
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An alternative catalytic cycle for selective methane oxidation to methanol with Cu clusters in zeolites†
Mario
Gallego
 ,
Avelino
Corma
and
Mercedes
Boronat
*
,
Avelino
Corma
and
Mercedes
Boronat
*
Instituto de Tecnología Química, Universitat Politècnica de València – Consejo Superior de Investigaciones Científicas, Av de los Naranjos s/n, 46022 Valencia, Spain. E-mail: boronat@itq.upv.es
First published on 31st January 2024
Abstract
The partial oxidation of methane to methanol catalyzed by Cu-exchanged zeolites involves at present a three-step procedure that requires changing reaction conditions along the catalytic cycle. In this work we present an alternative catalytic cycle for selective methane conversion to methanol using as active species small Cu5 clusters supported on CHA zeolite. Periodic DFT calculations show that molecular O2 is easily activated on Cu5 clusters producing bi-coordinated O atoms able to dissociate homolytically a CH bond from CH4 and to react with the radical-like non-adsorbed methyl intermediate formed producing methanol, while competitive overoxidation to CO2 is energetically disfavored. The present mechanistic study opens a new avenue to design catalytic materials based on their ability to stabilize radical species.
Introduction
The availability and low cost of methane, the main component of natural gas and biogas, makes it attractive as potential raw material to obtain other valuable chemicals, but its efficient transformation into liquid products such as methanol, easier to transport and able to act as versatile chemical feedstock, is extremely challenging. The current industrial process for methane upgrading requires the production of synthesis gas (CO + H2) via steam reforming but research efforts are focusing on finding an alternative, low temperature pathway to transform methane into methanol.1–3 Trying to mimic the selectivity afforded by the methane monooxygenase (pMMO) enzyme, Cu-exchanged zeolites have been successfully applied to partially oxidize methane to methanol with molecular O2, yet at the expense of low methane conversion to avoid over oxidation to CO2.4–12 Experimental and computational studies agree that the active species are dimeric [Cu2O]2+ or trimeric [Cu3O3]2+ Cu–oxo clusters stabilized by electrostatic interaction with framework Al sites.13–17 Moreover, a positive role of mixed [Cu2AlO3]2+ clusters formed by reaction of Cu cations with extra framework Al (EFAL) species in Cu-MOR has been recently reported.18–20A different approach to achieve high selectivity in oxidation reactions relies on the use of metal clusters of low atomicity as active species. Indeed, the electronic and catalytic properties of clusters composed by a precise small number of atoms are different from those of larger nanoparticles and isolated metal cations, and can be further modulated by interactions with organic ligands or inorganic supports.21–25 More specifically, it has been computationally predicted and experimentally confirmed that Cu5 clusters are able to activate molecular O2 and generate reactive O species while avoiding deep irreversible oxidation,26–29 which makes them promising candidates for a variety of catalytic applications.30–32 Previous computational work in our group indicates that O species bicoordinatively adsorbed at the edges of isolated Cu5 clusters favour the homolytic dissociation of the methane C–H bond and the selective formation of methanol following a radical rebound mechanism similar to that described for Cu-exchanged zeolites.6,33,34 However, the reactivity trends found for isolated metal clusters in gas phase might change due to their interaction with the organic or inorganic materials employed to stabilize them prior to catalytic application. Periodic DFT calculations of Cu5 and Cu7 clusters stabilized within the microporous structure of a CHA zeolite indicate that the morphology and electronic properties of the supported clusters varies with the Al content in the framework, and this is reflected in the catalytic activity towards O2 dissociation. A compromise between activity and stability against deep oxidation was found for Cu5 clusters encapsulated in a CHA zeolite containing two Al atoms per unit cell.35 In the present work, we use this catalyst model to investigate the complete mechanism of methane oxidation to methanol on a realistic material, considering not only the complete catalytic cycle but also the most important competitive pathways, namely catalyst deactivation by formation of surface methoxy groups and undesired overoxidation of methanol.
Computational details
Periodic density functional theory (p-DFT) calculations were performed with the VASP 5.2 code,36,37 using the PBE functional38 and including Grimme's D3 correction39 for dispersion interactions. The valence density was expanded in a plane wave basis set with a kinetic energy cutoff of 600 eV, and the effect of the core electrons in the valence density was taken into account by means of the projected augmented wave (PAW) formalism.40 All calculations are spin-polarized and, except otherwise stated, the optimizations converged to a doublet state with only one unpaired electron. Electronic energies were converged to 10−6 eV and geometries were optimized until forces on atoms were <0.01 eV A−1. Integration in the reciprocal space was carried out at the Γ k-point of the Brillouin zone.The catalyst was represented by means of a Cu5 cluster stabilized within the cavity of a zeolite with the CHA structure, that crystalizes in a hexagonal unit cell with Al2Si34O72 composition and lattice parameters a = b = 13.8026 Å, c = 15.0753 Å, α = β = 90° and γ = 120°. In all calculations, the positions of all atoms in the system were fully optimized without restrictions, and all stationary points were characterized by frequency calculations to confirm their nature and to obtain the thermal corrections to calculate Gibbs energies. Transition states were located using the DIMER algorithm,41,42 and the frequency calculations were used in these structures to confirm that the imaginary frequency corresponds to the desired reaction coordinate. The Hessian matrix and vibrational frequencies were calculated using density functional perturbation theory (DFPT).43
Results and discussion
Pathways for selective methane oxidation to methanol
Based on our previous theoretical investigation of O2 dissociation on zeolite-supported Cu5 clusters (Table S1, ESI†)35 and on the weak interaction of methane with Cu5 clusters,34 the catalyst model selected to start the mechanistic study consists of a partly oxidized Cu5O2 cluster stabilized within a cha cage containing two Al atoms in the framework (structure 1 in Fig. 1 and Fig. S1, ESI†). Due to the location of the Cu5 cluster close to the 8-ring window connecting two cha cages, CH4 only interacts favorably with one of the Cu atoms, forming structure 2 in Fig. 1 with an optimized Cu–C distance of 2.264 Å and a calculated Gibbs free interaction energy of −33 kJ mol−1. Since the two O atoms are not equivalent, four possible pathways labeled A to D were explored to convert methane into methanol on Cu5O2. | ||
| Fig. 1 Optimized geometries of minima and transition state structures involved in the oxidation of methane to methanol on a Cu5O2 cluster supported on CHA zeolite following different pathways A to D. Si and O atoms in the framework depicted as yellow and red wires, Al, Cu and reactant O depicted as light green, brown and red balls, respectively. Relative Gibbs energies at 478.15 K given in brackets in kJ mol−1. | ||
The first route considered, labeled A, proceeds through formation of a radical-like intermediate (structure 3 in Fig. 1 and Table S2, ESI†) in which the methyl group formed after the C–H bond dissociation is not interacting with the Cu cluster. The C–H bond length increases from 1.096 Å in reactant 2 to 1.461 Å in TS(2 → 3) and 1.906 Å in intermediate 3, while the O–H distance follows the opposite trend and decreases from 3.039 Å in 2 to 1.120 Å in TS(2 → 3) and finally 1.000 Å in 3. The calculated activation Gibbs energy is 102 kJ mol−1 (see Table 1) and intermediate 3 is 90 kJ mol−1 less stable than 2.
In a second step, the non-adsorbed methyl fragment reacts with the adsorbed hydroxyl group forming a methanol molecule mono-coordinated to the Cu5 cluster at a Cu–O distance of 2.065 Å (structure 4 in Fig. 1 and Table S2, ESI†). The optimized C–O distance in TS(3 → 4) is 2.077 Å, and due to the low stability of intermediate 3, the calculated activation Gibbs energy for the CO bond formation step is only 46 kJ mol−1 (see Table 1). An alternative pathway B involving the same O atom occurs without breaking the Cu–C coordination existing in reactant 2. In the transition state for C–H dissociation, TS(2 → 6), the optimized Cu–C, C–H and O–H distances are 1.981, 1.443 and 1.290 Å, respectively, and in intermediate 6 the methyl group is attached to Cu with a Cu–C bond length of 1.904 Å. This additional interaction forces the geometry of the O atom involved in the H transfer, leading to a higher activation Gibbs energy for C–H dissociation of 126 kJ mol−1, while it stabilizes significantly structure 6 (see relative energies in Table S1, ESI†). Such stabilization of 6 leads to a higher activation barrier, 94 kJ mol−1, for C–O bond formation through the same transition state structure involved in pathway A yielding adsorbed methanol 4. Comparison of the Gibbs energy profiles for routes A and B at 478.15 K (light and dark blue lines in Fig. 2a) shows that the main difference between them is the stability of the methyl intermediate, and that the highest barrier for the overall process is in both cases below 130 kJ mol−1.
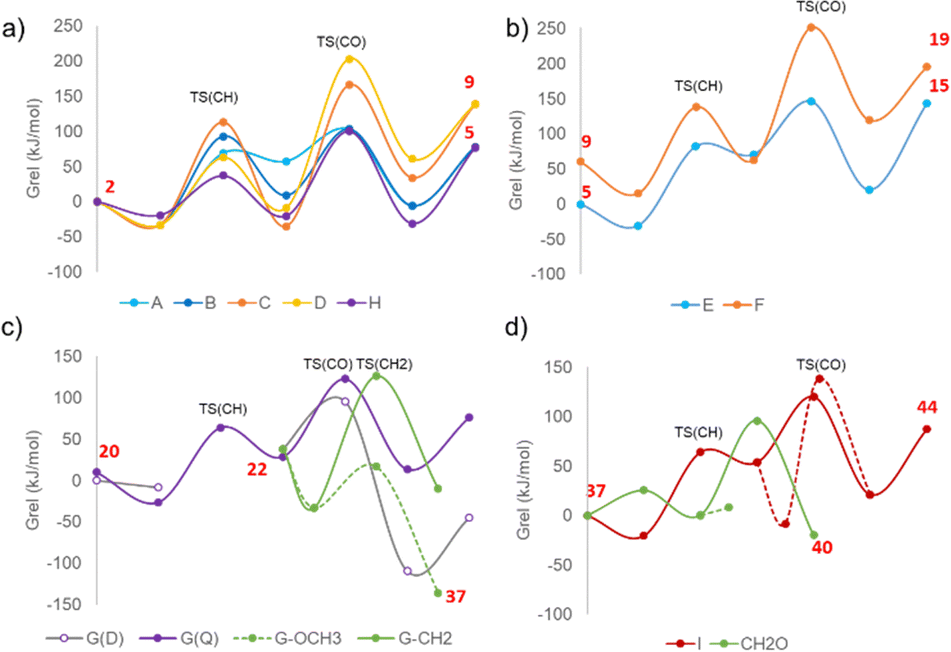 | ||
| Fig. 2 Calculated Gibbs energy profiles at 478.15 K for methane oxidation and some competitive processes on (a) Cu5O2, (b) Cu5O, (c) Cu5O3 and (d) Cu5–OH–OCH3 clusters supported on CHA zeolite. The transition states for methane C–H bond dissociation (TS(CH)) and C–O bond formation (TS(CO)) are indicated on the plots. The red numbers correspond to some relevant structures involved in the mechanisms. | ||
The two routes involving the other O atom of the Cu5O2 cluster also differ in the nature of the transition state for the C–H bond dissociation, with a non-adsorbed methyl group in TS(2 → 7) following pathway C and with methyl interacting with Cu in TS(2 → 10) following pathway D, which is reflected in the calculated activation Gibbs energies, 147 and 96 kJ mol−1, respectively. However, none of the two intermediates formed is a radical-like species, and the methyl group is in both cases attached to the Cu5O2 cluster, bi-coordinated in 7 with Cu–C distances of 1.991 and 2.076 Å, and mono-coordinated in 10 with a Cu–C distance of 1.911 Å. As a result of these interactions, intermediate 7 is 27 kJ mol−1 more stable than 10, and the activation Gibbs energies for the subsequent C–O bond formation step are really high in both cases, 203 and 213 kJ mol−1 following pathways C and D, respectively. The optimized C–O distances in TS(7 → 8) and TS(10 → 11) are similar, 2.143 and 2.130 Å, and the reaction product is methanol adsorbed either on an axial (structure 8) or an apical (structure 11) Cu atom, the former system being 27 kJ mol−1 more stable than the latter. The Gibbs energy profiles for route C (orange) and D (yellow) in Fig. 2a are less favorable than those described for pathways A and B, mostly due to the high barriers found for the reaction of adsorbed methyl and hydroxyl groups.
After the first CH4 oxidation and CH3OH desorption, two non-equivalent Cu5O structures, 5 and 9, appear as the initial species that further react with a second CH4 molecule following pathways E and F (see Fig. 2b and 3). In the Eley–Rideal pathway labelled E, CH4 does not interact with the Cu5O cluster, as indicated by the optimized value of 3.739 Å for the Cu–C distance in structure 12. The C–H bond dissociation produces a metastable radical-like intermediate 13 with the CH and HO distances evolving from 1.376 and 1.170 Å in TS(12 → 13) to 1.974 and 0.994 Å in intermediate 13, and with not too high activation Gibbs energies for CH bond breaking and CO bond formation, 113 and 75 kJ mol−1, respectively. In contrast, when following pathway F, CH4 adsorbs directly on the Cu5O cluster with an optimized Cu–C distance of 2.291 Å in structure 16, which decreases to 2.027 Å in TS(16 → 17) and to only 1.932 Å in the mono-coordinated methyl intermediate 17. During the C–H bond dissociation step the cluster rearranges and becomes planar, a geometry that remains during the formation of the new CO bond making this step energetically difficult, with a Gibbs energy of activation of 189 kJ mol−1 (see Table 1).
 | ||
| Fig. 3 Optimized geometries of minima and transition state structures involved in methane oxidation following pathways E and F on Cu5O clusters and pathway G on a Cu5O3 cluster supported on CHA zeolite. Si and O atoms in the framework depicted as yellow and red wires, Al, Cu, reactant O, C and H atoms depicted as light green, brown, red, gray and white balls, respectively. Relative Gibbs energies at 478.15 K given in brackets in kJ mol−1. | ||
While the results presented up to now indicate that CH4 could be oxidized to CH3OH through pathways A or B followed by E with activation Gibbs energies below 130 kJ mol−1, the highly exothermic adsorption of O2 on the Cu5O structure 5 obtained at the end of the first cycle, and its subsequent dissociation with an activation energy of only 13 kJ mol−1 produces a stable Cu5O3 system (structure 20 in Fig. 3 and Fig. S2, ESI†) whose reactivity should be explored. Further oxidation of Cu5O3 to Cu5O5 is not energetically favored (Fig. S2 in the ESI†) and due to the high O coverage on the cluster CH4 does not adsorb on any Cu atom, but interacts with a bi-coordinated O atom with an optimized O–H distance of 2.470 Å (structure 21 in Fig. 3), allowing only an Eley–Rideal mechanism labelled G. The high O coverage leads to a stabilization of the quadruplet state for the system. For structure 20 the doublet (D) state is 10 kJ mol−1 more stable than the quadruplet (Q), and the order is reversed in structure 21 for which the (D) state is 18 kJ mol−1 less stable than the quadruplet (Q). This is relevant because the transition state for C–H bond dissociation TS(21 → 22) yielding a non-adsorbed methyl radical (structure 22) could only be localized on the Q potential energy surface. The corresponding activation Gibbs energy is only 90 kJ mol−1, and intermediate 22 is 55 kJ mol−1 less stable than reactant 21. The Gibbs energy difference between 22 and 21 on the D energy surface is quite similar, 46 kJ mol−1, as well as all the optimized geometries, suggesting easy crossing between both surfaces. From that point, the transition state for CO bond formation TS(22 → 23) and the product CH3OH are clearly more stable on the D potential energy surface (see gray and violet Gibbs energy profiles in Fig. 2c), and the calculated activation barrier for the second step of the process is only 57 kJ mol−1.
Desorption of the methanol product from structure 23 leaves a Cu5O2 cluster, labeled 24 in Fig. 3, that is 37 kJ mol−1 less stable than the catalyst model 1 considered initially. To check whether the different conformation of the Cu atoms in 1 and 24 might lead to relevant reactivity differences an additional pathway H was explored on this system (Fig. S3, ESI†). CH4 does not adsorb on the cluster in the reactant structure 25, but after the dissociation of the CH bond through TS(25 → 26) with a calculated activation Gibbs energy of 64 kJ mol−1 (see Table 1), the resulting methyl group remains mono-coordinated to a Cu atom with an optimized Cu–C bond length of 2.049 Å. The relatively high stability of intermediate 26 results in an activation Gibbs energy of 129 kJ mol−1 for the CO bond formation step producing methanol, which after desorbing leaves a Cu5O system (structure 28) 36 kJ mol−1 less stable than 5 and 24 kJ mol−1 more stable than 9. Altogether, the Gibbs energy profile for pathway H is similar to that of pathway B as depicted in Fig. 2a, and confirms that CH4 oxidation to CH3OH can proceed on Cu5 clusters supported on CHA zeolite with activation energies below ∼130 kJ mol−1. Interestingly, the smoothest energy profile with the lowest activation barriers is obtained on the Cu5O3 cluster with the highest O coverage (Fig. 2c).
Pathways for competing processes
Since the main drawback of the methane to methanol reaction is the easy over-oxidation of the product to CO2, the feasibility of this and other undesired processes was investigated. Two main competing reactions were explored, the dissociation of a second C–H bond in adsorbed CH3OH or CH3 starting the unselective oxidation to CO2, and the formation of surface methoxy (CH3O) groups leading to catalyst blocking because they are not able to abstract a H from co-adsorbed OH to form CH3OH.34 As a consequence of the small size of the Cu5 cluster and its structural deformation due to interactions with the zeolite framework or with adsorbed species, CH3OH is usually obtained mono-coordinated to one Cu atom and far from any additional O atom able to abstract H, with the only exception of structure 8 involved in pathway C (Fig. 1 and 4a) in which the shortest O–H distance is 4.196 Å. Dissociation of the C–H bond is possible but endothermic by 54 kJ mol−1 and requires surpassing an activation barrier of 167 kJ mol−1. | ||
| Fig. 4 Optimized geometries of minima and transition state structures involved in competitive processes on Cu5O2 clusters supported on CHA zeolite. (a) CH bond dissociation in adsorbed methanol. (b) CH bond dissociation and methoxy formation from adsorbed methyl intermediates. Si and O atoms in the framework depicted as yellow and red wires, Al, Cu and reactant O depicted as light green, brown and red balls, respectively. Relative Gibbs energies at 478.15 K given in brackets in kJ mol−1. | ||
Alternatively, the adsorbed methyl groups formed as intermediates in pathways B, C and D (structures 6, 7 and 10, in Fig. 1 and 4b) are always closer to the co-adsorbed O atom, with O–H distances of 4.101, 4.063 and 4.131 Å, respectively. The reaction is endothermic when starting from structures 7 or 10, with the CH2 species being 27 and 97 kJ mol−1 less stable than the initial CH3 groups and with calculated activation Gibbs energies of 176 and 149 kJ mol−1, respectively. However, starting from structure 6 the process is slightly exothermic and involves an activation Gibbs energy of 105 kJ mol−1, indicating that formation of structure 30 with one CH2 and two OH groups could compete with methanol production (see energy profiles in Fig. S4a, ESI†). On the other hand, migration of a methyl group from a Cu atom in 6, 7 and 10 to a proximal O atom yielding methoxy intermediate structures 33 or 34 in Fig. 4 is endothermic by 66, 99 and 72 kJ mol−1, respectively, and requires surpassing high activation barriers of 199, 134 and 109 kJ mol−1 (see Table 1). Therefore, the probability to form undesired methoxy species on Cu5O2 clusters supported on CHA zeolite seems to be low.
The situation is different on Cu5O3. The non-adsorbed methyl radical present in structure 22 might adsorb on the Cu5O3 cluster forming a new Cu–C bond and generating a system 71 kJ mol−1 more stable (structure 35 in Fig. 5a). Dissociation of one C–H bond in this system to produce structure 36 is endothermic and involves a high activation Gibbs energy of 159 kJ mol−1, but migration of the methyl group to one of the three surface O atoms requires a much lower barrier of 50 kJ mol−1 and produces a very stable methoxy intermediate (structure 37 in Fig. 5a). Then, dissociation of a C–H bond in the methoxy group only requires 25 kJ mol−1 and yields formaldehyde (structure 38) which may either desorb from the cluster or transfer a H to a neighbouring OH group to form H2O and an adsorbed OCH group (structure 40) with an activation Gibbs energy of 96 kJ mol−1 (green profile in Fig. 2d).
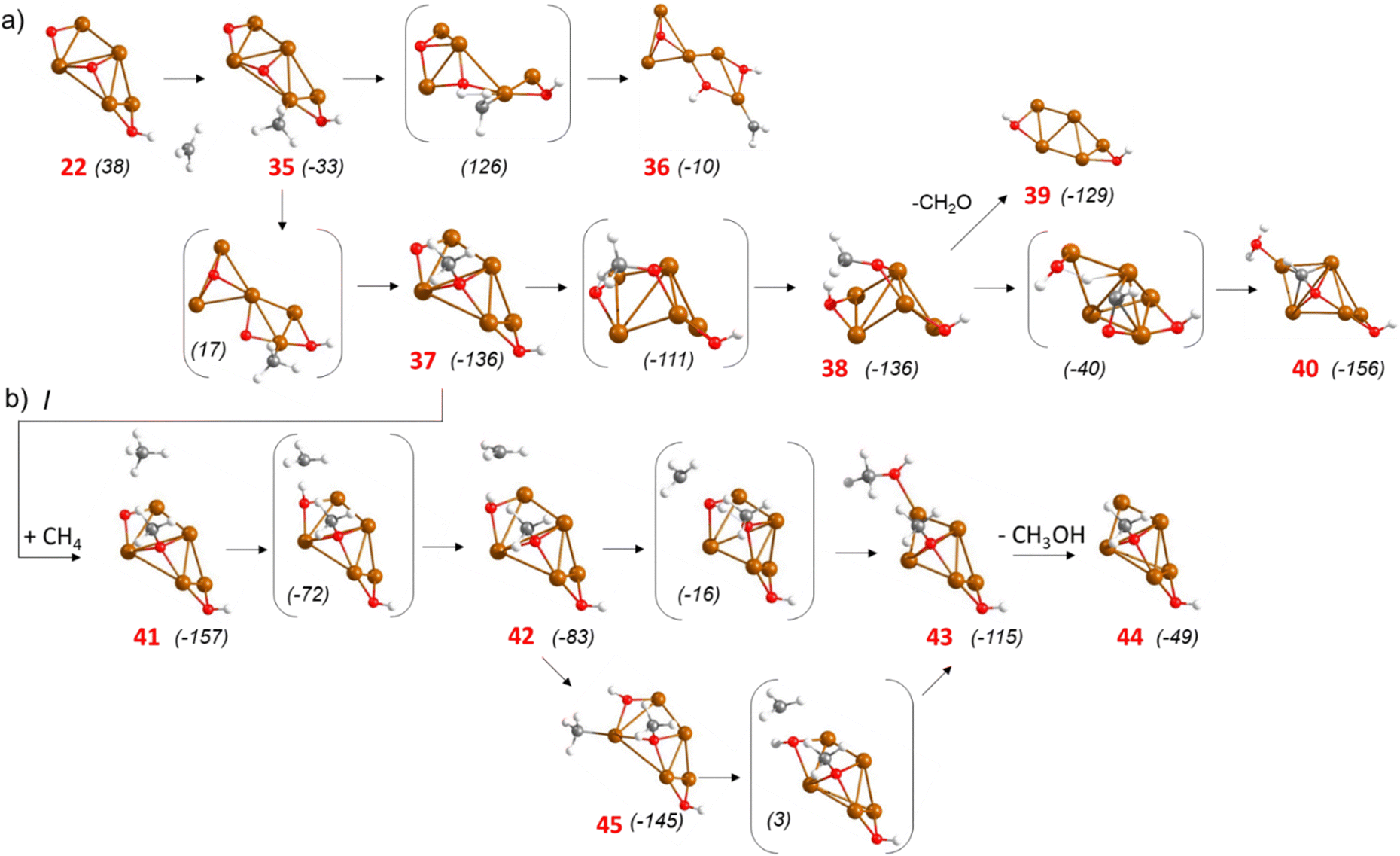 | ||
| Fig. 5 Optimized geometries of minima and transition state structures involved in (a) competing processes on a Cu5O3 cluster supported on CHA zeolite and (b) pathway I for methane oxidation on Cu5–OH–OCH3 catalytic system. Al, Cu and reactant O depicted as light green, brown and red balls, respectively. Relative Gibbs energies at 478.15 K given in brackets in kJ mol−1. | ||
These data suggest that, once formed, methanol is stable against further oxidation, but some methoxy intermediates possibly appearing on Cu5 clusters with three adsorbed O atoms are prone to break their C–H bonds and generate either aldehyde or more oxidized products. On the other hand, besides the methoxy and a hydroxyl group, structure 37 contains a bi-coordinated O atom at the cluster edge that reacts with an additional methane molecule following pathway I depicted in Fig. 5b. The activation Gibbs energy for the first C–H bond breaking is only 85 kJ mol−1 and formation of methanol from a non-adsorbed methyl radical 42 or from a more stable mono-coordinated methyl (structure 45) requires surpassing barriers of 67 and 142 kJ mol−1, respectively (red profile in Fig. 2d).
To facilitate the comparative discussion of all the processes investigated, kinetic constants k calculated at 478.15 K for all elementary steps are summarized in Table 2, and the most relevant reactions contributing to methane conversion are represented in Scheme 1. The results presented indicate that methanol can be produced efficiently via pathway A through a radical-like intermediate, with the kinetic constant k for the second best pathway B being two orders of magnitude lower. Only a bifurcation in path B leading to an adsorbed CH2 group might compete when starting from Cu5O2 system (narrow orange arrow in Scheme 1). After the first methanol desorption leaving Cu5O, a second catalytic cycle producing methanol and regenerating the Cu5 cluster (path E) is also favourable. Alternatively, the fast adsorption and dissociation of O2 leading to Cu5O3 also produces methanol efficiently (path G). Even the competing formation of a methoxy group (Cu5O(OH)OCH3 system) might produce methanol (path I), as well as formaldehyde as a by-product (wide orange arrow in Scheme 1).
| Model | Pathway | k (CH) (s−1) | k (CO) (s−1) | k (OCH3) (s−1) | k (CH2) (s−1) |
|---|---|---|---|---|---|
| a To formaldehyde. | |||||
| Cu5O2 | A | 71.8 | 9.40 × 10+7 | ||
| B | 1.71 × 10−1 | 5.37 × 10+2 | 1.82 × 10−9 | 43.4 | |
| C | 1.12 × 10−3 | 6.65 × 10−10 | 2.29 × 10−2 | 1.12 × 10−3 | |
| D | 3.25 × 10+2 | 5.37 × 10−11 | 12.3 | 6.77 × 10−4 | |
| H | 7.60 × 10+6 | 4.69 × 10−1 | |||
| Cu5O | E | 4.51 | 6.39 × 10+4 | ||
| F | 2.84 × 10−1 | 2.89 × 10−8 | |||
| Cu5O3 | G (Q) | 1.47 × 10+3 | 5.37 × 10+2 | 3.44 × 10+7 | 4.26 × 10−5 |
| I | 5.15 × 10+3 | 4.78 × 10+5 | 1.85 × 10+10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
||
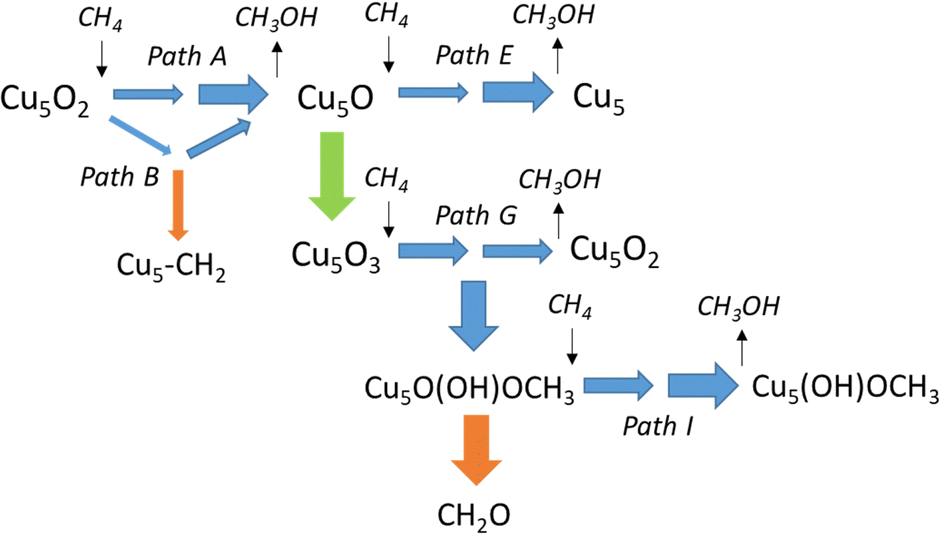 | ||
| Scheme 1 Most relevant pathways for methane oxidation to methanol on a Cu5O2 cluster supported on CHA zeolite. The width of the arrows represents the order of magnitude of the kinetic constant for each elementary step. Blue arrows are steps leading to methanol, orange arrow indicates catalyst blocking by methoxy formation, green arrow indicates the addition of O2 to the cluster. | ||
Conclusions
The mechanism of the selective oxidation of methane to methanol and of the most relevant competing reactions that decrease the selectivity of this challenging reaction has been theoretically investigated using periodic DFT calculations. The proposed catalyst consists of small Cu5 clusters stabilized within a CHA zeolite with a high Si/Al ratio of 17. The high stability against deep oxidation of such clusters facilitates the dissociation of the methane C–H bond and desorption of the methanol formed, making difficult its over-oxidation to CO2. Among the different pathways explored, the one proceeding through a radical-like non-adsorbed methyl intermediate is the most efficient, allowing to close the catalytic cycle with Gibbs activation energies lower than 115 kJ mol−1, of the same order as those reported experimentally for Cu-exchanged zeolites. The present findings open an avenue to design catalytic materials based on their ability to stabilize radical species.Author contributions
M. B. and A. C. conceptualized the study. M. G. carried out the DFT calculations supervised by M. B. All the authors discussed the results and contributed to the preparation, writing and revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish Government through MCIN PID2020-112590GB-C21 and TED2021-130739B-I00 (MCIN/AEI/FEDER,UE). We thank Red Española de Supercomputación (RES) and Servei d’Informatica of the University of Valencia for computational resources and technical support. M. G. thanks the Spanish MCIN for his fellowship PRE2018-083547.Notes and references
- P. Del Campo, C. Martínez and A. Corma, Activation and Conversion of Alkanes in the Confined Space of Zeolite-type Materials, Chem. Soc. Rev., 2021, 50, 8511–8595 RSC.
- A. I. Olivos-Suarez, A. Szécsényi, E. J. M. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, Strategies for the Direct Catalytic Valorization of Methane Using Heterogeneous Catalysis: Challenges and Opportunities, ACS Catal., 2016, 6, 2965–2981 CrossRef CAS.
- E. V. Kondratenko, T. Peppel, D. Seeburg, V. A. Kondratenko, N. Kalevaru, A. Martin and S. Wohlrab, Methane Conversion into Different Hydrocarbons or Oxygenates: Current Status and Future Perspectives in Catalyst Development and Reactor Operation, Catal. Sci. Technol., 2017, 7, 366–381 RSC.
- R. Balasubramanian and A. C. Rosenzweig, Structural and Mechanistic Insights into Methane Oxidation by Particulate Methane Monooxygenase, Acc. Chem. Res., 2007, 40, 573–580 CrossRef CAS PubMed.
- M. H. Mahyuddin, Y. Shiota, A. Staykov and K. Yoshizawa, Theoretical Overview of Methane Hydroxylation by Copper−Oxygen Species in Enzymatic and Zeolitic Catalysts, Acc. Chem. Res., 2018, 51, 2382–2390 CrossRef CAS PubMed.
- M. H. Mahyuddin, Y. Shiota and K. Yoshizawa, Methane Selective Oxidation to Methanol by Metal-Exchanged Zeolites: a Review of Active Sites and their Reactivity, Catal. Sci. Technol., 2019, 9, 1744–1768 RSC.
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, Selective Oxidation of Methane by the Bis(μ-oxo)dicopper Core Stabilized on ZSM-5 and Mordenite Zeolites, J. Am. Chem. Soc., 2005, 127, 1394–1395 CrossRef CAS PubMed.
- M. J. Wulfers, S. Teketel, B. Ipek and R. F. Lobo, Conversion of methane to methanol on copper- containing small-pore zeolites and zeotypes, Chem. Commun., 2015, 51, 4447–4450 RSC.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. Van Bokhoven, Selective Anaerobic Oxidation of Methane Enables Direct Synthesis of Methanol, Science, 2017, 356, 523–527 CrossRef CAS PubMed.
- K. Narsimhan, K. Iyoki, K. Dinh and Y. Román-Leshkov, Catalytic Oxidation of Methane into Methanol over Copper-Exchanged Zeolites with Oxygen at Low Temperature, ACS Cent. Sci., 2016, 2, 424–429 CrossRef CAS PubMed.
- K. T. Dinh, M. M. Sullivan, K. Narsimhan, P. Serna, R. J. Meyer, M. Dincă and Y. Román-Leshkov., Continuous Partial Oxidation of Methane to Methanol Catalyzed by Diffusion-Paired Copper Dimers in Copper-Exchanged Zeolites, J. Am. Chem. Soc., 2019, 141, 11641–11650 CrossRef CAS.
- R. J. Brenneis, E. P. Johnson, W. Shi and D. L. Plata, Atmospheric- and Low-Level Methane Abatement via an Earth-Abundant Catalyst, ACS Environ. Au, 2022, 2, 223–231 CrossRef CAS PubMed.
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, A [Cu2O]2 Core in Cu-ZSM-5, the Active Site in the Oxidation of Methane to Methanol, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908–18913 CrossRef CAS PubMed.
- S. Grundne, M. A. C. Markovit, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Single-site Trinuclear Copper Oxygen Clusters in Mordenite for Selective Conversion of Methane to Methanol, Nat. Commun., 2015, 6, 7546–7554 CrossRef PubMed.
- G. Li, P. Vassilev, M. Sanchez-Sanchez, J. A. Lercher, E. J. M. Hensen and E. A. Pidko, Stability and Reactivity of Copper Oxo-Clusters in ZSM-5 Zeolite for Selective Methane Oxidation to Methanol, J. Catal., 2016, 338, 305–312 CrossRef CAS.
- D. K. Pappas, A. Martini, M. Dyballa, K. Kvande, S. Teketel, K. A. Lomachenko, R. Baran, P. Glatzel, B. Arstad and G. Berlier, et al., The Nuclearity of the Active Site for Methane to Methanol Conversion in Cu-Mordenite: A Quantitative Assessment, J. Am. Chem. Soc., 2018, 140, 15270–15278 CrossRef CAS PubMed.
- U. Engedahl, H. Grönbeck and A. Hellman, First-Principles Study of Oxidation State and Coordination of Cu-Dimers in Cu-SSZ-13 during Methane-to-Methanol Reaction Conditions, J. Phys. Chem. C, 2019, 123, 26145–26150 CrossRef CAS.
- M. Dyballa, D. K. Pappas, K. Kvande, E. Borfecchia, B. Arstad, P. Beato, U. Olsbye and S. Svelle, On How Copper Mordenite Properties Govern the Framework Stability and Activity in the Methane-to-Methanol Conversion, ACS Catal., 2019, 9, 365–375 CrossRef CAS.
- I. Lee, M. S. Lee, L. Tao, T. Ikuno, R. Khare, A. Jentys, T. Huthwelker, C. N. Borca, A. Kalinko, O. Y. Gutiérrez, N. Govind, J. L. Fulton, J. Z. Hu, V. A. Glezakou, R. Rousseau, M. Sanchez-Sanchez and J. A. Lercher, Activity of Cu−Al−Oxo Extra-Framework Clusters for Selective Methane Oxidation on Cu-Exchanged Zeolites, JACS Au, 2021, 1, 1412–1421 CrossRef CAS PubMed.
- L. Tao, E. Khramenkova, I. Lee, T. Ikuno, R. Khare, A. Jentys, J. L. Fulton, A. A. Kolganov, E. A. Pidko, M. Sanchez-Sanchez and J. A. Lercher, Speciation and Reactivity Control of Cu-Oxo Clusters via Extraframework Al in Mordenite for Methane Oxidation, J. Am. Chem. Soc., 2023, 145, 17710–17719 CrossRef CAS PubMed.
- L. Liu and A. Corma, Metal Catalysts for Heterogeneous Catalysis: from Single Atoms to Nanoclusters and Nanoparticles, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- E. Fernández and M. Boronat, Sub Nanometer Clusters in Catalysis, J. Phys.: Condens. Matter, 2019, 31, 013002 CrossRef.
- C. Dong, Y. Li, D. Cheng, M. Zhang, J. Liu, Y. G. Wang, D. Xiao and D. Ma, Supported Metal Clusters: Fabrication and Application in Heterogeneous Catalysis, ACS Catal., 2020, 10, 11011–11045 CrossRef CAS.
- H. Rong, S. Ji, J. Zhang, D. Wang and Y. Li, Synthetic Strategies of Supported Atomic Clusters for Heterogeneous Catalysis, Nat. Commun., 2021, 11, 5884–58817 CrossRef.
- J. Jasik, A. Fortunelli and S. Vajda, Exploring the materials space in the smallest particle size range: from heterogeneous catalysis to electrocatalysis and photocatalysis, Phys. Chem. Chem. Phys., 2022, 24, 12083–12115 RSC.
- E. Fernández, M. Boronat and A. Corma, Trends in the Reactivity of Molecular O2 with Copper Clusters: Influence of Size and Shape, J. Phys. Chem. C, 2015, 119, 19832–19846 CrossRef.
- P. Concepción, M. Boronat, S. García-García, E. Fernández and A. Corma, Enhanced Stability of Cu Clusters of Low Atomicity against Oxidation. Effect on the Catalytic Redox Process, ACS Catal., 2017, 7, 3560–3568 CrossRef.
- A. Zanchet, P. López-Caballero, A. O. Mitrushchenkov, D. Buceta, M. A. López-Quintela, A. W. Hauser and M. P. de Lara-Castells, On the Stability of Cu5 Catalysts in Air Using Multireference Perturbation Theory, J. Phys. Chem. C, 2019, 123, 27064–27072 CrossRef CAS PubMed.
- D. Buceta, S. Huseyinova, M. Cuerva, H. Lozano, L. J. Giovanetti, J. M. Ramallo-Lopez, P. Lopez-Caballero, A. Zanchet, A. O. Mitrushchenkov, A. W. Hauser, G. Barone, C. Huck-Iriart, C. Escudero, J. C. Hernandez-Garrido, J. J. Calvino, M. Lopez-Haro, M. P. de Lara-Castells, F. G. Requejo and M. A. Lopez-Quintela, Stability and Reversible Oxidation of Sub-Nanometric Cu5 Metal Clusters: Integrated Experimental Study and Theoretical Modeling, Chem. – Eur. J., 2023, e202301517, DOI:10.1002/chem.202301517.
- P. Lopez-Caballero, A. W. Hauser and M. P. de Lara-Castells, Exploring the Catalytic Properties of Unsupported and TiO2-Supported Cu5 Clusters: CO2 Decomposition to CO and CO2 Photoactivation, J. Phys. Chem. C, 2019, 123, 23064–23074 CrossRef CAS PubMed.
- E. Fernández, M. Boronat and A. Corma, The Crucial Role of Cluster Morphology on the Epoxidation of Propene Catalyzed by Cu5: A DFT Study, J. Phys. Chem. C, 2020, 124, 21549–21558 CrossRef.
- E. Fernández, M. Boronat and A. Corma, The 2D or 3D Morphology of Sub-Nanometer Cu5 and Cu8 Clusters Changes the Mechanism of CO Oxidation, Phys. Chem. Chem. Phys., 2022, 24, 4504–4514 RSC.
- H. A. Doan, X. Wang and R. Q. Snurr, Computational Screening of Supported Metal Oxide Nanoclusters for Methane Activation: Insights into Homolytic versus Heterolytic C−H Bond Dissociation, J. Phys. Chem. Lett., 2023, 14, 5018–5024 CrossRef CAS PubMed.
- M. Gallego, A. Corma and M. Boronat, Sub-nanometer Copper Clusters as Alternative Catalysts for the Selective Oxidation of Methane to Methanol with Molecular O2, J. Phys. Chem. A, 2022, 126, 4941–4951 CrossRef CAS PubMed.
- M. Gallego, A. Corma and M. Boronat, Influence of the zeolite support on the catalytic properties of confined metal clusters: a periodic DFT study of O2 dissociation on Cun clusters in CHA, Phys. Chem. Chem. Phys., 2022, 24, 30044–30050 RSC.
- J. Hafner, Ab-Initio Simulations of Materials Using VASP: Density-Functional Theory and Beyond, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Henkelman and H. Jónsson, A Dimer Method for Finding Saddle Points on High Dimensional Potential Surfaces using only First Derivatives, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef CAS.
- A. Heyden, A. T. Bell and F. J. Keil, Efficient Methods for Finding Transition States in Chemical Reactions: Comparison of Improved Dimer Method and Partitioned Rational Function Optimization Method, J. Chem. Phys., 2005, 123, 224101 CrossRef PubMed.
- S. Baroni, S. de Gironcoli, A. Dal Corso and P. Giannozzi, Phonons and Related Crystal Properties from Density-Functional Perturbation Theory, Rev. Mod. Phys., 2001, 73, 515–562 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Calculated energies and optimized geometries of multiple O2 adsorption and dissociation on Cu5. Relative energies and Gibbs energies of all structures involved in the study. Gibbs energy profiles for competing processes. See DOI: https://doi.org/10.1039/d3cp05802f |
| This journal is © the Owner Societies 2024 |