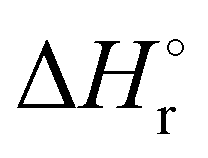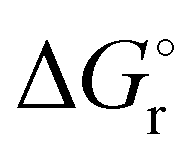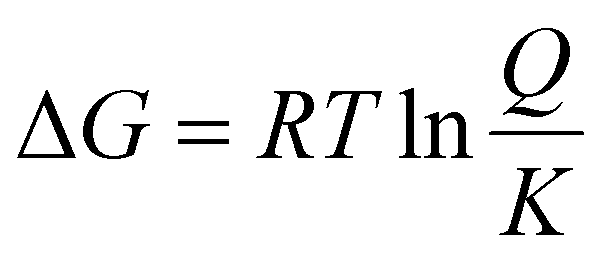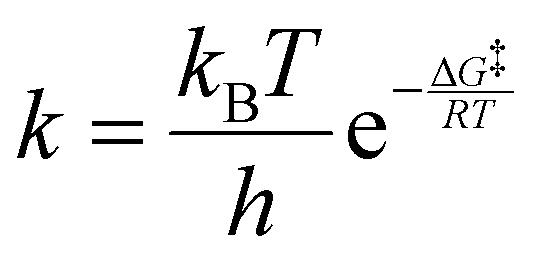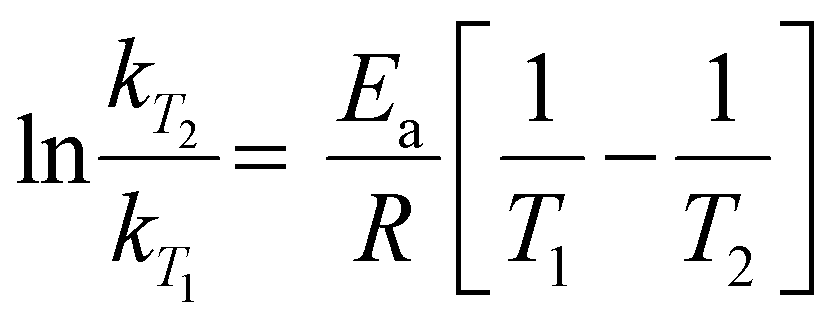Structural and thermochemical investigation of 1,3-bis(λ4-boraneyl)-1λ4,3λ4-imidazolidine adduct for chemical hydrogen storage†
Received
6th December 2023
, Accepted 16th May 2024
First published on 16th May 2024
Abstract
The structure, thermochemical properties and reaction pathways of a cyclic amine diborane complex (1,3-bis(λ4-boraneyl)-1λ4,3λ4-imidazolidine) were investigated using quantum chemical calculations. Structural and thermochemical analysis revealed that the simultaneous and spontaneous elimination of both hydrogen molecules from this complex is predicted to occur under thermoneutral conditions. This observation is further supported by the investigation of the BH3-catalysed dehydrogenation pathway. The calculated thermochemical parameters indicate that the energy requirements for hydrogen release from this complex are minimal, suggesting efficient hydrogen release capability under suitable conditions. Additionally, the activation barriers, ∼75 and ∼20 kJ mol−1 for the first and second dihydrogen release from the catalysed dehydrogenation reactions of this compound exhibit moderate kinetics, confirmed by kinetic studies. These findings and the ability of the system to easily release two molecules of dihydrogen emphasize the potential of 1,3-bis(λ4-boraneyl)-1λ4,3λ4-imidazolidine as a highly effective hydrogen storage material.
Introduction
Hydrogen is increasingly studied as a replacement for fossil fuels given these resources are rapidly depleting and there are grave environmental impacts associated with their burning.1 A cleaner, abundant and more sustainable energy source (hydrogen) is being actively pursued.2 However, hydrogen has a lower gravimetric density and complications associated with compressibility thus its application as an energy carrier is limited by ability to store it.3,4 One approach to rectify this issue is storing hydrogen chemically and generating it thermally or by reaction on demand.5–7 The adsorption of molecular H2 on metal cluster is of interest in hydrogen storage. A DFT study on chemisorption of H2 on neutral cobalt dimers showed that a significant quenching of the magnetic moment and a linear weakening of the metal–metal bond strength during H2 chemisorption.8 Ammonia borane, with a hydrogen content of 19.6 wt% has attracted attention in this context.9 The substitution effect of primary amines plays an important role in understanding the hydrogen elimination reaction because the exothermicity of the hydrogen release reaction is reduced by substitution on the nitrogen atom. The predicted lower reaction enthalpies and higher complexation energies for the H2 evolution of methyl ammonia borane suggested that the introduction of a methyl group on the nitrogen atom improves the reversibility of the system and inhibits the formation of unwanted byproducts such as BH3 and NH3.10
Derivatives of ammonia borane such as hydrazine borane7 and ethylene diamine bisborane6 were reported to have low dehydrogenation temperatures and high hydrogen purity. Hydrazine borane (HB) with a gravimetric capacity of 15.4 wt% H has been considered as a promising chemical hydrogen storage material. A synthesis method of HB was reported in 2012, utilizing a two-step process involving salt metathesis and solvent extraction-drying, resulting in an 80% yield and 99.6% purity. This method allowed for the production of HB within three days. The obtained HB was fully characterized using various techniques, including NMR, IR, XRD, TGA and DSC.11 Moreover, the transition metal catalyst rhodium(III) chloride was found to provide the highest catalytic performance in the metal catalysed hydrolysis of HB at room temperature. HB releases around 3.0 equivalent H2 in the presence of RhCl3 during hydrolysis with a H2 generation rate turn over frequency (TOF) of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. The catalytic hydrolysis of HB is stoichiometric, indicating that HB is capable of generating H2 effectively even just by adding water to solid HB.12 A very recent study demonstrates that MoOx-promoted NiPd nanocatalyst exhibits an excellent catalytic activity with almost 100% H2 selectivity in hydrogen release from HB in alkaline conditions at 323 K with a total TOF of 405 h−1.13 Therefore, the development of a metal catalyst with high durability and efficiency for the dehydrogenation of HB promotes the application of HB as a promising hydrogen storage material.
000 h−1. The catalytic hydrolysis of HB is stoichiometric, indicating that HB is capable of generating H2 effectively even just by adding water to solid HB.12 A very recent study demonstrates that MoOx-promoted NiPd nanocatalyst exhibits an excellent catalytic activity with almost 100% H2 selectivity in hydrogen release from HB in alkaline conditions at 323 K with a total TOF of 405 h−1.13 Therefore, the development of a metal catalyst with high durability and efficiency for the dehydrogenation of HB promotes the application of HB as a promising hydrogen storage material.
The catalytic effect on the hydrogen evolution reaction of ethylene diamine bisborane (EDAB) has been tested with a variety of ionic liquids, namely [BMMIM]Cl, [BMMIM][OAc], [BMIM][OAc] and [BMIM][OMs]. The mixture of 1-butyl-2,3-dimethylimidazolium chloride and EDAB can release about 6.5 wt% of hydrogen at 413 K, rivalling conventional hydrogen storage systems such as pressure tanks.14 A recent comparative study on the thermal dehydrogenation of EDAB with methanesulfonate-based deep eutectic solvents (DESs) and methanesulfonate anion based ionic liquids showed that the EDAB/DES system released 3.2 eq. of hydrogen with a lower induction period compared to 3.7 eq. of hydrogen for the ionic liquid-based solvent at the same temperature (378 K).15
Diammoniate diborane, [(NH3)2BH2]+[BH4]− (DADB), an ionic isomer of ammonia borane (AB), is a stable crystalline solid at room conditions. A comparative investigation of DADB with AB revealed that both compounds showed two distinct exothermic decomposition steps at below 473 K and the decomposition products of DADB and AB were very similar based on the ex situ XRD and FTIR analyses. However, DADB demonstrated several advantages such as a lower dehydrogenation temperature (about 283 K lower), foam-free reaction and absence of an induction period during thermal decomposition compared to AB leading to DADB being touted as a candidate for hydrogen storage applications.16
Hydrazine bisborane (HBB), another ammonia borane related boron–nitrogen–hydrogen compound which contains 16.8 wt% of theoretical hydrogen capacity was first reported in the early 1950's. An experimental investigation showed that HBB dehydrogenates at low temperature around 373 K and prevents formation of unwanted volatile by-products like ammonia and diborane during the hydrogen elimination process, confirmed by a very high purity (>99%) of dehydrogenation for HBB.17 According to a complete characterization result of HBB by XRD, NMR, FTIR, Raman, TGA and DSC the risk of the explosion of pristine HBB during thermolysis conditions (>423 K and with 10 °C min−1 heat ramp) makes this adduct not suitable for solid-state chemical hydrogen storage. However, as aqueous HBB dehydrogenates in the presence of an active and selective metal-based catalyst, it has been evidenced that aqueous HBB can be used for liquid-state chemical hydrogen storage.18
Cyclic amine borane systems appear to be more suitable for chemical hydrogen storage as these can offer stability, near thermoneutral evolution of hydrogen and the potential for recycling. A theoretical study conducted at the G4MP2 level of theory examined the hydrogen elimination reaction in cyclic amine boranes such as aziridine, azetidine, pyrrolidine, and piperidine boranes, both with and without the presence of a BH3 catalyst. The results showed that BH3, which forms as a result of B–N bond cleavage, acts as a catalyst, reducing the energy barrier for dehydrogenation reactions. This indicates the important role of BH3 as a catalyst in facilitating hydrogen release from cyclic amine boranes.19 Investigation of the partial in situ dehydrogenation of cyclic amine diborane complex 1,4-bis(borane) piperazine (PPZBB) via gas electron diffraction demonstrated that the release of two equivalents of H2 from PPZBB is experimentally feasible for this system. This observation was supported computationally by the predicted BH3-catalysed reaction potential energy surface as well as the thermochemical properties of the hydrogen release reactions. The predicted activation energies and dehydrogenation reaction energies for the release of the first and second H2 molecules suggested a favourable, spontaneous and exergonic reaction.20 PPZBB could be a more efficient chemical hydrogen storage material due to the release of two equivalents of H2 per PPZBB molecule than the previously studied cyclic amine borane complexes with only one BH3 group.21,22
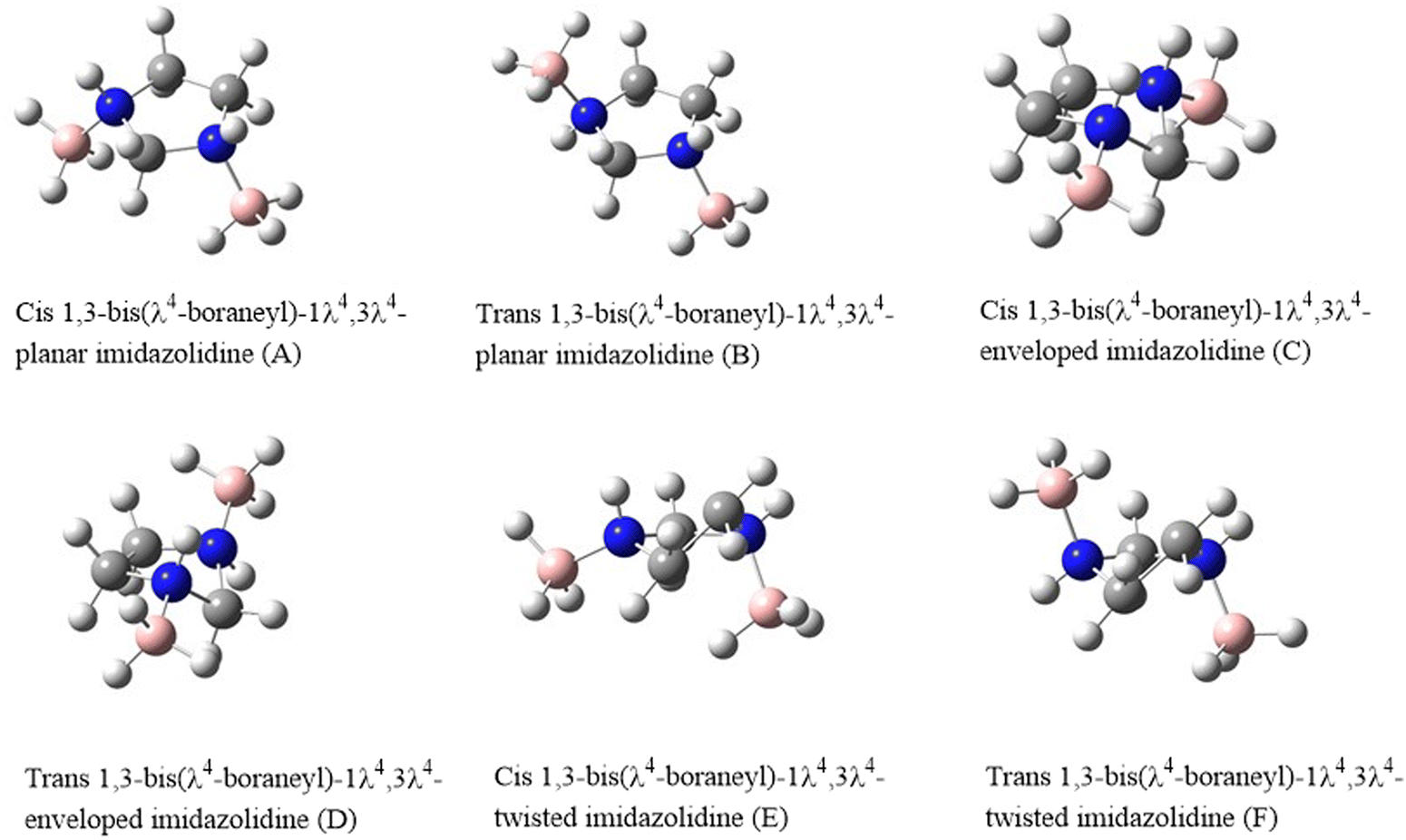
The main scope of this work is to investigate the structural parameters of the various conformers of 1,3-bis(λ4-boraneyl)-1λ4,3λ4-imidazolidine (IMBB) via ab initio electronic structure calculations. The structures of the possible conformers and stereoisomers of IMBB are shown in Fig. 1. The naming of each structure is based on the relative position of the BH3 units to the imidazolidine ring. We have also studied the thermochemistry and kinetics for the dehydrogenation reaction of the most stable conformer of IMBB. This work reports for the first time, the investigation of IMBB as a potential candidate for chemical hydrogen storage.
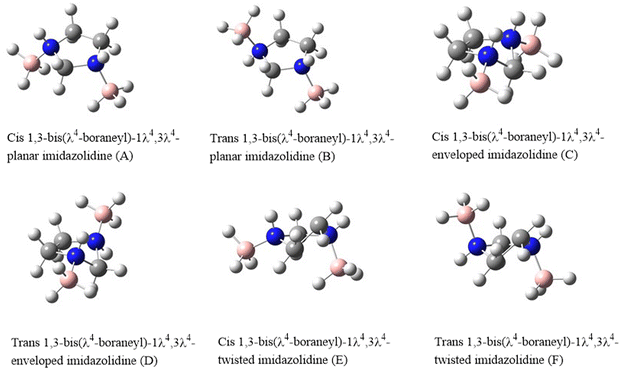 |
| | Fig. 1 Structures of the proposed conformers and stereoisomers of IMBB. | |
Computational methods
All electronic structure calculations were carried out using the Gaussian 1623 software suite. Two different conformers of IMBB, each with three stereoisomers, were identified and initially a series of calculations were carried out for each structure using the hybrid meta exchange–correlation functional (M06-2X)24 with 6-31G*25 basis set. The calculated Cartesian coordinates and the optimized structures for all structures are provided in the ESI† (Tables S1–S6 and Fig. S1–S6 respectively). The convergence of all calculations to minima on the ground-state potential energy surface was verified via vibrational frequency analysis. The stable lowest energy stereoisomer of the trans conformer of IMBB (Fig. 1; B) was used for further studies. The equilibrium structure was optimised using second-order Moller-Plesset (MP2)26 perturbation theory and M06-2X theory with 6-31G*, 6-311G*, 6-311+G*25,27,28 and cc-pVQZ29 basis sets to incorporate the effects of electron correlation on the geometric parameters. A series of geometry calculations were also conducted for dehydrogenated species of IMBB by removing a hydrogen on the BH3 above the plane and hydrogen on the corresponding nitrogen first and then a hydrogen on the BH3 below the plane and hydrogen on the corresponding nitrogen and vice versa using the MP2 and the M06-2X theories with 6-31G*, 6-311G*, 6-311+G* and cc-pVQZ basis sets. The convergence of all calculations to minima on the ground-state potential energy surface was verified via vibrational frequency analysis. The calculated Cartesian coordinates for all the molecules are provided in the ESI† (Tables S7–S31).
The rB–N bond dissociation energies, (B–N)BDEs were calculated at the CBS-QB3 level utilizing total electronic energies and the thermochemical parameters were calculated at 298.15 K at CBS-QB3 level using CBS-QB3 enthalpies and CBS-QB3 free energies. The calculated CBS-QB3 enthalpies and CBS-QB3 free energies are provided in the ESI† (Table S32). The (B–N)BDEs and the thermochemical parameters were also predicted at 298.15 K using CCSD(T) level by employing the total atomization energies and heat of formations as described by Curtiss et al.30 Extrapolations to complete basis set have been carried out for the CCSD(T) method using Dunning's31 correlation consistent basis sets, cc-pVnZ with n = 2,3,4. Extrapolations were performed using a mixed Gaussian/exponential formula (eqn (1)) suggested by Peterson et al.32 in order to obtain CCSD(T) energies for the CBS limit extrapolation. Atomization energies have been shown to be in closer agreement with experiment up through cc-pV5Z in the mixed extrapolation approach compared to other extrapolation methods. The thermal correction for the enthalpy, entropy, Gibbs free energy and zero-point energies were calculated at the MP2/cc-pVTZ level. The calculated energies and corrections for enthalpy (H), Gibbs free energy (G) and zero-point energies used in the calculation of the (B–N)BDEs are provided in the ESI† (Table S33) and the above parameters used in the calculation of the thermochemical properties are provided in the ESI† (Table S34).
| | | E(n) = Ecbs + Ae−(n−1) + Be−(n−1)2 | (1) |
where
n = 2 (cc-pVDZ), 3 (cc-pVTZ) and 4 (cc-pVQZ)
To study the impact of incorporating counterpoise corrections on the accuracy of bond dissociation energies of IMBB, the (B–N)BDE calculations were performed using CCSD(T) with counterpoise corrections for this complex. Extrapolations to complete basis set with counterpoise corrections have been performed using the cc-pVnZ (n = 2, 3, 4) correlation consistent basis sets. The extrapolations were performed using the mixed Gaussian expression above.
Thermochemical parameters were also predicted at elevated temperatures (653 K and 823 K) using the CBS-QB3 level by employing CBS-QB3 enthalpies and CBS-QB3 free energies. As provided in the ESI† Tables S35–S36, the calculated CBS-QB3 enthalpies and CBS-QB3 free energies were used for computing the thermochemical properties at 653 K and 823 K.
Transition-state structures are often located using the method of the synchronous transit-guided quasi-Newton (STQN).33 However, in this study there many molecules involved in the BH3-catalysed dehydrogenation reaction steps. Consequently, the STQN method cannot be used to obtain the TS for the pathway of the dehydrogenation reaction of IMBB. Instead, the transition-state structures were obtained by a normal eigenvalue-following technique that follows the reaction path from the equilibrium geometry to the TS structure by specifying which vibrational mode should lead to a reaction given sufficient kinetic energy. The potential energy surfaces for the uncatalysed and BH3-catalysed hydrogen release from IMBB have been constructed using CBS-QB3 relative energies. Intrinsic reaction coordinate calculations34 were also carried out at the B3LYP/6-31G* level to confirm the connections between the TSs and local minima. The calculated Cartesian coordinates for all molecules and optimized structures of the reactants and products for the uncatalysed and catalysed dehydrogenation reactions of IMBB are provided in the ESI† (Tables S37–S48 and Fig. S7–S12).
The feasibility of the uncatalysed and BH3-catalysed dehydrogenation reaction of IMBB at elevated temperatures were checked using transition-state theory. The calculations were performed at two different temperatures: 653 K and 823 K. The energies of activated complexes (TSs) and reactants for both the uncatalysed and catalysed pathways obtained at the CBS-QB3 level were employed for calculating the activated thermodynamic parameters. The CBS-QB3 enthalpies and CBS-QB3 free energies employed in the determination of activated thermochemical properties are provided in the ESI† (Table S49).
Results and discussion
Conformational analysis and ab initio electronic structure calculations
The relative energy values and molecular symmetry adopted by each structure of IMBB at M06-2X/6-31G* level of theory is shown in Table 1. Out of six possible structures, three minima were identified on the potential energy surface. The structures, trans planar IMBB (B), trans enveloped IMBB (D) and trans twisted IMBB (F) are stereoisomers; molecules that have the same molecular formula and connectivity of atoms but differ in their spatial arrangement. Similarly, the remaining three structures [cis planar IMBB (A), cis enveloped IMBB (C) and cis twisted IMBB (E)] are also stereoisomers due to their disparate spatial arrangements. For the trans stereoisomers, following geometry optimization of the three starting stereoisomers, the structures optimized to the same envelope structure which was then used for further analysis as the lowest energy conformer and will be referred to as IMBB from this point on. The three cis stereoisomers optimised to two different structures, twisted and envelope, with an energy difference of 0.2 kJ mol−1. However, both stereoisomers were ∼13.6 kJ mol−1 higher in energy than the trans conformer and further calculations in relation to dehydrogenation were not performed for these structures.
Table 1 Energy and molecular symmetry for each conformer and stereoisomer of IMBB following geometry optimisation
| No. |
Conformers |
Relative energy (M06-2X/6-31G*) (kJ mol−1) |
Symmetry |
| 1 |
A |
13.7 |
C
s
|
| 2 |
B |
0.0 |
C
1
|
| 3 |
C |
13.7 |
C
s
|
| 4 |
D |
0.0 |
C
1
|
| 5 |
E |
13.9 |
C
1
|
| 6 |
F |
0.0 |
C
1
|
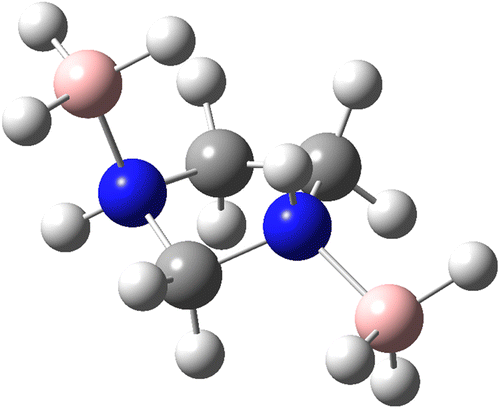
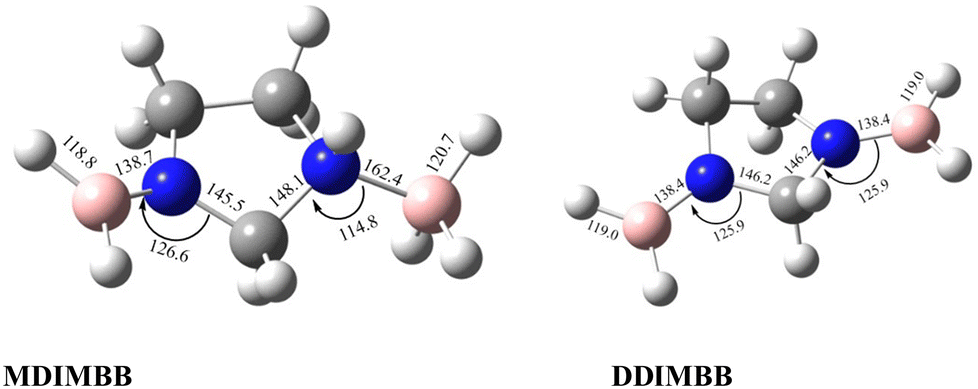
To analyse the effects of methods and basis sets on the geometry of the system, the MP2 and M06-2X methods with assorted basis sets containing additional polarization, diffuse functions and Dunning's correlation consistent were employed. The results for selected structural parameters of IMBB are tabulated in Table 2 while a full list of the parameters is provided in the ESI† (Table S50). The optimized structure of the IMBB at M06-2X/cc-pVQZ is shown in Fig. 2. The results for selected structural parameters for the dehydrogenated counterparts of IMBB, namely mono-dehydrogenated IMBB (MDIMBB) and di-dehydrogenated IMBB (DDIMBB) by the removal of a hydrogen on the BH3 above the ring and hydrogen on the corresponding nitrogen first and then a hydrogen on the BH3 below the ring and hydrogen on the corresponding nitrogen is tabulated in Table 3. The corresponding optimized structures of each dehydrogenated species at M06-2X/cc-pVQZ are shown in Fig. 3. The results for selected structural parameters for the dehydrogenated species of IMBB by the removal of a hydrogen on the BH3 below the ring and hydrogen on the corresponding nitrogen first and then a hydrogen on the BH3 above the ring and hydrogen on the corresponding nitrogen is tabulated in Table 4. The corresponding optimized structures of each dehydrogenated species at M06-2X/cc-pVQZ are shown in Fig. 4.
Table 2 The optimized structural parameters for the IMBB at different levels of theory and basis set
| Parameter |
MP2/6-31G* |
MP2/6-311G* |
MP2/6-311+G* |
M06-2X/6-31G* |
M06-2X/cc-pVQZ |
| All distances are in pm; bond angles are in degrees. Detailed structural parameters are provided in ESI Table S50. |
|
rB–N |
165.0 |
164.1 |
164.1 |
164.7 |
163.3 |
|
rN–C |
148.6 |
148.6 |
148.6 |
148.3 |
148.0 |
|
rB–H |
121.1 |
121.3 |
121.5 |
121.1 |
120.6 |
| ∠C–N–B |
113.3 |
113.1 |
113.3 |
112.9 |
113.1 |
| ∠N–B–H |
104.8 |
105.3 |
105.2 |
104.8 |
105.2 |
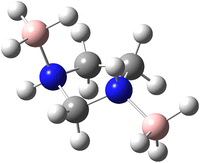 |
| | Fig. 2 The optimized structure of IMBB at M06-2X/cc-pVQZ level. | |
Table 3 Optimized structural parameters for MDIMBB and DDIMBB at different levels of theory and basis set by the removal of H on the BH3 above the ring and H on the corresponding N followed by a H on the BH3 below the ring and H on the corresponding N. (The first value is for parameters relating to the N center with BH3 above the ring; the second value is for parameters relating to the N center with BH3 below the ring)
|
MDIMBB
|
MP2/6-31G* |
MP2/6311G* |
MP2/6311+G* |
M06-2X/6-31G* |
M06-2X/cc-pVQZ |
| All distances are in pm; bond angles are in degrees. |
|
rB–N |
139.7/164.1 |
139.5/163.3 |
139.8/163.3 |
139.2/163.7 |
138.7/162.4 |
|
rN–C |
146.0/148.7 |
145.8/148.8 |
145.8/148.9 |
145.8/148.4 |
145.5/148.1 |
|
rB–H |
119.4/121.2 |
119.6/121.4 |
119.6/121.5 |
119.4/121.2 |
118.8/120.7 |
| ∠C–N–B |
126.7/115.0 |
127.0/115.0 |
127.1/114.9 |
126.4/114.8 |
126.6/114.8 |
| ∠N–B–H |
118.7/104.8 |
118.5/105.3 |
118.5/105.2 |
118.6/104.8 |
118.6/105.2 |
|
DDIMBB
|
MP2/6-31G* |
MP2/6-311G* |
MP2/6-311+G* |
M06-2X/6-31G* |
M06-2X/cc-pVQZ |
|
rB–N |
139.4 |
139.2 |
139.5 |
138.9 |
138.4 |
|
rN–C |
146.6 |
146.7 |
146.8 |
146.3 |
146.2 |
|
rB–H |
119.6 |
119.8 |
119.8 |
119.5 |
119.0 |
| ∠C–N–B |
125.9 |
125.9 |
125.9 |
125.8 |
125.9 |
| ∠N–B–H |
118.9 |
118.7 |
118.7 |
118.7 |
118.8 |
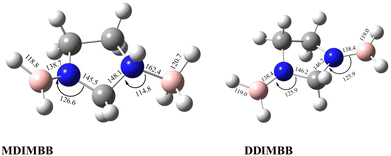 |
| | Fig. 3 Optimized structures of the two dehydrogenated counterparts of IMBB (MDIMBB and DDIMBB) with geometrical parameters at M06-2X/cc-pVQZ by the removal of H2 above then below the ring. | |
Table 4 The optimized structural parameters for the MDIMBB and DDIMBB at different levels of theory and basis set by the removal of H on the BH3 below the ring and H on the corresponding N followed by H on the BH3 above the ring and H on the corresponding N. (The first value is for parameters relating to the N center with BH3 below the ring; the second value is for parameters relating to the N center with BH3 above the ring)
|
MDIMB
|
MP2/6-31G* |
MP2/6-311G* |
MP2/6-311+G* |
M06-2X/6-31G* |
M06-2X/cc-pVQZ |
| All distances are in pm; bond angles are in degrees. |
|
rB–N |
139.7/164.1 |
139.5/163.3 |
139.6/164.2 |
139.2/163.7 |
138.7/162.4 |
|
rN–C |
146.0/148.7 |
145.8/148.8 |
146.5/148.8 |
145.8/148.4 |
145.5/148.1 |
|
rB–H |
119.4/121.2 |
119.6/121.4 |
119.7/121.4 |
119.4/121.2 |
118.8/120.7 |
| ∠C–N–B |
126.7/115.0 |
127.0/115.0 |
125.1/113.9 |
126.4/114.8 |
126.6/114.8 |
| ∠N–B–H |
118.7/104.8 |
118.5/105.3 |
118.6/105.2 |
118.6/104.8 |
118.6/105.2 |
|
DDIMBB
|
MP2/6-31G* |
MP2/6-311G* |
MP2/6-311+G* |
M06-2X/6-31G* |
M06-2X/cc-pVQZ |
|
rB–N |
139.4 |
139.2 |
139.5 |
138.9 |
138.4 |
|
rN–C |
146.6 |
146.7 |
146.8 |
146.3 |
146.2 |
|
rB–H |
119.6 |
119.8 |
119.8 |
119.5 |
119.0 |
| ∠C–N–B |
125.9 |
125.9 |
125.9 |
125.8 |
125.9 |
| ∠N–B–H |
118.9 |
118.7 |
118.7 |
118.7 |
118.8 |
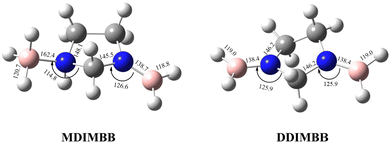 |
| | Fig. 4 The optimized structures of the two dehydrogenated counterparts of IMBB (MDIMBB and DDIMBB) with geometrical parameters at M06-2X/cc-pVQZ by the removal of hydrogen below then above the ring. | |
It should be noted that IMBB, along with its dehydrogenated counterparts MDIMBB and DDIMBB, all exhibit C1 symmetry. Most calculated bond lengths and bond angles for IMBB as well as all the dehydrogenated counterparts did not change significantly with change in method or improvement in basis set and are within ∼1 pm or 1° for each molecule. The rB–N bond distances are comparable to that of PPZBB in which rB–N was found to be 163.3 pm at M06-2X/cc-pVQZ level.20 Therefore, it can be concluded that the ring size does not affect rB–N significantly. It is also noticeable that from the geometry optimization of the dehydrogenated species of IMBB, removing hydrogen from the B–N above the ring followed by hydrogen from the B–N below the ring and vice versa does not make any significant change to the structural parameters. This is important as the ring is not planar and the envelope may have made a difference to the structural parameters and therefore the energies obtained. Fortunately, this was not the case and therefore only one pathway was studied further.
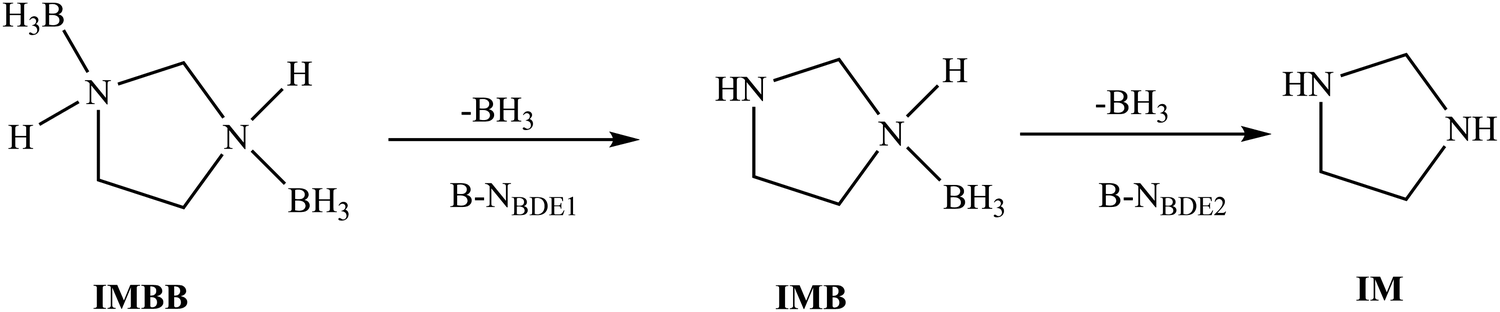
Bond dissociation energies (BDEs)
The sequence of bond breaking for the two rB–N bonds in IMBB is shown in Fig. 5. Table 5 shows the calculated bond dissociation energies for the first rB–N bond to form imidazolidine borane (IMB) (B–NBDE1) and then the second rB–N bond to form the free imidazolidine (IM) (B–NBDE2) at CCSD(T) level with and without counterpoise (CP) corrections and CBS-QB3 level at 298.15 K. The B–NBDE is reported at extrapolation to CBS (as outlined above) for the CCSD(T) method in this table. The energy required to break the first rB–N bond is lower than the second rB–N bond as observed at both levels of theory. A similar observation was made in the previously studied compound 1,4-(bis)borane piperazine (PPZBB).20 These two B–NBDEs are higher than the rB–N bond dissociation reported for NH3BH3 at CCSD(T)/CBS level.35 Based on the results obtained for IMBB, it was observed that the B–NBDEs are lower for IMBB compared to PPZBB at CCSD(T)/CBS level.20 This outcome is expected due to the decrease in the size of the nitrogen-containing ring in IMBB, which leads to a reduction in inductive effects and consequently a decrease in the strength of the dative bond. Therefore, the bond dissociation energies were found to be lower in IMBB than those in PPZBB. Furthermore, it has been observed that the inclusion of counterpoise corrections results in a larger deviation from the uncorrected approach at the CCSD(T)/CBS-extrapolations. These values should be more accurate as counterpoise corrected (B–N)BDE improves the accuracy by eliminating the artificial stabilization introduced by basis set superposition error (BSSE).36 The inclusion of the counterpoise correction ensures that computational results may be in good agreement with experimental value to break the rB–N bonds. Therefore, the accurate counterpoise corrected (B–N)BDEs are used to subsequently predict the reaction pathways of IMBB.
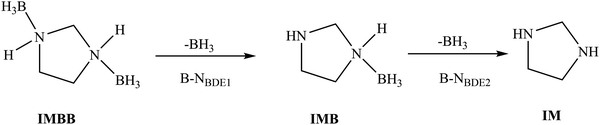 |
| | Fig. 5 Reaction scheme for the sequential bond dissociation of the rB–N bonds in IMBB. | |
Table 5 Bond dissociation energies for IMBB predicted at CCSD(T) level (with and without CP corrections) and CBS-QB3 level at 298.15 K
| Property |
CCSD(T)/CBS |
CBS-QB3 |
| Units in kJ mol−1. |
| B–NBDE1 |
139.8 |
136.3 |
| B–NBDE2 |
146.0 |
145.3 |
| B–NBDE1 (CP) |
208.6 |
— |
| B–NBDE2 (CP) |
227.0 |
— |
Thermochemical studies
Thermochemical properties such as dehydrogenation enthalpy (ΔHr), Gibbs free energy (ΔGr) and entropy (ΔSr) were predicted at both the CCSD(T) and CBS-QB3 levels of theory at 298.15 K. Table 6 presents the calculated thermochemical parameters of IMBB for the release of the first and second H2 molecule at 298.15 K. To simplify this discussion only the accurate CCSD(T) method is discussed because the Dunning's correlation consistent basis set aug-cc-pCV(T+d)Z is used to minimize the errors below the chemical accuracy limit. This method predicts thermochemical details with only approximately 2.092 kJ mol−1 error.37Table 6 demonstrates that the reaction energies for the release of the first and second H2 molecules are remarkably similar. This similarity indicates a spontaneous, exothermic and simultaneous process in which both H2 molecules are released near thermoneutral conditions, as opposed to a sequential release of one molecule followed by another. The negative value of 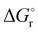 and positive value of
and positive value of 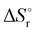 along with the products having lower Gibbs free energy compared to the reactants (as shown in Fig. 6 and 7), suggested a fast, exergonic and favourable dehydrogenation reaction for IMBB.
along with the products having lower Gibbs free energy compared to the reactants (as shown in Fig. 6 and 7), suggested a fast, exergonic and favourable dehydrogenation reaction for IMBB.
Table 6 Thermochemical parameters for IMBB predicted at CCSD(T) and CBS-QB3 level of theory at 298.15 K
| Property |
CCSD(T)/CBS |
CBS-QB3 |
| First |
Second |
First |
Second |
Units in kJ mol−1 except 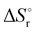 which is in J K−1 mol−1. First and Second implies the reaction to release the first and second H2 molecule from IMBB. which is in J K−1 mol−1. First and Second implies the reaction to release the first and second H2 molecule from IMBB. |
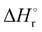
|
−13.5 |
−9.4 |
−17.8 |
−14.5 |
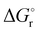
|
−51.7 |
−49.3 |
−54.6 |
−51.4 |
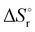
|
+128.1 |
+133.8 |
+123.7 |
+123.9 |
 |
| | Fig. 6 Energy profile diagram for the dehydrogenation of IMBB without the catalyst BH3 at 298.15 K using CBS-QB3. Relative Gibbs free energies (in brackets) in kJ mol−1. | |
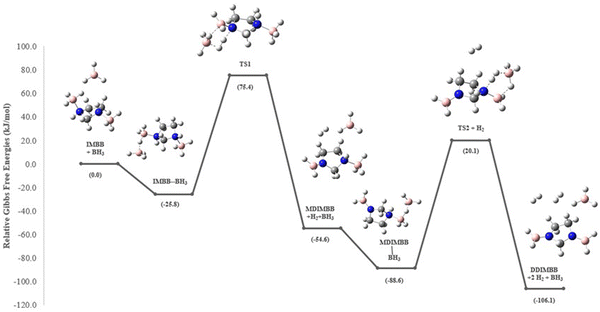 |
| | Fig. 7 Energy profile diagram for the dehydrogenation of IMBB with the catalyst BH3 at 298.15 K using CBS-QB3. Relative Gibbs free energies (in brackets) in kJ mol−1. | |
The impact of temperature on the thermochemical parameters of the dehydrogenation reaction of IMBB is evident, as highlighted in Table 7. The energies involved in the release of the first and second molecules of H2 at temperatures of 653 K and 823 K exhibit a notable similarity. This suggests that both H2 molecules are released under thermoneutral conditions at high temperatures just as they are at 298.15 K. By utilizing eqn (2) and (3), the equilibrium constant at 298.15 K and the reaction quotients at 653 K and 823 K were computed based on the free energies provided in Table 7. In these equations, ΔG° represents the standard temperature (298.15 K) free energy change for the reaction, ΔG indicates the free energy change for the reaction at higher temperatures, R represents the universal gas constant, T denotes the temperature (in Kelvin), and Q represents the reaction quotient at elevated temperatures.
| | | ΔG° = −RTlnK | (2) |
| | 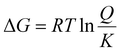 | (3) |
Table 7 Thermochemical parameters for IMBB predicted at CBS-QB3 level of theory at 298.15 K, 653.00 K and 823.00 K
| Property |
298.15 K |
653.00 K |
823.00 K |
| First |
Second |
First |
Second |
First |
Second |
| Units in kJ mol−1 except ΔSr which is in J K−1 mol−1. First and second indicates the reaction to release the first and second H2 molecule from IMBB. |
| ΔHr |
−17.8 |
−14.5 |
−13.5 |
−10.4 |
−13.1 |
−10.0 |
| ΔGr |
−54.6 |
−51.4 |
−100.9 |
−97.7 |
−123.7 |
−120.5 |
| ΔSr |
123.7 |
123.9 |
133.8 |
133.7 |
134.4 |
134.3 |
Table 8 displays the resulting equilibrium constant and reaction quotients. Notably, the computed equilibrium constant for the first and second hydrogen release reaction at 298.15 K is 3.68 × 109 and 1.01 × 109 respectively. The thermochemical studies reveal that the dehydrogenation of IMBB is predicted to occur spontaneously at all three temperatures, with ΔG < 0, ΔS > 0, and K > Q. Furthermore, it is worth mentioning that the enthalpies for the dehydrogenation of IMBB were less exothermic at all three temperatures compared to our previously studied compound pyrrole borane.38
Table 8 Equilibrium constant and reaction quotients predicted at different temperatures for the dehydrogenation of IMBB
| Temperature/K |
Equilibrium constant/s−1 |
Ratios |
|
K
1 and Q1 are the equilibrium constant and reaction quotient respectively for the reaction to release the first equivalent of H2. K2 and Q2 are the equilibrium constant and reaction quotient respectively for the reaction to release the second equivalent of H2. |
|
|
K
1
|
K
2
|
K
1/K2 |
| 298.15 |
3.68 × 109 |
1.01 × 109 |
3.64 |
|
|
Reaction quotients/s−1 |
|
|
|
Q
1
|
Q
2
|
Q
1/Q2 |
| 653.00 |
31.23 |
15.48 |
2.02 |
| 823.00 |
51.84 |
22.76 |
2.28 |
Reaction energy pathway for hydrogen release from IMBB
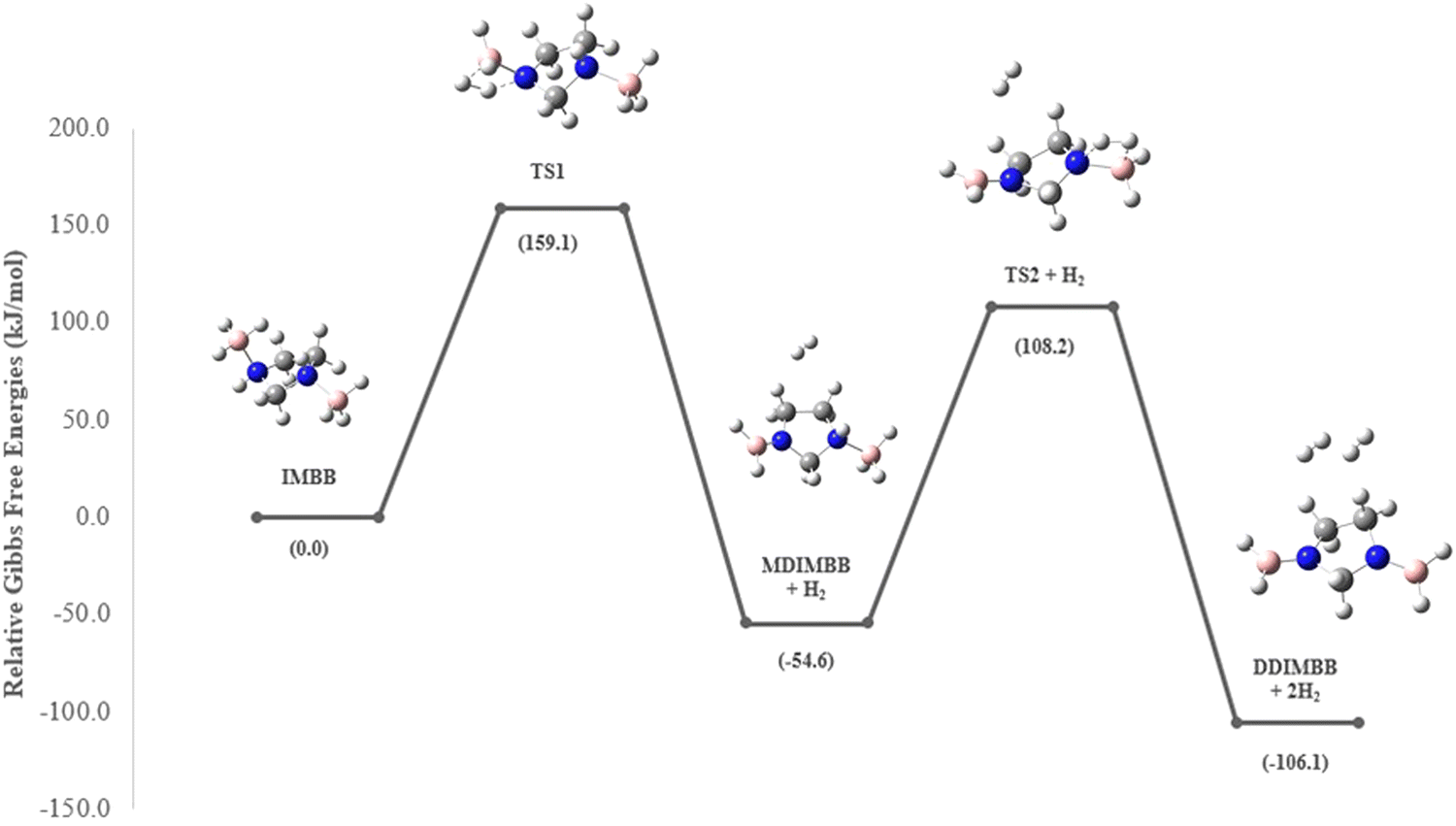
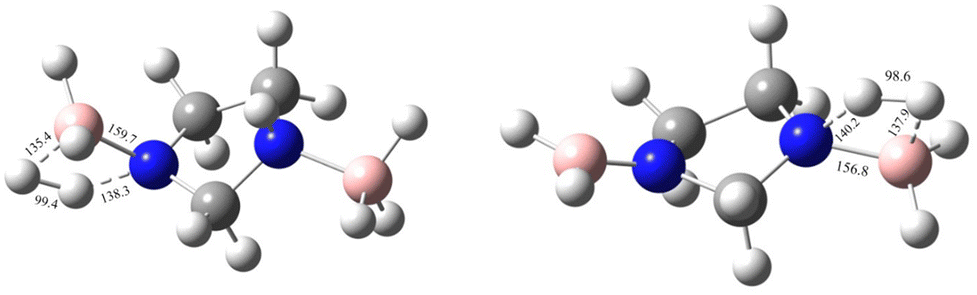
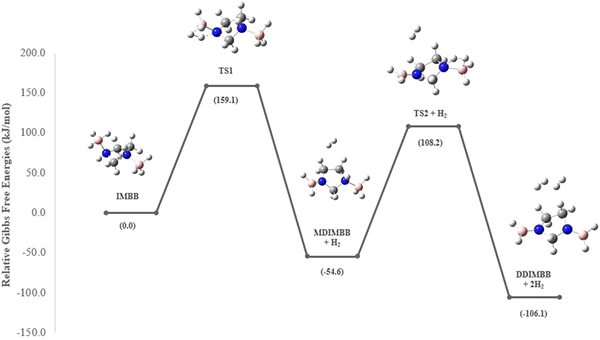
Sun et al. carried out a quantitative estimation of the activation barrier for a simple diborane system [hydrazine bisborane (HBB)] using Kissinger's method39,40 and it has been reported that the activation energy of the first-step dehydrogenation of HBB is determined to be approximately 106.4 kJ mol−1, indicating moderate kinetics for the first dehydrogenation step for HBB.17 In the present study the capacity of IMBB to release two equivalents of hydrogen in the presence or absence of BH3 catalyst in a favourable manner has been demonstrated. The energy pathway for the dehydrogenation reaction has been predicted at CBS-QB3 level. The BH3-uncatalysed and BH3-catalysed energy profile diagrams are depicted in Fig. 6 and 7. The transition-state structures (TS) of the dehydrogenation reaction without and with catalyst are shown in Fig. 8 and 9. The dehydrogenation pathways with and without BH3 were found to be near exothermic. For the BH3-uncatalysed dehydrogenation of IMBB, the activation barriers at 298.15 K for the release of first and second equivalents of dihydrogen are 159.1 kJ mol−1 and 108.2 kJ mol−1 respectively. It has been observed that B–NBDE1 and B–NBDE2 at CCSD(T)/CBS level with counterpoise correction are higher than the corresponding energy barriers indicating that the dehydrogenation of IMBB favourable over rB–N bond dissociation even without a catalyst (Table 5, Fig. 6). While the uncatalysed dehydrogenation of IMBB showed the potential for H2 evolution, the predicted activation barriers for a favourable reaction are still relatively high. Therefore, it is crucial to investigate the dehydrogenation of IMBB in the presence of a catalyst with the aim of further reducing the reaction barriers.
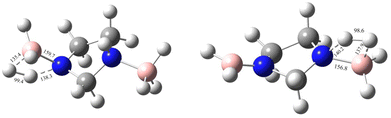 |
| | Fig. 8 The first and second transition-state structures with geometrical parameters for the uncatalysed dehydrogenation of IMBB at the CBS-QB3 level. | |
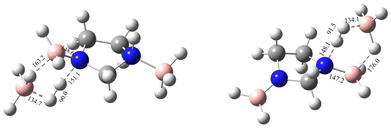 |
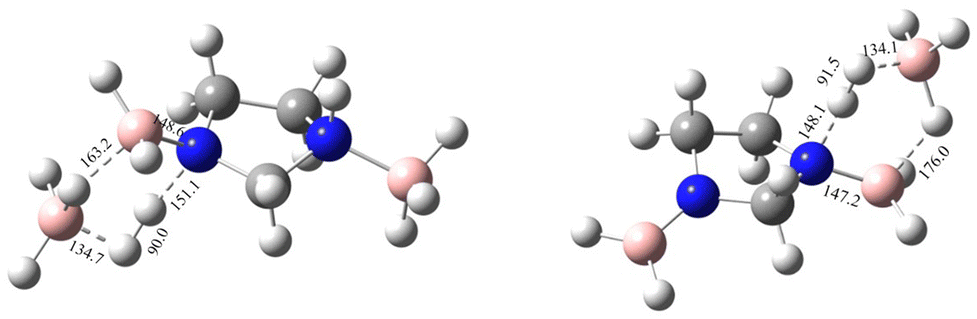
| | Fig. 9 The first and second transition-state structures with geometrical parameters for the catalysed dehydrogenation of IMBB with BH3 at the CBS-QB3 level. | |
It has been shown experimentally that PPZBB releases two equivalents of dihydrogen in the presence of a BH3 catalyst. This result is supported by the computed BH3-catalysed reaction energy pathway of the hydrogen release reaction. The predicted rB–N bond dissociation energies for PPZBB at CCSD(T)/CBS level (170.3 kJ mol−1 and 182.0 kJ mol−1) are much higher than the respective barrier heights (28.1 kJ mol−1 and 7.9 kJ mol−1) suggesting that the dehydrogenation of the PPZBB is favoured over dissociation in the presence of a BH3 catalyst.20 For IMBB, the interaction between IMBB and the BH3 catalyst resulted in the formation of an adduct intermediate depicted in the ESI† (Fig. S14) in a barrier-less manner, with an energy difference of approximately −25.8 kJ mol−1 compared to the initial reactants. Then the adduct passes through the transition-state, TS1 giving the mono-dehydrogenated product MDIMBB, the catalyst BH3 and the first H2 molecule. Again, as can be seen from Fig. 7, the condensation of the BH3 with MDIMBB leads to formation of the second adduct intermediate shown in the ESI† (Fig. S15). The reaction then progresses to generate the second H2 molecule alongside the di-dehydrogenated product DDIMBB through a second transition-state, TS2. The calculated (B–N)BDEs and respective activation barriers for the BH3-catalysed dehydrogenation of IMBB clearly demonstrates that the dehydrogenation process will be favoured over dissociation of the IMBB adduct due to the reaction barriers (75.4 kJ mol−1 and 20.1 kJ mol−1) being significantly lower than the dissociation energies (208.6 kJ mol−1 and 227.0 kJ mol−1) (Fig. 7, Table 5).
It is worth mentioning that TS1, with a higher barrier than TS2, is expected to be the rate-determining step during the H2 elimination process of IMBB. Therefore, once the first hydrogen is eliminated, subsequent dehydrogenation occurs more rapidly and does not significantly affect the overall rate.
Transition-state theory was used to examine the feasibility of the uncatalysed and BH3-catalysed dehydrogenation reactions of IMBB at 653 K and 823 K. While compounds such as azetidine borane and 1,4-(bis)boranepiperazine show experimental dehydrogenation at approximately 393 K in previous studies conducted, cyclic amine boranes like pyrrolidine, piperidine, and morpholine borane did not exhibit hydrogen release even at elevated temperatures ranging from 370 to 400 K. Consequently, for our study, higher temperatures of 653 and 823 K have been selected.20
Computed activated thermodynamic parameters, as presented in Table 9, were used to predict the rate information. Rate constants at various temperatures were computed using the calculated free energies (Table 9) for the uncatalysed and catalysed hydrogen elimination reactions of IMBB, utilizing the Eyring equation (eqn (4)) and Arrhenius equation (eqn (5)) as shown below:
| | 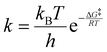 | (4) |
| | 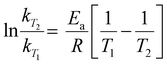 | (5) |
Table 9 Computed activated thermodynamic parameters for uncatalysed and catalysed hydrogen elimination of IMBB at 298.15 K using CBS-QB3 level
| Property |
Uncatalysed dehydrogenation |
Catalysed dehydrogenation |
| First |
Second |
First |
Second |
| Units in kJ mol−1 except ΔS‡r which is in J K−1 mol−1. ΔH‡r is the change in enthalpy of activation and ΔS‡r is the change in entropy of activation. First and second indicates the reaction to release the first and second H2 molecule from IMBB. |
| ΔH‡r |
161.6 |
162.8 |
33.5 |
31.9 |
| ΔG‡r |
159.1 |
162.8 |
75.4 |
74.7 |
| ΔS‡r |
8.4 |
−0.088 |
−140.4 |
−143.7 |
In the equations, k represents the rate constant, kB represents the Boltzmann constant (1.38 × 10−23 J K−1), T denotes temperature (in Kelvin), h represents Planck's constant (6.63 × 10−34 J s), ΔG‡ signifies the free energy of activation (change in free energies of activated complexes; TSs and reactants), R is the universal gas constant (8.314 J mol−1 K−1), and T1 and T2 refer to two different temperatures in Kelvin.
Table 10 illustrates the computed rate constants for the uncatalysed and catalysed hydrogen release from IMBB at different temperatures. At a temperature of 298.15 K, the predicted rate constants using eqn (4) for the first and second dihydrogen eliminations in the uncatalysed pathway of IMBB are 8.41 × 10−16 and 1.85 × 10−16, respectively. Conversely, for the release of the first and second dihydrogen molecules in the catalysed pathway of IMBB, the rate constants are 3.90 × 10−1 and 5.00 × 10−1, respectively. Importantly, the rate constants for both pathways of IMBB increase as the temperature rises.
Table 10 Calculated rate constants for the uncatalysed and catalysed dehydrogenation at different temperatures
| Temp./K |
Rate constants/s−1 |
| Uncatalysed dehydrogenation |
Catalysed dehydrogenation |
|
k
1
|
k
2
|
k
1/k2 |
k
3
|
k
4
|
k
3/k4 |
|
k
1 and k2 are rate constants for the first and second hydrogen release reaction for the uncatalysed dehydrogenation of IMBBB respectively. K3 and k4 are rate constants for the first and second hydrogen release reaction for the catalysed dehydrogenation of IMBBB respectively. |
| 298.15 |
8.41 × 10−16 |
1.85 × 10−16 |
4.55 |
3.90 × 10−1 |
5.00 × 10−1 |
7.80 × 10−1 |
| 653.00 |
1.91 |
2.33 × 10−3 |
8.21 × 102 |
1.51 × 103 |
5.57 |
2.72 × 102 |
| 823.00 |
8.85 × 102 |
4.38 × 10−1 |
2.02 × 103 |
6.35 × 103 |
8.47 |
7.50 × 102 |
Furthermore, it is observed that the rate constant for the first dihydrogen elimination is higher than the rate constant for the second dihydrogen release in both the uncatalysed and catalysed pathways at all three temperatures. This observation reinforces the notion that the first dihydrogen release reaction is immediately followed by the second dihydrogen release at these elevated temperatures.
Conclusions
The molecular structure of IMBB and its mono and di-dehydrogenated forms were determined employing quantum chemical methods. Structural investigation suggested that no significant differences were observed in geometrical parameters for each molecule when altering the basis set and method. The predicted rB–N bond length for IMBB closely matches that of PPZBB from a previous study. The dehydrogenation reaction energies for the release of the first and second equivalents of dihydrogen from IMBB are very similar indicating a simultaneous and spontaneous evolution of both molecules of H2. This process is predicted to be less exothermic than that for pyrrole borane and should occur at all temperatures investigated. However, the practical temperature suitable for the dehydrogenation process should be confirmed through future experimental investigations. The reaction barriers for the BH3-catalysed dehydrogenation processes are found to be much lower than the corresponding rB–N bond dissociation energies. This observation suggests that IMBB is more likely to undergo dehydrogenation across rB–N than dissociation of this bond. Transition-state theory indicates that dihydrogen elimination in the presence of a BH3 catalyst is expected to be a faster reaction at higher temperatures. Furthermore, the higher rate constant for the first dihydrogen release compared to the second dihydrogen release reinforces the postulation of simultaneous release of both dihydrogen molecules at elevated temperatures. IMBB has the advantage of being able to release two equivalents of dihydrogen per IMBB molecule, giving greater potential as a hydrogen storage material compared to previously studied monosubstituted systems. The findings should be confirmed through experimental work in the future. Additionally, the structure of the parent molecule and its dehydrogenated counterpart continues to be of general interest in the field.
Author contributions
SLM conceptualized the project, advised on methodology, administered the project, provided access to software, supervised FRMI, advised on writing and editing of the manuscript. FRMI undertook the data curation and analysis, wrote the original draft of the manuscript, edited the manuscript and prepared all images for publication.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
FRMI acknowledges the Accelerating Higher Education Expansion and Development (AHEAD) operation in Sri Lanka for their generous financial support through the PhD Scholarship and the University of Canterbury School of Physical and Chemical Sciences for their Research Support Scholarship that has provided essential funding. FRMI and SLM thank Dr Aliyu Ja'o for his invaluable guidance and discussion.
References
- M. Hoel, Depletion of Fossil Fuels and the Impacts of Global Warming, Fuel Energy Abstr., 1996, 37, 460 Search PubMed.
- L. Barreto, A. Makihira and K. Riahi, The Hydrogen Economy in the 21st Century: A Sustainable Development Scenario, Int. J. Hydrogen Energy, 2003, 28, 267–284 CrossRef CAS.
- U. Eberle, M. Felderhoff and F. Schüth, Chemical and Physical Solutions for Hydrogen Storage, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS PubMed.
- L. Schlapbach and A. Züttel, Hydrogen-Storage Materials for Mobile Applications, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- T. J. Groshens and R. A. Hollins, New Chemical Hydrogen Storage Materials Exploiting the Self-Sustaining Thermal Decomposition of Guanidinium Borohydride, Chem. Commun., 2009, 3089–3091 RSC.
- D. Neiner, A. Karkamkar, M. Bowden, Y. Joon Choi, A. Luedtke, J. Holladay, A. Fisher, N. Szymczak and T. Autrey, Kinetic and Thermodynamic Investigation of Hydrogen Release from Ethane 1,2-Di-Amineborane, Energy Environ. Sci., 2011, 4, 4187–4193 RSC.
- T. Hügle, M. F. Kühnel and D. Lentz, Hydrazine Borane: A Promising Hydrogen Storage Material, J. Am. Chem. Soc., 2009, 131, 7444–7446 CrossRef PubMed.
- C. D. Zeinalipour-Yazdi, DFT Study of the Coverage-Dependent Chemisorption of Molecular H2 on Neutral Cobalt Dimers, Surf. Sci., 2017, 656, 54–59 CrossRef CAS.
- T. B. Marder, Will We Soon Be Fueling Our Automobiles with Ammonia-Borane?, Angew. Chem., Int. Ed., 2007, 46, 8116–8118 CrossRef CAS PubMed.
- C. H. Sun, X. D. Yao, A. J. Du, A. L. Li, S. Smith and G. Q. Lu, Computational Study of Methyl Derivatives of Ammonia Borane for Hydrogen Storage, Phys. Chem. Chem. Phys., 2008, 10, 6104–6106 RSC.
- R. Moury, G. Moussa, U. B. Demirci, J. Hannauer, S. Bernard, E. Petit, A. Van Der Lee and P. Miele, Hydrazine Borane: Synthesis, Characterization, and Application Prospects in Chemical Hydrogen Storage, Phys. Chem. Chem. Phys., 2012, 14(5), 1768–1777 RSC.
- S. Karahan, M. Zahmakran and S. Özkar, Catalytic Hydrolysis of Hydrazine Borane for Chemical Hydrogen Storage: Highly Efficient and Fast Hydrogen Generation System at Room Temperature, Int. J. Hydrogen Energy, 2011, 36, 4958–4966 CrossRef CAS.
- Q. Yao, K. Yang, W. Nie, Y. Li and Z. H. Lu, Highly Efficient Hydrogen Generation from Hydrazine Borane via a MoOx-Promoted NiPd Nanocatalyst, Renewable Energy, 2020, 147, 2024–2031 CrossRef CAS.
- S. Sahler, H. Konnerth, N. Knoblauch and M. H. G. Prechtl, Hydrogen Storage in Amine Boranes: Ionic Liquid Supported Thermal Dehydrogenation of Ethylene Diamine Bisborane, Int. J. Hydrogen Energy, 2013, 38, 3283–3290 CrossRef CAS.
- D. K. Mishra, G. Pugazhenthi and T. Banerjee, Ionic Liquid-Based Deep Eutectic Solvent as Reaction Media for the Thermal Dehydrogenation of Ethylene Diamine-Bis-Borane, ACS Sustainable Chem. Eng., 2020, 8, 4910–4919 CrossRef CAS.
- Z. Fang, J. Luo, X. Kang, H. Xia, S. Wang, W. Wen, X. Zhou and P. Wang, Facile Solid-Phase Synthesis of the Diammoniate of Diborane and Its Thermal Decomposition Behavior, Phys. Chem. Chem. Phys., 2011, 13, 7508–7513 RSC.
- W. Sun, Q. Gu, Y. Guo, Z. Guo, H. Liu and X. Yu, Hydrazine Bisborane as a Promising Material for Chemical Hydrogen Storage, Int. J. Hydrogen Energy, 2011, 36, 13640–13644 CrossRef CAS.
- S. Pylypko, E. Petit, P. G. Yot, F. Salles, M. Cretin, P. Miele and U. B. Demirci, Key Study on the Potential of Hydrazine Bisborane for Solid- and Liquid-State Chemical Hydrogen Storage, Inorg. Chem., 2015, 54, 4574–4583 CrossRef CAS PubMed.
- K. Sen, T. Banu, T. Debnath, D. Ghosh and A. K. Das, Catalytic Role of Borane and Alane in Hydrogen Release from Cyclic Amine Adducts CnH2n + 1N·XH3 [X = B, Al; N = 2–5]: A Theoretical Interpretation, RSC Adv., 2014, 4, 21924–21938 RSC.
-
A. M. Ja’o, Experimental and Theoretical Study of New Compounds for Hydrogen Storage, PhD thesis, University of Canterbury, 2019.
- A. M. Ja’O, S. L. Masters, D. A. Wann, C. D. Rankine, J. P. F. Nunes and J. C. Guillemin, Direct Experimental Observation of in Situ Dehydrogenation of an Amine-Borane System Using Gas Electron Diffraction, J. Phys. Chem. A, 2019, 123, 7104–7112 CrossRef PubMed.
- A. M. Ja’o, D. A. Wann, C. D. Rankine, J. P. F. Nunes, J. C. Guillemin and S. L. Masters, Structural and Thermochemical Studies of Pyrrolidine Borane and Piperidine Borane by Gas Electron Diffraction and Quantum Chemical Calculations, Struct. Chem., 2021, 32, 205–213 CrossRef.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 16 (Revision C.01), Gaussian, Inc., Wallingford, CT, 2019 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Function, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, Self-Consistent Molecular Orbital Methods. XXIII. A Polarization-Type Basis Set for Second-Row Elements, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- M. J. Frisch, M. Head-Gordon and J. A. Pople, A Direct MP2 Gradient Method, Chem. Phys. Lett., 1990, 166, 275–280 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self-Consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, Self-Consistent Molecular Orbital Methods 25. Supplementary Functions for Gaussian Basis Sets, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- D. E. Woon and T. H. Dunning, Gaussian Basis Sets for Use in Correlated Molecular Calculations. V. Core-Valence Basis Sets for Boron through Neon, J. Chem. Phys., 1995, 103, 4572–4585 CrossRef CAS.
- L. A. Curtiss, K. Raghavachari, P. C. Redfern and J. A. Pople, Assessment of Gaussian-2 and Density Functional Theories for the Computation of Enthalpies of Formation, J. Chem. Phys., 1997, 106, 1063–1079 CrossRef CAS.
- T. H. Dunning, Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- D. Feller, K. A. Peterson and J. Grant Hill, On the Effectiveness of CCSD(T) Complete Basis Set Extrapolations for Atomization Energies, J. Chem. Phys., 2011, 135, DOI:10.1063/1.3613639.
- C. Peng and H. B. Schlegel, Combining Synchronous Transit and Quasi-Newton Methods to Find Transition States, Isr. J. Chem., 1993, 33, 449–454 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, Reaction Path Following in Mass-Weighted Internal Coordinates, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- M. T. Nguyen, V. S. Nguyen, M. H. Matus, G. Gopakumar and D. A. Dixon, Molecular Mechanism for H2 Release from BH3NH3, Including the Catalytic Role of the Lewis Acid BH3, J. Phys. Chem. A, 2007, 111, 679–690 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- J. Zhang and E. F. Valeev, Prediction of Reaction Barriers and Thermochemical Properties with Explicitly Correlated Coupled-Cluster Methods:
A Basis Set Assessment, J. Chem. Theory Comput., 2012, 8, 3175–3186 CrossRef CAS PubMed.
- F. R. M. Irfan and S. L. Masters, A Computational Investigation of Pyrrole Borane as a Potential Hydrogen Storage System, Comput. Theor. Chem., 2023, 1225, 114156 CrossRef.
- H. E. Kissinger, Reaction Kinetics in Differential Thermal Analysis, Anal. Chem., 1957, 29, 1702–1706 CrossRef CAS.
- F. Wang, Y. Liu, M. Gao, K. Luo, H. Pan and Q. Wang, Formation Reactions and the Thermodynamics and Kinetics of Dehydrogenation Reaction of Mixed Alanate Na2LiAlH6, J. Phys. Chem. C, 2009, 113, 7978–7994 CrossRef CAS.
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*
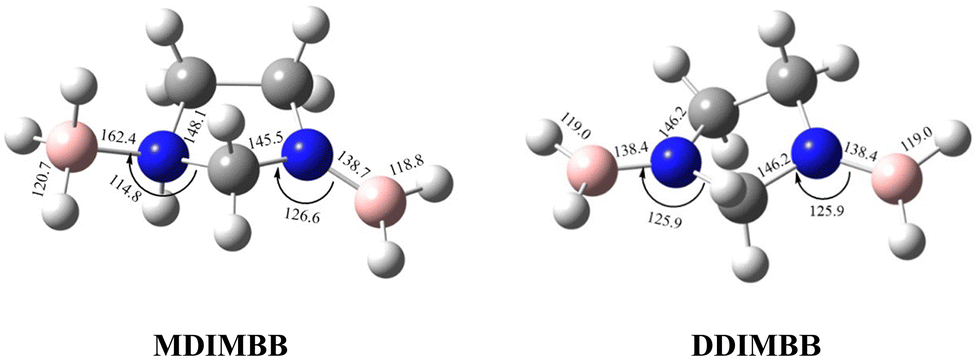
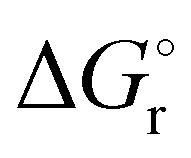 and positive value of
and positive value of 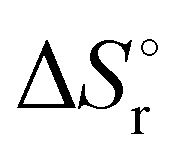 along with the products having lower Gibbs free energy compared to the reactants (as shown in Fig. 6 and 7), suggested a fast, exergonic and favourable dehydrogenation reaction for IMBB.
along with the products having lower Gibbs free energy compared to the reactants (as shown in Fig. 6 and 7), suggested a fast, exergonic and favourable dehydrogenation reaction for IMBB.
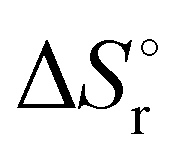 which is in J K−1 mol−1. First and Second implies the reaction to release the first and second H2 molecule from IMBB.
which is in J K−1 mol−1. First and Second implies the reaction to release the first and second H2 molecule from IMBB.