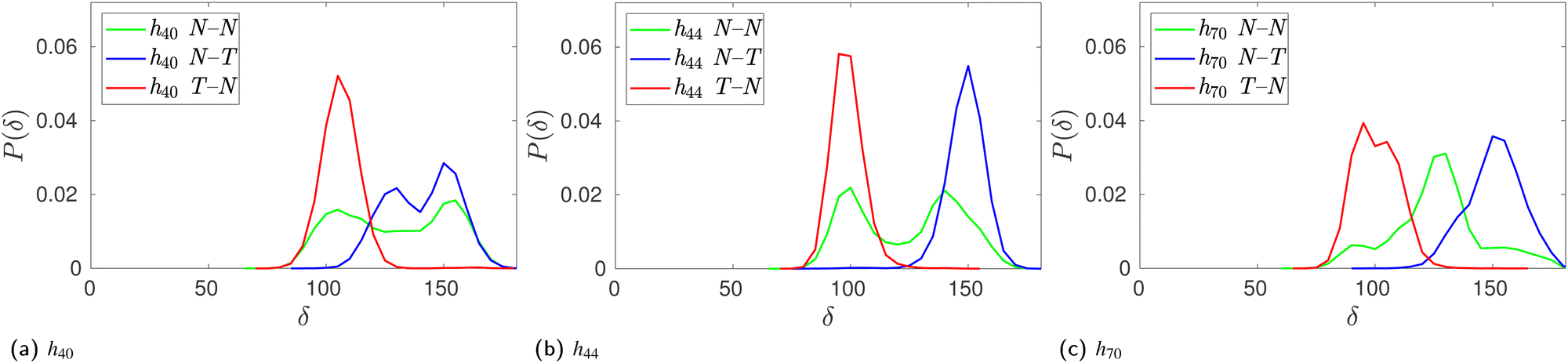 | ||
| Fig. 9 Effect of glucose tilting on the angle between three consecutive glycosidic oxygen atoms, δ, of HP-β-CDs when two adjacent glucose units are native-like or untilted (N–N, green curve), an untilted glucose is followed by a tilted glucose (N–T, blue curve), and a tilted glucose is followed by an untilted glucose (T–N, red curve): (a) h40; (b) h44; and (c) h70. | ||
3.5 Enhanced CD–water hydrogen bonding improves aqueous solubility of HP-β-CDs
So far we have been discussing the effect of structural changes in HP-β-CDs. To understand how the substitution of 2-HP enhances the solubility of HP-β-CDs, it is necessary to examine the interactions between CD and water. Table S2 (ESI†) lists the number of intramolecular CD–CD and intermolecular CD–water hydrogen bonds and the CD–water interaction energy along with its electrostatic and dispersion contributions for β-CD and HP-β-CDs. In native β-CD, the secondary rim forms 5.6 hydrogen bonds, to which its rigidity is attributed. The number of CD–water hydrogen bonds is ∼32, predominantly contributed by the interactions of the primary hydroxyl groups with the surrounding water medium. The average number of hydrogen bonds for h04 is exactly the same as for the native CD, whereas for h05 and h07, the number of CD–CD hydrogen bonds is a little lower, but the number of CD–water hydrogen bonds is a little higher than for the native CD. With substitution at the secondary rim, there is a dramatic increase of 7–14 CD–water hydrogen bonds; concurrently, the number of CD–CD hydrogen bonds reduces to only 1–3. The maximum number of CD–water hydrogen bonds is observed for h77; however, there is no clear-cut trend in the increase in CD–water hydrogen bonds or the decrease in CD–CD hydrogen bonds in h7x systems. Similarly, it is indeed puzzling to see the increase in the number of CD–CD hydrogen bonds when the number of substitutions is increased in the secondary hydroxyl rim in hCD0x systems.Therefore, we closely inspected the components of the CD molecules that form intramolecular hydrogen bonds (Fig. S3, ESI†). In h0x, all intramolecular hydrogen bonds are mainly within the secondary rim, with a few instances of the substituted 2-HP groups hydrogen bonding among them and with the primary rim. The number of hydrogen bonds within the secondary rim, counterintuitively, increases when the substitution in the secondary rim increases in hx0 systems. This increase results from the higher incidence of the favoured orientation of the O3 and O2 atoms that facilitates the O3–O2 hydrogen bond;4 it is to be noted that substituted O2 cannot donate hydrogen bonds. We also observed some unusual hydrogen bonds in hx0 systems, i.e., among the primary hydroxyl groups and between the primary hydroxyl group and the ring oxygen, unseen in native β-CD, arising because of the structural deformation and conformational changes in the glucose units. Such unusual hydrogen bonding involving ring oxygen is found in other HP-β-CDs, like h22, h23, h32, h43, h72, and h75. The formation of a hydrogen bond between the primary and secondary rims seen in h32 is also a clear consequence of the tilting of glucose units.
Every substituted 2-HP group in all HP-β-CDs forms on average about 2 hydrogen bonds with the water medium. The 2-HP group is found to point outward of the cavity, existing in the trans conformation with respect to the O2 or O6 atoms to which it is attached in the secondary and primary rim, respectively. As a result, the O2 and O6 atoms point inward to the CD cavity and are not water accessible; therefore, they do not participate in hydrogen bonding, unlike their unsubstituted counterparts. The partial substitution of the secondary rim, as in h40, disrupts the hydrogen bond belt of the secondary hydroxyl rim, as is evident from the drop in the number of O2–O3 hydrogen bonds from 5.6 to 1 in the native β-CD. This allows both the 2-HP groups and O3 atoms to form hydrogen bonds with water leading to an ∼2–3 increase in the number of hydrogen bonds with water for every 2-HP substitution at the secondary rim, whereas in the native β-CD, O3 can only form 1 hydrogen bond with water. On the other hand, every O6 atom in the primary hydroxyl rim of native β-CD forms on average two hydrogen bonds with water; however, upon substitution only at the primary rim, the O6 atoms become inaccessible to water. So, the hydrogen bonds between water and the 2-HP groups replace the hydrogen bonds between water and O6 atoms in β-CD; hence, no drastic increase in the number of CD–water hydrogen bonds is observed in h0x systems.
The average interaction energy of all CD molecules with water along with their electrostatic and dispersion contributions are tabulated in Table S2 (ESI†). The average interaction energy of a single α-glucose molecule (Gromos56A6CarboR) in an SPC/E water box is determined to be −254 kJ mol−1, which is comparable to the value of −286 kJ mol−1 obtained for CHARMM36 α-glucose molecule in SPC/E water.59 The difference is attributable to the difference in the number of glucose–water hydrogen bonds; the former forms 8.5 hydrogen bonds and the latter forms 9.4 hydrogen bonds with water. The interaction energy of native CD is −1125 kJ mol−1, which is about 4.4 times more attractive that of a single glucose unit. The dominant contribution (∼86%) to the interaction energies arises from the electrostatic interactions. The interaction energy of all HP-β-CDs is more favorable than the native-CD, leading to the increase in aqueous solubility of HP-β-CDs reported experimentally. Since the guest p-nitrophenol does not form any hydrogen bonds with HP-β-CDs (Table S3, ESI†), there is no significant change in the number of CD–water hydrogen bonds (Fig. 10a) and the CD interaction energy (Fig. 10b) in the presence of the guest.
 | ||
| Fig. 10 Similarity between (a) the average number of hydrogen bonds per HP-β-CD molecule, 〈nHB〉, and (b) the average CD–water interaction energies, 〈εCD–water〉, for HP-β-CDs in the absence (x-axis) and the presence (y-axis) of the guest p-nitrophenol inside the CD cavity. The extent of correlation for the average CD–water interaction energies of HP-β-CDs in the absence (red) and the presence (blue) of the guest p-nitrophenol inside the CD cavity with (c) the average number of CD–water hydrogen bonds, 〈nHBCD–water〉, and (d) the average solvent accessible surface area, 〈SASA〉. | ||
Interestingly, when 2-HP is substituted only in the primary rim (h0x), there is a clear trend of an increase in the dispersion interaction, while the extent of the change in the electrostatic interaction is comparatively small (Table S2, ESI†). On the other hand, when the substitution is only at the secondary rim, i.e., (hx0) systems, there is no significant change in the dispersion interaction, while the alteration in the electrostatic interaction is significant. This trend is similar to that observed for the number of CD–water hydrogen bonds. In fact, we see a strong correlation between the number of hydrogen bonds formed by the CD with the surrounding water molecules and its interaction energy (Fig. 10c); such a correlation has been observed earlier for cavity-bound water4 and for various osmolytes in 20% aqueous solutions.59 A point to note is that from the slope of the linear fit in Fig. 10c, we shouldn't interpret 38.7 kJ mol−1 as the energy for a single CD–water hydrogen bond because the interaction energy, εCD–water, was actually computed as the sum of the dispersion and electrostatic interactions between the CD and all the water molecules in the system. We have also examined the correlation between the average CD–water interaction energy with the average solvent access surface area (SASA) of HP-β-CDs in the presence and absence of a guest within the CD cavity (Fig. 10d). In the presence of a guest, there is a slight increase in the SASA as the cavity opens, yet the cavity is not completely solvent accessible as the guest is occupying the cavity. As expected, the SASA is also well correlated to the average CD–water interaction energy; however, the correlation between the average CD–water interaction energy and the average number of CD–water hydrogen bonds is stronger. Overall, our findings suggest that the solubility and structural stability of CDs depends on the conflicting interplay of hydrogen bonding of CD with water and within itself; therefore, care is required in fine-tuning them to obtain the CD structure with the desired properties required for the application.
4 Conclusions
The 2-HP substitution of CD enhances the interaction energy of CD in solution corroborating the increase in aqueous solubility of HP-β-CDs. The solubility enhancement is affected by the drop in the number of CD–CD hydrogen bonds in the secondary rim and the corresponding increase in the number of CD–water hydrogen bonds. The breach of the O2–O3 hydrogen bond belt in the secondary hydroxyl rim escalates the conformational flexibility of CD and its constituent glucose units. Furthermore, the key structural parameters change dramatically, sampling a broader conformational space, finally leading to the distortion of the CD structure and closure of its inner cavity. As opposed to the earlier conceived notion, sugar puckering does not influence the cavity closure. It is the glucose tilting that brings about this effect. However, in the presence of a guest molecule, the interaction energy of CD in solution remains unchanged due to the absence of a hydrogen bond between the host and guest, thus the number of CD–water hydrogen bonds remains unaffected. The tilting of the glucose units is prohibited in the presence of a guest thereby leading to reopening of the cavity. However, the conformational changes of the glucose units are independent of the cavity occupancy and remain the same even in the presence of a guest molecule.Our detailed investigation on the structural changes in HP-β-CDs with different degrees of substitution has revealed that substitution in the secondary hydroxyl rim is required for solubility enhancement. Substitution at the primary hydroxyl rim does not enhance solubility as substitution at the primary rim makes O6 atoms inaccessible to water, so hydrogen bonds between O6 and water molecules are only replaced by those between the 2-hydroxypropyl group and water. As a result, the CD–water interactions remain unaltered. The strategic substitution in the primary rim, however, aids in improving the stability of the CD structure. For example, in the case of partial substitution at both hydroxyl rims of CDs, the HP-β-CDs with alternate glucose units substituted in either rim deforms less than that with the same glucose units di-substituted.
The lack of good crystals of HP-β-CDs had inhibited the experimental investigation of HP-β-CDs. The higher tilting of glucose units along the glycosidic plane in HP-β-CDs not only deforms the CD cavity, but could also promote agglomeration at higher concentrations, thereby preventing the formation of high-quality crystals; it may not be a desired feature for therapeutics as well. Although the inclusion of a guest leads to stabilisation of the CD cavity leading to the formation of CD-guest crystals in most cases, the most structurally deformed systems like h72, h73, h74 and h75, however, fail to encapsulate any guest molecules in their cavities. Our study clearly points out the importance of hydrogen bonding interactions and delineates the conflicting mechanism underpinning the cavity shape and structure vs. CD aqueous solubility. It is the intramolecular interactions that promote an open cavity, while the intermolecular hydrogen bonding with water is required for high solubility. It is necessary for materials scientists to understand this trade-off when manufacturing HP-β-CDs of the desired degree for their application.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by Women Leading IITM Initiative Grant 2021–2023. We also thank High Performance Computing Environment (HPCE), IIT Madras for providing the computational resources.Notes and references
- W. Saenger, Cyclodextrin inclusion compounds in research and industry, Angew. Chem., Int. Ed. Engl., 1980, 19(5), 344–362 CrossRef.
- J. Szejtli, Introduction and general overview of cyclodextrin chemistry, Chem. Rev., 1998, 98(5), 1743–1754 CrossRef CAS PubMed.
- W. Saenger, J. Jacob, K. Gessler, T. Steiner, D. Hoffmann and H. Sanbe, et al., Structures of the common cyclodextrins and their larger analogues beyond the doughnut, Chem. Rev., 1998, 98(5), 1787–1802 CrossRef CAS PubMed.
- A. A. Sandilya, U. Natarajan and M. H. Priya, Molecular view into the cyclodextrin cavity: Structure and hydration, ACS Omega, 2020, 5(40), 25655–25667 CrossRef CAS PubMed.
- D. W. Armstrong, J. R. Faulkner Jr and S. M. Han, Use of hydroxypropyl-and hydroxyethyl-derivatized β-cyclodextrins for the thin-layer chromatographic separation of enantiomers and diastereomers, J. Chromatogr. A, 1988, 452, 323–330 CrossRef CAS PubMed.
- W. J. Shieh and A. R. Hedges, Properties and applications of cyclodextrins, J. Macromol. Sci., Part A: Pure Appl. Chem., 1996, 33(5), 673–683 CrossRef.
- Z. Xiao, Y. Zhang, Y. Niu, Q. Ke and X. Kou, Cyclodextrins as carriers for volatile aroma compounds: a review, Carbohydr. Polym., 2021, 269, 118292 CrossRef CAS PubMed.
- S. S. Jambhekar and P. Breen, Cyclodextrins in pharmaceutical formulations I: Structure and physicochemical properties, formation of complexes, and types of complex, Drug Discovery Today, 2016, 21(2), 356–362 CrossRef CAS PubMed.
- N. Zafar, H. Fessi and A. Elaissari, Cyclodextrin containing biodegradable particles: From preparation to drug delivery applications, Int. J. Pharm., 2014, 461(1–2), 351–366 CrossRef CAS PubMed.
- B. Gidwani and A. Vyas, A comprehensive review on cyclodextrin-based carriers for delivery of chemotherapeutic cytotoxic anticancer drugs, BioMed Res. Int., 2015, 2015, 198268 Search PubMed.
- N. Qiu, X. Li and J. Liu, Application of cyclodextrins in cancer treatment, J. Inclusion Phenom. Macrocyclic Chem., 2017, 89(3–4), 229–246 CrossRef CAS.
- J. Gebhardt, C. Kleist, S. Jakobtorweihen and N. Hansen, Validation and comparison of force fields for native cyclodextrins in aqueous solution, J. Phys. Chem. B, 2018, 122(5), 1608–1626 CrossRef CAS PubMed.
- C. Cézard, X. Trivelli, F. Aubry, F. Djedani-Pilard and F. Y. Dupradeau, Molecular dynamics studies of native and substituted cyclodextrins in different media: 1. Charge derivation and force field performances, Phys. Chem. Chem. Phys., 2011, 13(33), 15103–15121 RSC.
- J. Szejtli, The properties and potential uses of cyclodextrin derivatives, J. Inclusion Phenom. Mol. Recognit. Chem., 1992, 14(1), 25–36 CrossRef CAS.
- M. Malanga, J. Szemán, É. Fenyvesi, I. Puskás, K. Csabai and G. Gyémánt, et al., Back to the future: a new look at hydroxypropyl β-cyclodextrins, J. Pharm. Sci., 2016, 105(9), 2921–2931 CrossRef CAS PubMed.
- S. Concha-Santos, S. Pérez-Casas, P. Brocos and Á. Piñeiro, Testing the effect of the cavity size and the number of molecular substitutions in host–guest complexes formed by 2-hydroxypropyl-cyclodextrins and n-octyl-β-D-glucopyranoside, J. Chem. Thermodyn., 2013, 67, 112–119 CrossRef CAS.
- Á. Buvári-Barcza, D. Bodnár-Gyarmathy and L. Barcza, Hydroxypropyl-β-cyclodextrins: Correlation between the stability of their inclusion complexes with phenolphthalein and the degree of substitution, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 18(3), 301–306 CrossRef.
- J. Pitha, C. T. Rao, B. Lindberg and P. Seffers, Distribution of substituents in 2-hydroxypropyl ethers of cyclomaltoheptaose, Carbohydr. Res., 1990, 200, 429–435 CrossRef CAS PubMed.
- C. T. Rao, B. Lindberg, J. Lindberg and J. Pitha, Substitution in β-cyclodextrin directed by basicity: preparation of 2-O-and 6-O-[(R)-and (S)-2-hydroxypropyl] derivatives, J. Org. Chem., 1991, 56(3), 1327–1329 CrossRef CAS.
- C. Yuan, B. Liu and H. Liu, Characterization of hydroxypropyl-β-cyclodextrins with different substitution patterns via FTIR, GC–MS, and TG–DTA, Carbohydr. Polym., 2015, 118, 36–40 CrossRef CAS PubMed.
- G. Dufour, B. Evrard and P. De Tullio, Rapid quantification of 2-hydroxypropyl-β-cyclodextrin in liquid pharmaceutical formulations by 1H nuclear magnetic resonance spectroscopy, Eur. J. Pharm. Sci., 2015, 73, 20–28 CrossRef CAS PubMed.
- N. M. Davies, G. Wang and I. G. Tucker, Evaluation of a hydrocortisone/hydroxypropyl-β-cyclodextrin solution for ocular drug delivery, Int. J. Pharm., 1997, 156(2), 201–209 CrossRef CAS.
- T. Loftsson, M. E. Brewster and M. Masson, Role of cyclodextrins in improving oral drug delivery, Am. J. Drug Delivery, 2004, 2(4), 261–275 CrossRef CAS.
- H. Matsuda and H. Arima, Cyclodextrins in transdermal and rectal delivery, Adv. Drug Delivery Rev., 1999, 36(1), 81–99 CrossRef CAS PubMed.
- T. O. Carpenter, A. Gerloczy and J. Pitha, Safety of parenteral hydroxypropyl β-cyclodextrin, J. Pharm. Sci., 1995, 84(2), 222–225 CrossRef CAS PubMed.
- C. L. Kear, J. Yang, D. A. Godwin and L. A. Felton, Investigation into the mechanism by which cyclodextrins influence transdermal drug delivery, Drug Dev. Ind. Pharm., 2008, 34(7), 692–697 CrossRef CAS PubMed.
- J. Pitha and J. Pitha, Amorphous water-soluble derivatives of cyclodextrins: nontoxic dissolution enhancing excipients, J. Pharm. Sci., 1985, 74(9), 987–990 CrossRef CAS PubMed.
- K. Harata, C. T. Rao and J. Pitha, Crystal structure of 6-O-[(R)-2-hydroxypropyl] cyclomaltoheptaose and 6-O-[(S)-2-hydroxypropyl] cyclomaltoheptaose, Carbohydr. Res., 1993, 247, 83–98 CrossRef CAS PubMed.
- K. Harata, C. T. Rao, J. Pitha, K. Fukunaga and K. Uekama, Crystal structure of 2-O-[(S)-2-hydroxypropyl] cyclomaltoheptaose, Carbohydr. Res., 1991, 222, 37–45 CrossRef CAS PubMed.
- A. Tačić, I. Savić, V. Nikolić, I. Savić, S. Ilić-Stojanović, D. Ilić, S. Petrović, M. Popsavin and A. Kapor, Inclusion complexes of sulfanilamide with β-cyclodextrin and 2-hydroxypropyl-β-cyclodextrin, J. Inclusion Phenom. Macrocyclic Chem., 2014, 80, 113–124 CrossRef.
- J. Li, S. Zhang, Y. Zhou, S. Guan and L. Zhang, Inclusion complexes of fluconazole with β-cyclodextrin and 2-hydroxypropyl-β-cyclodextrin in aqueous solution: preparation, characterization and a structural insight, J. Inclusion Phenom. Macrocyclic Chem., 2016, 84, 209–217 CrossRef CAS.
- F. Hirayama, Z. Wang and K. Uekama, Effect of 2-hydroxypropyl-β-cyclodextrin on crystallization and polymorphic transition of nifedipine in solid state, Pharm. Res., 1994, 11, 1766–1770 CrossRef CAS PubMed.
- J. S. Kim, Study of flavonoid/hydroxypropyl-β-cyclodextrin inclusion complexes by UV-Vis, FT-IR, DSC, and X-Ray diffraction analysis, Prev. Nutr. Food Sci., 2020, 25(4), 449 CrossRef CAS PubMed.
- J. A. Hamilton and L. Chen, Crystal structures of inclusion complexes of β-cyclodextrin with S-(+)-and (R)-(−)-fenoprofen, J. Am. Chem. Soc., 1988, 110(13), 4379–4391 CrossRef CAS.
- M. G. Usha and R. J. Wittebort, Structural and dynamical studies of the hydrate, exchangeable hydrogens, and included molecules in β and γ-cyclodextrins by powder and single-crystal deuterium magnetic resonance, J. Am. Chem. Soc., 1992, 114(5), 1541–1548 CrossRef CAS.
- T. Aree and S. Jongrungruangchok, Enhancement of antioxidant activity of green tea epicatechins in β-cyclodextrin cavity: Single-crystal X-ray analysis, DFT calculation and DPPH assay, Carbohydr. Polym., 2016, 151, 1139–1151 CrossRef CAS PubMed.
- T. Aree, β-Cyclodextrin encapsulation of nortriptyline HCl and amitriptyline HCl: Molecular insights from single-crystal X-ray diffraction and DFT calculation, Int. J. Pharm., 2020, 575, 118899 CrossRef CAS PubMed.
- K. Betlejewska-Kielak, E. Bednarek, A. Budzianowski, K. Michalska and J. K. Maurin, Comprehensive characterisation of the flurbiprofen/β-cyclodextrin inclusion complex using X-ray techniques and NMR spectroscopy, J. Mol. Struct., 2023, 1285, 135450 CrossRef CAS.
- J. Blanchard and S. Proniuk, Some important considerations in the use of cyclodextrins, Pharm. Res., 1999, 16(12), 1796 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: An advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 4(1), 17 CAS.
- W. Khuntawee, M. Karttunen and J. Wong-ekkabut, A molecular dynamics study of conformations of beta-cyclodextrin and its eight derivatives in four different solvents, Phys. Chem. Chem. Phys., 2017, 19(35), 24219–24229 RSC.
- H. S. Hansen and P. H. Hünenberger, A reoptimized GROMOS force field for hexopyranose-based carbohydrates accounting for the relative free energies of ring conformers, anomers, epimers, hydroxymethyl rotamers, and glycosidic linkage conformers, J. Comput. Chem., 2011, 32(6), 998–1032 CrossRef CAS PubMed.
- W. Plazinski, A. Lonardi and P. H. Hünenberger, Revision of the GROMOS 56A6CARBO force field: Improving the description of ring-conformational equilibria in hexopyranose-based carbohydrates chains, J. Comput. Chem., 2016, 37(3), 354–365 CrossRef CAS PubMed.
- A. K. Malde, L. Zuo, M. Breeze, M. Stroet, D. Poger and P. C. Nair, et al., An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0, J. Chem. Theory Comput., 2011, 7(12), 4026–4037 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, Molecular dynamics with coupling to an external bath, J. Chem. Phys., 1984, 81(8), 3684–3690 CrossRef CAS.
- S. Nosé, A molecular dynamics method for simulations in the canonical ensemble, Mol. Phys., 1984, 52(2), 255–268 CrossRef.
- W. G. Hoover, Canonical dynamics: Equilibrium phase-space distributions, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef PubMed.
- M. Parrinello and A. Rahman, Polymorphic transitions in single crystals: A new molecular dynamics method, J. Appl. Phys., 1981, 52(12), 7182–7190 CrossRef CAS.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes, J. Comput. Phys., 1977, 23(3), 327–341 CrossRef CAS.
- H. L. Strauss and H. M. Pickett, Conformational structure, energy, and inversion rates of cyclohexane and some related oxanes, J. Am. Chem. Soc., 1970, 92(25), 7281–7290 CrossRef CAS.
- H. S. Hansen and P. H. Hünenberger, Using the local elevation method to construct optimized umbrella sampling potentials: calculation of the relative free energies and interconversion barriers of glucopyranose ring conformers in water, J. Comput. Chem., 2010, 31(1), 1–23 CrossRef CAS PubMed.
- Á. Buvári-Barcza, E. Rák, Á. Mészáros and L. Barcza, Complex formation of hydroxypropyl-β-cyclodextrins with p-nitrophenol, in Proceedings of the Eighth International Symposium on Cyclodextrins: Budapest, Hungary, March 31–April 2, 1996. Springer; 1996. pp. 187–188.
- B. A. Barczane and L. Barcza, β-cyclodextrin inclusion complexes: effect of the host substitution on molecular recognition, Acta Pharm. Hung., 2000, 70(3–6), 82–88 CAS.
- S. H. Choi, E. N. Ryu, J. J. Ryoo and K. P. Lee, FT-Raman spectra of o-, m-, and p-nitrophenol included in cyclodextrins, J. Inclusion Phenom. Macrocyclic Chem., 2001, 40, 271–274 CrossRef CAS.
- A. Buvari-Barcza, E. Rak, A. Meszaros and L. Barcza, Inclusion complex formation of p-nitrophenol and p-nitrophenolate with hydroxypropyl-β-cyclodextrins, J. Inclusion Phenom. Mol. Recognit. Chem., 1998, 32(4), 453–459 CrossRef CAS.
- Á. Buvári-Barcza and L. Barcza, Influence of the guests, the type and degree of substitution on inclusion complex formation of substituted β-cyclodextrins, Talanta, 1999, 49(3), 577–585 CrossRef PubMed.
- D. Cremer and J. A. Pople, General definition of ring puckering coordinates, J. Am. Chem. Soc., 1975, 97(6), 1354–1358 CrossRef CAS.
- T. Steiner and W. Saenger, Closure of the cavity in permethylated cyclodextrins through glucose inversion, flipping, and kinking, Angew. Chem., Int. Ed., 1998, 37(24), 3404–3407 CrossRef CAS PubMed.
- S. Sundar, A. A. Sandilya and M. H. Priya, Unraveling the influence of osmolytes on water hydrogen-bond network: From local structure to graph theory analysis, J. Chem. Inf. Model., 2021, 61(8), 3927–3944 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp05354g |
| This journal is © the Owner Societies 2024 |