
SAXS unveils porous anodes for potassium-ion batteries: dynamic evolution of pore structures in Fe@Fe2O3/PCNFs composite nanofibers†
Ruiqi
Shao
a,
Yingjie
Dong
a,
Qingqing
Wu
a,
Haiting
Shi
*a,
Jinxi
Bao
a,
Feng
Tian
b,
Tianyu
Li
a and
Zhiwei
Xu
 *a
*a
aState Key Laboratory of Separation Membranes and Membrane Processes, School of Textile Science and Engineering, Tiangong University, Tianjin 300387, China. E-mail: shihaiting@tiangong.edu.cn; xuzhiwei@tiangong.edu.cn
bShanghai Synchrotron Radiation Facility, Zhangjiang Lab, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201204, China
First published on 23rd January 2024
Abstract
The porous structure of composite nanofibers plays a key role in improving their electrochemical performance. However, the dynamic evolution of pore structures and their action during ion intercalation/extraction processes for negative electrodes are not clear. Herein, porous carbon composite nanofibers (Fe@Fe2O3/PCNFs) were prepared as negative electrode materials for potassium-ion batteries. Electrochemical test findings revealed that the composites had good electrochemical characteristics, and the porous structure endowed composite electrodes with pseudo-capacitive behaviors. After 1500 discharge/charge cycles at a current density of 1000 mA g−1, the specific capacity of the potassium-ion batteries was 144.8 mAh g−1. We innovatively used synchrotron small-angle X-ray scattering (SAXS) technique to systematically investigate the kinetic process of potassium formation in composites and showed that the kinetic process of potassium reaction in composites can be divided into four stages, and the pores with smaller average diameter distribution are more sensitive to changes in the reaction process. This work paves a new way to study the deposition kinetics of potassium in porous materials, which facilitates the design of porous structures and realizes the development of alkali metal ion-anode materials with high energies.
1. Introduction
The development of lithium-ion batteries for sustainable and clean energy storage is limited by the scarcity of lithium resources.1,2 Therefore, it is urgent to find a new type of battery energy storage system that is rich in resources and similar to a lithium-ion battery.3 Sodium/potassium batteries are considered the most promising materials to replace lithium-ion batteries.4–10 Although potassium ion batteries are a very promising energy storage system, the development of their practical applications has a long way to go. Advanced electrode materials are an important component in optimising electrochemical performance and facilitating effective energy storage.11 However, the larger radius of Na+/K+ compared to Li+ leads to severely slow reaction kinetics, fragmented electrode structures, and much poorer rate capability and cyclability.12–14 Therefore, there is an urgent need to investigate suitable anode materials for potassium and sodium ion batteries. In addition, potassium ions15 are more readily and reversibly embedded in graphite materials compared to sodium ions.16,17 Thus, extensive research should focus on developing potassium ion batteries with high energy and power density and long service life.10Compared to traditional insertion reaction electrode materials, transition metal oxides have higher capacity and rechargeable electrical capacity and are widely sourced and non-toxic, thus attracting much attention.18,19 Anode electrodes used for potassium ion batteries are mainly metal oxides (CuO,20,21 Ti6O11, and Co3O422–24), transition metal sulfides (SnS2, Sb2S3, FeS2, NiS, Cu2S, and CoS), transition metal selenides (FeSe2, ZnSe, and NiSe2), and so on. Fe2O3, a conversion-based metal oxide, has high theoretical capacity but large volume change and low conductivity. Considering the high-volume expansion rate and low conductivity of the Fe2O3 material itself, research should focus on improving the electrochemical performance of iron oxide as an anode material for potassium ion batteries. It is generally believed that structural damage can be reduced by adjusting nano-structures or surface properties.25,26
The rational design of carbonaceous composites plays an important role in improving their potassium storage performance. The fine tuning of composite nanostructures can improve the diffusion of potassium ions, enhance reaction kinetics, shorten the diffusion distance, provide multiple channels between the electrolyte and the active material, etc., thus improving electrochemical performance. Moreover, the carbonaceous composites with different dimensional characteristics showed satisfactory electrochemical performance when used as anodes for potassium ion batteries.27 For example, Wu et al. assembled highly dispersed cobalt nanodots embedded in N, P, S co-doped hexapod carbons by polymer coating and high-temperature carbonisation processes using 3D hexapod-shaped ZIF-67 as a precursor to efficiently increase the specific surface area, which provided a complementary range of contact between the electrolyte and the electrode, and showed excellent multiplicative capacity and cycling stability after 450 cycles.28 Furthermore, it has been demonstrated that creating a porous structure is a successful strategy for enhancing electrochemical characteristics including capacity, charge/discharge efficiency, and cycling life.29–32 Guo et al.33 used a self-etching strategy to construct interconnected mesoporous carbon structures, and the richly ordered mesoporous structure could provide a short transport path for K+. Xu et al.34 prepared N-doped porous carbon nanofiber electrodes with abundant micropore structures in the electrode. The cell was able to give a capacity of 635 mAh g−1 at 50 mA g−1 for 50 cycles thanks to the synergistic impact of pore confinement and N doping. It has been established that macropores (dp > 50 nm) can facilitate the penetration of K+ and serve as a buffer space as well as electrolyte reservoirs, significantly improving the structural stability and capacity;35 mesopores (2 nm < dp < 50 nm) reduce the ion diffusion distance and prevent mechanical deterioration;36 micropores (dp < 2 nm) could provide an active site for the adsorption of K ions primarily in the high-potential zone, thereby increasing the capacity.37
The above-mentioned studies have shown that porous structures and the control of the pore size in the porous carbon skeleton are important for improving the electrochemical performance of potassium ion batteries.38–40 Therefore, various approaches have been used to explore the mechanism of pore structure. The potassium intercalation mechanism in hard carbons was revealed by Zhang et al.41 The porous electrode was characterized by in situ XRD assisted by a high-resolution mapping technique under two successive cycles of potassiation and depotassiation, revealing the potassium ion storage mechanism. Li et al.42 used quantitative kinetic analysis to analyze the capacitance- and diffusion-controlled charge storage in carbon foam electrodes. It was demonstrated that the adsorption of potassium ions by the porous electrode mainly depends on the porous structure and large specific surface area, and the adsorption mechanism significantly increases the capacity of the functional electrode. Chen et al.43 reveal the potassium storage mechanism of the chitin-derived hierarchical porous nitrogen-doped carbon microsphere electrodes via simulation calculations. This was attributed to the hierarchical porous microstructure and the nitrogen-internal carbon structure. Although the researchers uncovered the porous electrodes’ mode of operation via density functional theory, there is still a gap in exploring the changes in pore structure and specific mechanisms of action during charging and discharging. Due to the high-quality light source with a continuous spectrum, high intensity and collimation, synchrotron small-angle x-ray scattering (SAXS) was confirmed to be an advanced technique to explore the microscopic dynamic structure of matter and the processes of various transients.44 Our group has recently45 used SAXS to characterize the pore structure information of protein separators in batteries and explored the inhibition mechanism of separators. Based on our previous experience,46 we believed that characterizing the dynamic evolution of pore structures in ion battery electrodes during the charge/discharge process via the synchrotron radiation technique could demonstrate electrochemical performance's connections with pore structure systematically.
In this work, Fe2O3 nanofibers with a porous structure were prepared by electrospinning technology and the micropores, mesopores and macropores were produced by the decomposition of the pore-forming agent PMMA. The graded pores and high specific surface area could improve the diffusion rate of ions, thus improving electrochemical performance. In addition, by adjusting the calcination temperature, the introduction of elemental Fe into Fe2O3 porous carbon nanofibers can boost the material's conductivity, which is conducive to the formation of three-dimensional conductive networks. Fe@Fe2O3/PCNFs have a hierarchical pore structure and a 3D conductive network. The unique shape can facilitate the quick transport of potassium ions as well as respond to the severe volume variations of active materials. To investigate the action mechanisms of pore structures throughout the charge–discharge cycle, SAXS was used to further investigate the influence of the variation in structural scale, certain surface area, dispersion of pore sizes, and interface of nanoparticles or nanopores on the electrochemical performance of the Fe@Fe2O3/PCNFs in detail. It will provide a direction for the development of porous electrode structures in ion batteries.
2. Experimental section
2.1. Fabrication of porous Fe@Fe2O3/PCNFs composites
Polyacrylonitrile (PAN) (Mw ≈ 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Aladdin), N,N-dimethylformamide (DMF) (≥ 99.9%, Aladdin), iron(III) acetylacetonate (FeAcAc) (98%, Aladdin), and poly(methyl methacrylate) (PMMA) (Universal shooting level, Aladdin) were used without purification. First, 8 g of DMF solution was thoroughly mixed with 1 g each of PAN and PMMA at 60 °C for 12 h. Then, 1 g of FeAcAc was added to the mixture by stirring for 12 h to ensure homogenous dispersion of FeAcAc in the solution. At a feed rate of 0.0010 mm s−1, the precursor solution was then injected into a syringe. The end of the needle formed a stable Taylor cone, when the applied voltage between the needle and the collector was 14 KV, and the electrospun nanofibers were collected on a drum collector with respect to the needle, 15 cm away. Because of the color of FeAcAc salt itself, the fiber film was light red. After the fiber film was peeled from the collector, it needed to be dried in the oven at 60 °C for 24 h to remove excess organic solvents. Then, the fiber film was cut into a suitable size and put between two graphite plates for pre-oxidation, and it was stabilized by heating in an oven at 280 °C for 3 h in air. The stabilized film was carbonized in a tube furnace at 600 °C for 2 h in an argon environment to obtain the porous Fe@Fe2O3/PCNFs composite film. The raising rate of the temperature from room temperature to 600 °C was 3 °C min−1. After carbonization, the film became black, and the conductivity of the film had been improved.
000, Aladdin), N,N-dimethylformamide (DMF) (≥ 99.9%, Aladdin), iron(III) acetylacetonate (FeAcAc) (98%, Aladdin), and poly(methyl methacrylate) (PMMA) (Universal shooting level, Aladdin) were used without purification. First, 8 g of DMF solution was thoroughly mixed with 1 g each of PAN and PMMA at 60 °C for 12 h. Then, 1 g of FeAcAc was added to the mixture by stirring for 12 h to ensure homogenous dispersion of FeAcAc in the solution. At a feed rate of 0.0010 mm s−1, the precursor solution was then injected into a syringe. The end of the needle formed a stable Taylor cone, when the applied voltage between the needle and the collector was 14 KV, and the electrospun nanofibers were collected on a drum collector with respect to the needle, 15 cm away. Because of the color of FeAcAc salt itself, the fiber film was light red. After the fiber film was peeled from the collector, it needed to be dried in the oven at 60 °C for 24 h to remove excess organic solvents. Then, the fiber film was cut into a suitable size and put between two graphite plates for pre-oxidation, and it was stabilized by heating in an oven at 280 °C for 3 h in air. The stabilized film was carbonized in a tube furnace at 600 °C for 2 h in an argon environment to obtain the porous Fe@Fe2O3/PCNFs composite film. The raising rate of the temperature from room temperature to 600 °C was 3 °C min−1. After carbonization, the film became black, and the conductivity of the film had been improved.
2.2 Material characterization
SEM (Hitachi S4800), TEM (Hitachi H7650), and energy-dispersive X-ray spectrometer (EDS) with TEM were used to evaluate the surface structure and morphology of the composite fiber film. EDS also captured the element mapping. Bruker Smart 1000 diffractometer with Cu Kα (λ = 1.5418 Å) radiation was used to study the X-ray diffraction (XRD) patterns of porous Fe@Fe2O3/PCNFs in a 2θ range from 20° to 80°. On the surfaces of the composite fiber film, chemical states of the constituent elements were determined by X-ray photoelectron spectroscopy (XPS) studies (NEXSA, Thermo Fisher). To examine the flaws in the porous nanofibers in the film, a Laser Confocal Raman Spectroscopy sample was taken (XploRA PLUS, Horiba, 532 nm argon-ion excitation). Using an automated gas sorption analyzer (Autosorb-iQ-C), N2 adsorption and desorption isotherms were performed to further examine the surface area and pore diameter of composites. A thermogravimetric analyzer (TG 209 F3 Tarsus) was performed to determine the Fe2O3 contents in the composite film by increasing the temperature from room temperature to 800 °C in the air at a heating rate of 10 °C min−1.2.3. Electrode measurements
Since the carbonized fiber film structure was retained and still had flexibility and bendability, the electrode could be prepared by punching. The electrical characteristics were characterized by galvanostatic cycling of 2025 coin-type half cells with the porous Fe@Fe2O3/PCNFs composite as the working electrode. PIBs (CR 2025) were assembled with a porous Fe@Fe2O3/PCNFs composite electrode, a separator (Whatman GF/A), and a metallic K sheet in an argon-filled glove box (O2 ≤ 0.1 ppm, H2O ≤ 0.1 ppm). The electrolyte for the test was 0.8 M KFSI in a mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) solution (1:1 volume). SIBs (CR 2025) were assembled by a working electrode composed of porous Fe2O3@Fe/CNF and a reference electrode composed of a Na-material foil. Then, 1 M NaClO4 in a mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) (1:1 volume) solution was used as the electrolyte. Galvanostatic discharging/charging was tested in the fixed voltage window between 0.01 and 3 V at 25 °C using a LAND tCT3001A system. The rate test of the cells was conducted under different current densities (10 cycles each at 100 mA g−1, 200 mA g−1, 500 mA g−1, 1000 mA g−1, and 2000 mA g−1). On a CHI 660E, measurements were made by cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS).
2.4. Synchrotron small-angle X-ray scattering
Synchrotron small-angle X-ray scattering technology is an important technology to detect the microstructure of materials, frequently used to investigate nanoscale materials (nanoparticles or nanoholes) with uneven electron densities' structural size, specific surface, pore size distribution, and interface data. Porous carbon nanofibers are heterogeneous porous media, and the internal pores are mainly distributed on the micro-scale. The porous Fe@Fe2O3/PCNFs electrodes were tested under different potentials to reveal the influence of the number and morphology of the holes of the porous negative electrode material on the performance of the battery. The SAXS dataset was obtained from the X-ray small-angle scattering beamline station (BL16B1) of the Shanghai Synchrotron Radiation Facility (SSRF) in China.3. Results and discussion
Fig. 1 illustrates the detailed preparation process of Fe@Fe2O3/PCNFs. PAN and PMMA with a certain mass ratio were added into DMF for heating and stirring, and iron acetylacetonate (FeAcAc) was added after complete dissolution, and then mixed evenly and allowed to stand still to prepare an electrospinning precursor solution. Polymer composite nanofibers were successfully prepared by electrospinning. Under the condition of the external electric field, when the needle extrudes the solution, a continuous polymer jet is released after the external electric field exceeds the surface tension of the solution. With the volatilization of the solvent, a nanofiber network is formed on the surface of the collector. After the nanofiber mesh is stripped from the collector, it needs to be pre-oxidized under air conditions to convert thermoplastic PAN into nonplastic cyclic or trapezoidal compounds. The structure of the nanofiber mesh is stabilized to prevent the structure of the nanofiber mesh from being damaged by excessive shrinkage in the subsequent calcination process. During the calcination of polymer composite fiber mesh, PMMA is used as a pore-forming agent. When the temperature rises to a certain extent, PMMA is separated from the composite fiber and decomposed to form pores, which can produce a porous structure without other means. At the same time, PAN and iron acetylacetonate begin to decompose, and iron acetylacetonate is oxidized to iron oxide nanoparticles. When the decomposition products of PAN and PMMA escape from the fiber, some iron oxide particles inside the fiber will be pushed to the surface of the fiber, making iron oxide particles evenly distributed inside and on the surface of the fiber. Due to the reducibility of carbon at high temperatures, part of iron oxide will be reduced to iron, so it will eventually evolve into Fe@Fe2O3/PCNFs. Fig. 1b represents the frequency distribution curves for the obtained nanofibers and nanoparticles. Fiber diameter and particle radius were obtained using software that measured the dimensions in the SEM image and then averaged them. According to this figure, the average diameter of the PAN/PMMA/FeAcAc electrospun nanofibers is around 1 micron, the average diameter of the calcined nanofibers is about 0.5 microns, and the average diameter of the Fe2O3 particles obtained by oxidation after calcination is around 64 nm.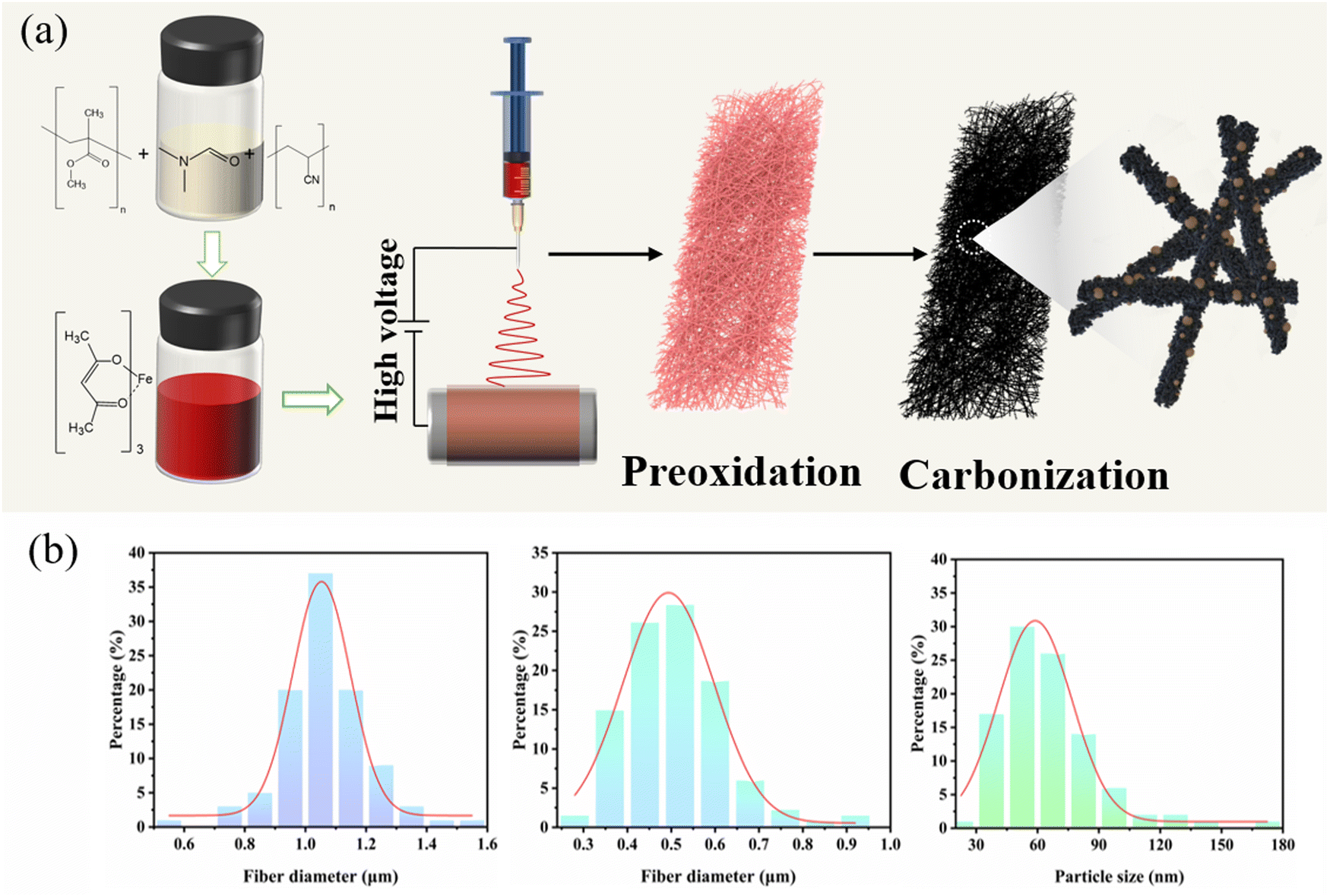 | ||
| Fig. 1 (a) Schematic illustration of the preparation process of Fe@Fe2O3/PCNFs. (b) Fiber diameter and distribution curves: PAN/PMMA/FeAcAc electrospun nanofibers, Fe@Fe2O3/PCNFs, and Fe@Fe2O3 nanoparticles. | ||
The SEM image in Fig. 2a shows the morphology of the PAN/PMMA/FeAcAc electrospun nanofibers. The nanofibers without heat treatment have uniform diameter distribution around 1 micron. The fiber surface is smooth without obvious nodules, which indicates that there is no phase separation of the polymer during spinning. From the SEM image of the Fe@Fe2O3/PCNFs in Fig. 2b, it can be observed that the surface of the nanofibers is rough, with obvious pore structures, and the nanoparticles on the surface and wrapped inside of the carbon fibers present uniform distribution. Although the diameter of the fibers after calcination treatment is reduced, the fiber structure is still maintained, and there is no phenomenon of fiber fracture, pulverization, or agglomeration of nanoparticles. It not only shows that the iron source compound can be uniformly dispersed in the spinning solution under the raw material ratio, but also shows that nanoparticles, pore structure, and fiber structure coexist well at the specific calcination temperature. The pore structure and nanoparticle distribution inside the nanofibers can be seen clearly in the TEM picture (Fig. 2c), particularly the uniform dispersion of particles on the carbon matrix's surface. In addition, the pores connected inside and outside the fiber can not only improve the material's precise surface area, and accelerate the infiltration of electrolytes, but also help to alleviate the increase in nanoparticle volume during the discharge procedure. The EDS mapping of C, O, and Fe from TEM (Fig. 2d) illustrates the uniform distribution of Fe@Fe2O3 nanoparticles in carbon nanofibers. The pattern of the chosen area's electron diffraction of Fe@Fe2O3/PCNFs (Fig. 2e) showed the polycrystalline characteristics of the material. The diffraction rings match well with the (220) and (311) planes of γ-Fe2O3 and the (110) plane of cubic Fe. According to the HRTEM images in Fig. 2f, the spacing of the two lattice stripes at approximately 0.20 nm and 0.25 nm corresponds to the (110) crystal face of iron and (311) crystal faces of iron oxide, respectively. Meanwhile, the lattice stripes of amorphous carbon around the nanoparticles can also be observed. Combined with the SEAD and HRTEM results, it can be judged that porous nanofibers with core–shell structure nanoparticles wrapped by a carbon matrix are finally formed after heat treatment, wherein the core of core–shell nanoparticles is iron and the shell is iron oxide. The formation of this structure is mainly due to the reduction of Fe2O3 induced by carbon formed after the carbonization of PAN/PMMA/FeAcAc electrospun nanofibers at high temperatures to Fe. Iron and carbon can be added to a substance to increase its electrical conductivity.
 | ||
| Fig. 2 (a) SEM image of pristine PAN/PMMA/FeAcAc electrospun nanofibers. (b) SEM image of Fe@Fe2O3/PCNFs. (c) Low-resolution TEM image of Fe@Fe2O3/PCNFs. (d) Results of EDS elemental mapping of Fe@Fe2O3/PCNFs corresponding to C, Fe, and O. (e) Selected area electron diffraction (SAED) pattern of Fe@Fe2O3/PCNFs. (f) HRTEM image of Fe@Fe2O3/PCNFs. | ||
The XRD pattern of the Fe@Fe2O3/PCNFs composite obtained by electrospinning and calcination is shown in Fig. 3a. The diffraction peaks at 30.2°, 35.6°, 43.2°, 53.7° and 57.2° matched with the (220), (311), (400), (422) and (511) crystal faces of standard γ-Fe2O3 (JCPDS No. 39-1346), respectively. In addition, through comparison, it is found that the diffraction peak situated at 44.3° corresponds to the (110) crystal plane of pure phase Fe (PDF#85-1410). This indicates that the composite of Fe and Fe2O3 is formed. The characteristic vibrational patterns of γ-Fe2O3 are observed in the Raman spectra (Fig. 3b) corresponding to the peaks of 210, 269, 430, 507 and 609 cm−1. The strong peaks at 1327 and 1580 cm−1 in Raman spectra are mainly caused by the carbonization, representing D and G bands of defect carbon, respectively. The D peak's intensity is primarily a reflection of the carbon matrix's flaws. The ID/IG value is about 1.1, which has the characteristics of amorphous graphite. These defects can provide more active sites for the reaction process. The composite made of Fe@Fe2O3/PCNFs is illustrated in Fig. 3c along with the N2 adsorption/desorption isotherm curve. It can be observed from the figure that a clear hysteresis loop appears in the isotherm when the relative pressure is high, indicating that the sample has mesoporous properties. During the calcination process, the pore-forming agent produces a pore structure on the surface and inside of the fiber. This structure is conducive to the uniform deposition of nanoparticles embedded in carbon nanofibers. It can not only effectively separate nanoparticles to prevent agglomeration, but also stabilize the fiber structure to alleviate the collapse of the structure caused by the growth of volume during charging and discharging. The test results of composite materials by the BET method show that the specific surface area is 348.125 m2 g−1, the pores' size is 3.8 nm (as shown in the illustration), and the total pore volume is 0.278 cm3 g−1. The mass fraction of γ-Fe2O3 and carbon in composites made of Fe@Fe2O3/PCNFs was calculated using TGA analysis (Fig. 3d). The weight loss was not noticed until the room temperature rose to about 300 °C, which indicates that the composite had good stability in the air. The oxidation of carbon is mostly responsible for the weight loss between 300 °C and 500 °C. When the temperature rises to 800 °C, the mass of the composite does not change significantly, and the mass percentage of the remaining material is about 23.4%. To further confirm the composition of the material, the XPS spectra of the obtained composite material were recorded. Four distinct characteristic peaks appear in Fig. 3e, which are indexed as Fe 2p, O 1s, N 1s and C 1s. The main peak in Fig. 3f can be well fitted by six peaks, of which the two typical characteristic peaks at 710.2 eV and 724.1 eV correspond to Fe 2p3/2 and Fe 2p1/2 of Fe3+, respectively. The 718.9 eV and 732.1 eV peaks are indexed to the satellite peaks of Fe 2p3/2 and Fe 2p1/2 for Fe3+. The 706.5 eV peak corresponds to the Fe 2p3/2 peak in Fe0, and the signal at 719.2 eV is the satellite peak for metallic iron. From these peaks, it can be inferred that the composite is mainly composed of Fe and Fe2O3. In the C 1s spectrum (Fig. 3g), the peaks at 284.8, 286.1, 287.4 and 288.9 eV can be assigned to C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–N/CC, C–O–C and O–CO, indicating that PAN is well carbonized. In the O 1s spectra (Fig. 3h), the main peak can be fitted by the Fe–O, CO/C–O, and C–O–C peaks at 529.8, 531.1 and 533.3 eV, which confirm that iron oxides and faulty carbon are present. The N 1s spectrum (Fig. 3i) consists of four peaks centered at 398.0, 399.9, 401.0 and 402.9 eV, matching pyridine N, pyrrole N, graphite N and N–O, respectively. Combined with TGA and XPS quantitative analyses, the proportion of Fe, Fe2O3 and carbon in the Fe@Fe2O3/PCNFs composite is 10.8%, 12.6% and 76.6%.
C, C–N/CC, C–O–C and O–CO, indicating that PAN is well carbonized. In the O 1s spectra (Fig. 3h), the main peak can be fitted by the Fe–O, CO/C–O, and C–O–C peaks at 529.8, 531.1 and 533.3 eV, which confirm that iron oxides and faulty carbon are present. The N 1s spectrum (Fig. 3i) consists of four peaks centered at 398.0, 399.9, 401.0 and 402.9 eV, matching pyridine N, pyrrole N, graphite N and N–O, respectively. Combined with TGA and XPS quantitative analyses, the proportion of Fe, Fe2O3 and carbon in the Fe@Fe2O3/PCNFs composite is 10.8%, 12.6% and 76.6%.
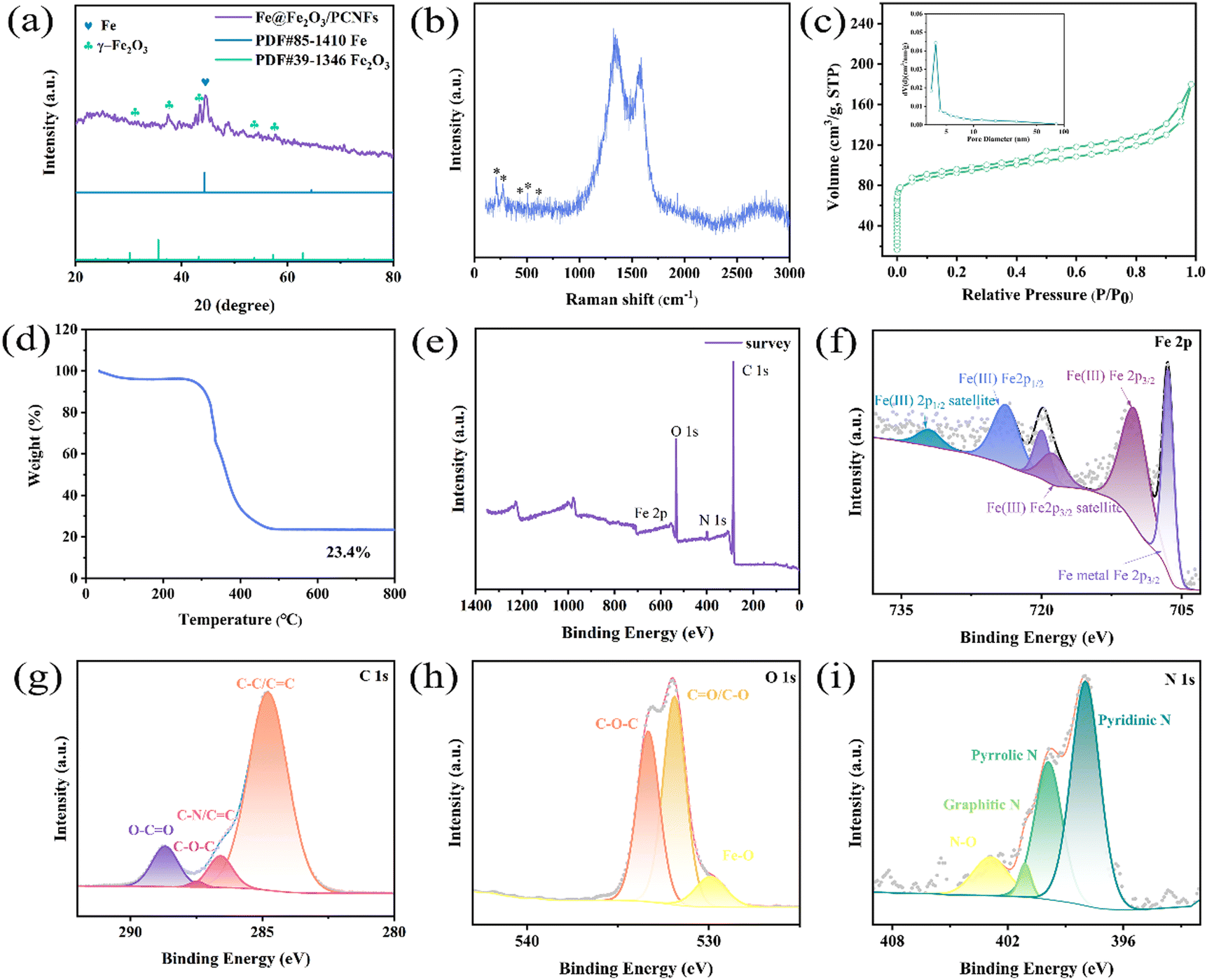 | ||
| Fig. 3 (a) XRD pattern, (b) Raman spectra, (c) N2 adsorption/desorption isotherm curve, and pore size distribution diagram (illustration). (d) The results of TGA. XPS spectra: (e) survey, (f) Fe 2p spectrum, (g) C 1s spectrum, (h) O 1s spectrum, and (i) N 1s spectrum of Fe@Fe2O3/PCNFs. | ||
The electrochemical performance of the Fe@Fe2O3/PCNFs composite was studied by using a coin cell. At a scan rate of 0.1 mV s−1, the cyclic voltammetry (CV) of Fe@Fe2O3/PCNFs in the range of 0.01–3.0 V is shown in Fig. 4a. During the first cycle, an obvious cathode peak appears at about 0.62 V, which represents the reduction reaction of Fe2O3. In the second and third cycles, the wide cathode peak at 0.73 V was also attributed to the reduction of Fe2O3. The CV curves obtained from the following two scans are highly coincident, and the obvious difference from the first cycle of the CV curve is due to the irreversible loss caused by the formation of SEI. The anodic peak at about 1.0 V was caused by the oxidation reaction of Fe during the first cycle of anodic scanning, but only a narrow peak at about 0.9 V could be observed in the following two cycles of scanning. It is worth noting that the phenomenon that the redox peaks in the CV curves of the last two circles are not obvious may be caused by the low redox kinetics. The reaction process is described in the following equation:
| Fe2O3 + 6K+ + 6e− → 2Fe + 3K2O | (1) |
| 2Fe + 3K2O → Fe2O3 + 6K+ + 6e− | (2) |
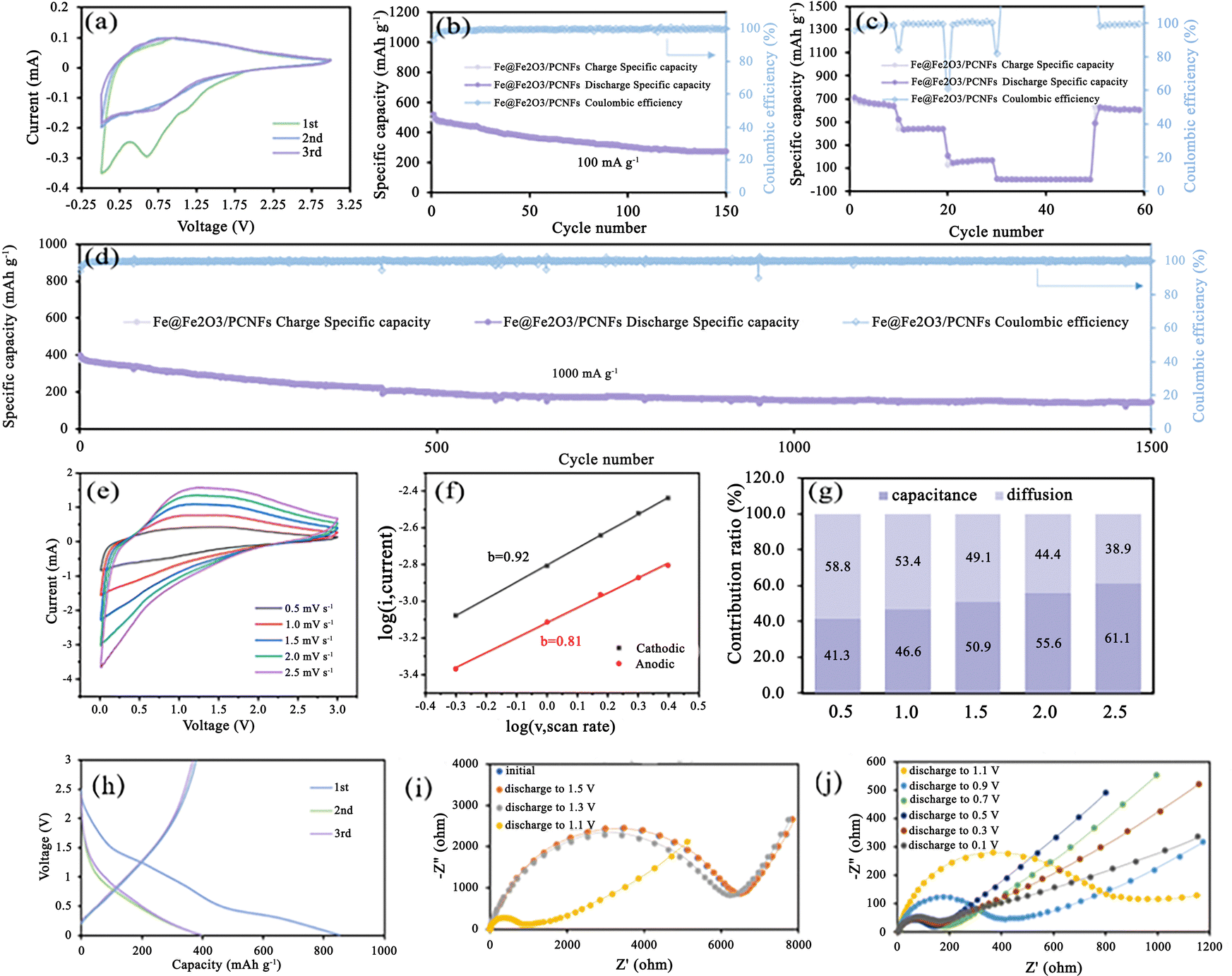 | ||
| Fig. 4 (a) CV curves at a scan rate of 0.1 mV s−1, (b) charge/discharge specific capacity, and Coulombic efficiency measured for 150 cycles at a current density of 100 mA g−1. (c) Rate capability at a current density of 100, 200, 500, 1000, 2000, and 100 mA g−1. (d) Long-term cycling performance at 1000 mA g−1. (e) CV curves at different scan rates of 0.5, 1.0, 1.5, 2.0, and 2.5 mV s−1. (f) The b value and (g) capacitance and diffusion contribution ratios at different scan rates of 0.5, 1.0, 1.5, 2.0, and 2.5 mV s−1 of the Fe@Fe2O3/PCNFs composite in K-ion cells. (h) The first three cycles of charge/discharge curves in potassium ion cells, (i) EIS spectra of the initial state, discharging to 1.5 V, 1.3 V, and 1.1 V and (j) EIS spectra when discharging to 1.1 V, 0.9 V, 0.7 V, 0.5 V, 0.3 V, and 0.1 V. | ||
Fig. 4a shows the charge/discharge curves for the first three cycles, which is in agreement with the findings of the CV measurement. An initial discharge capacity of 853 mAh g−1 was shown during the first cycle. It is inevitable to see significant capacity reduction after the first cycle. The coulombic efficiency of the next two laps was almost 100%; however, the charge–discharge curves of those two laps are heavily overlapping and do not show a significant capacity decline, remaining around 396 mAh g−1. Owing to the composite's strong structural stability and high conductivity, the K-ion cell has good cyclic performance, the initial discharge capacity is 487 mAh g−1 at 100 mA g−1, and a stable discharge capacity of 273 mAh g−1 was maintained after 150 cycles, with the coulombic efficiency remaining at 100% and a capacity retention rate of 56% (Fig. 4b). The irreversible capacity loss is caused by the development of the SEI layer and the irreversible phase transformation during the production of potassium. To further explore the rate performance of Fe@Fe2O3/PCNFs composites, we adopted the following methods: change the current density every 10 cycles, gradually increase the current density from 100 mA g−1 to 2000 mA g−1, and finally recover to 100 mA g−1. In general, the Fe@Fe2O3/PCNFs composites show good rate properties. Fe@Fe2O3/PCNFs composites had reversible capacities of 640.9, 209.6 and 167.8 mAh g−1 at current densities of 100, 200 and 500 mA g−1, respectively. When the current density drops to 100 mA g−1 from high current densities of 1000 and 2000 mA g−1, the specific capacity of the composites promptly increases to 627 mA g−1. After 60 cycles, the material retains 85.2% of the initial reversible capacity, showing favorable reversibility (Fig. 4c). Fe@Fe2O3/PCNFs composites' exceptional multiplicative properties can be linked to their distinctive porous fiber structure. Fig. 4d illustrates the long-cycle performance of the Fe@Fe2O3/PCNFs electrode at 1000 mA g−1 for the purpose of examining the cycling stability of the composite at high current densities. After 1500 discharging/charging cycles, 144.8 mAh g−1 specific capacity can still be obtained, accounting for 31.3% of the initial specific capacity. To investigate whether the Fe@Fe2O3/PCNFs electrode exhibits simultaneous redox conversion and surface capacitance behavior, composite-assembled coin cells are subjected to CV testing at different sweep speeds from 0.5 to 2.5 mV s−1 (Fig. 4e). The dynamic relationship between the current density i and the scanning speed v was studied as shown in (3), and then the results were converted to get formula (4):
| i = avb | (3) |
logi = loga + b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) logv logv | (4) |
Substitute different scanning speeds and the corresponding current peaks into the formula, and the fitting cathode and anode b values in Fig. 4f are 0.92 and 0.81 respectively. Because the calculated b value is between 0.5 and 1.0, at this time, the battery has a pseudo-capacitance property. To know the proportion of specific capacitance behavior and diffusion behavior in the total capacitance, the following formula was used to calculate the contribution rate of capacitance and diffusion behavior:
| i = avb = icap + idiff = k1v + k2v1/2 | (5) |
| i/v1/2 = k1v1/2 + k2 | (6) |
The capacitance contribution ratios obtained by scanning at rates of 0.5, 1.0, 1.5, 2.0 and 2.5 mV s−1 are 41.3%, 46.6%, 50.9%, 55.6% and 61.1%, respectively (Fig. 4g). The surface capacitance behavior of the composite increases with the increase in the scanning speed, which indicates the dominant role of the surface capacitance behavior. The discharge/charge curves for the composite's first three cycles in the potassium system are shown in Fig. 4h, and they are consistent with the CV data. The platform area of the first circle is not obvious corresponding to the transformation process. A high initial discharge capacity of 853 mAh g−1 is seen in the first cycle. After the first cycle, significant capacity degradation can be observed, which is unavoidable. However, the charging and discharging curves of the following two cycles are highly coincident, and there is no obvious capacity attenuation. The capacity is maintained at around 396 mAh g−1, and these two cycles' coulombic efficiency is very close to 100%. The electrode material in the potassium-based system has a considerably high initial discharge capacity during the first cycle. The first circle of the platform area corresponds to the reduction reaction stage in the CV curve. After three times of charging and discharging, 738 mAh g−1 is a stable value for the capacity. Fig. 4i and j's Nyquist diagram illustrates the impedance spectrum of the material used as the anode electrode in potassium-ion batteries. Semicircles are clearly visible at high frequencies, while vertical lines are seen at low frequencies at various discharge potentials. The size of the RF, or the battery's charge transfer resistance, is related to the semicircle diameter at high frequencies. The RF can also be thought of as the ion migration resistance at the electrode/electrolyte interface. In the deep discharge state, the decrease in the slope may be due to the continuous potassium reaction, leading to the growth of nanoparticle volume, the reduction in pore structure and the increase in ion diffusion resistance.
To further evaluate the electrochemical performance of Fe@Fe2O3/PCNFs composite electrode materials, sodium-ion batteries were also assembled. According to the previous studies on the storage of sodium ions for Fe2O3, it can be observed that Fe2O3 and Na occur in conversion reactions. In Fig. S1 (ESI†), at a scan rate of 0.1 mV s−1, in the initial discharge cycle, there are two cathode peaks, which represent the reduction reaction. The weak peak at about 1.1 V is assigned to the insertion reaction of Na. The rough 0.5 V peak is due to the growth of layers at the solid electrolyte interface (SEI) and the decrease of Fe2O3. On the second and third lap scans, the cathode peak at 1.1 V disappeared and the cathode peak at 0.5 V moved to 0.65 V. Notably, two mild anodic peaks of 0.8 V and 1.4 V, respectively, were produced in the first charge cycle. The two weak anodic peaks were attributed to the two-step oxidation reaction (Fe0 → Fe2+ and Fe2+ → Fe3+, respectively). The corresponding redox in the CV curve is as follows:
| Fe2O3 + 6Na+ + 6e− → 2Fe + 3Na2O | (7) |
| 2Fe + 3Na2O → Fe2O3 + 6Na+ + 6e− | (8) |
The CV curves obtained from the subsequent two laps of scanning overlap indicate that the material has good reversibility. The difference from the first circle is mainly due to the occurrence of irreversible reactions, such as partial insertion reaction of Na and the production of SEI membrane. With a starting capacity of 837 mAh g−1 at 100 mA g−1, after ten cycles, altering the current density results in 596.5 mAh g−1. Fig. S2 (ESI†) illustrates the rate performance of Fe@Fe2O3/PCNFs composites at 100 mA g−1 to 2000 mA g−1 current densities. After 10 cycles at 500 mA g−1, the specific capacity of 280 mAh g−1 can be achieved, and when the current density is increased once more, the capacity rapidly decreases. When the initial current density was reached, that is, after 60 cycles, it was stable at 680 mAh g−1, and the capacity retention rate was 81.2%. After 1500 cycles at a high current density of 1000 mA g−1, the Fe@Fe2O3/PCNFs composite anode is as shown in Fig. S4 (ESI†). Its long cycling performance is comparable to that of the majority of micro- and nano-structured α-Fe2O3 anode materials used in sodium-ion batteries, with a reversible capacity of 157.8 mAh g−1. In addition, we evaluated the CV curves of Fe@Fe2O3/PCNFs composites as anode electrode materials of sodium-ion batteries at different scanning speeds. Using the b value, the calculation formula of the above potassium ion battery cathode material was obtained (Fig. S6, ESI†). At this time, the corresponding cathode and anode b value is 0.82 and 0.85. When the b value is between 0.5 and 1.0, it has the battery behavior and the capacitance behavior. When the Fe@Fe2O3/PCNFs composite is used as the anode electrode material of sodium ion battery, it still has pseudo-capacitance properties. In order to compare with the potassium ion battery system, the contribution rates of the surface capacitance behaviors of the materials at different scanning speeds were calculated. The corresponding contribution rates of the surface capacitance behavior at 0.5, 1.0, 1.5, 2.0 and 2.5 mV s−1 were 52.9%, 60%, 64.3%, 67.3% and 71%, respectively. The capacitance behavior of composite materials also plays a leading role in the sodium-electric system. Compared with the potassium system, the sodium system has a slightly higher capacitance contribution rate at different sweep speeds. The high contribution rate of capacitive behavior mainly comes from the special porous structure: a large number of mesopores provide rich active surfaces for the composite materials; the three-dimensional carbon fiber network and elemental iron provide higher conductivity and fast conduction channels for electrons; the pore structure is conducive to the infiltration of the electrolyte and accelerates the conduction of ions in the pores.
The composite's discharge/charge curve for the first three cycles in the sodium-based system is shown in Fig. S8a (ESI†). The charging and discharging curves of sodium electric systems have no obvious charging and discharging platforms, which may be caused by the larger radii of sodium ions. Under various discharge potential states, it can be seen that the composite electrode displays less resistance in the sodium electrical system (Fig. S8b and c, ESI†). In the sodium electric system, the semicircle diameter in the high-frequency region decreases sharply when discharging at 0.5 V. The fundamental reason for this is that Fe2O3 goes through a reduction reaction to produce more iron, which further enhances the electrode's conductivity and lowers the charge transfer resistance. Ion diffusion in the pores and ion adsorption on the electrode surface are both represented by the linear slope at low frequencies. At the beginning of the discharge reaction, the slope increases linearly, which indicates that the speed of ion diffusion to the electrode surface increases, to some extent, which proves that the pore structure of the material is conducive to the reaction. Therefore, Fe@Fe2O3/PCNFs composite electrodes with a small semicircular diameter and a steep linear slope have good electrochemical properties in both potassium and sodium electric systems.
In order to further explore the role of pore structure in the electrochemical performances, SAXS was used to systematically investigate the dynamic change of pore size during the charging and discharging process. Fig. 5a depicts the layout of the SAXS beamline terminal station. 2θ represents the scattering angle, L denotes the separation between the sample and the detector, and r is the distance from the pixel to the center of the direct beam in the figure. In order to avoid the error caused by the air scattering signal, the incident X-ray beam needs to be placed in the vacuum tube. The vacuum tube's ion chamber serves as an upstream detector to keep track of the beam's intensity. The device's upstream and downstream tube’ lengths can be changed in order to achieve a varied distance between the sample and the detector. The downstream detector is acted by the photodiode installed on the beamstop, which primarily serves the purpose of detecting changes in the beam's intensity after passing through the sample. The SAXS data are collected using a Pilatus 2MF detector with an active area of 253.7 mm × 288.8 mm and a pixel of 1475 × 1479. Within the range of 0.06-3.50 nm−1, SAXS typically detects q (nm−1) values. Here q = 4sin/2θ is the modulus of the scattering vector q, where 2θ is the scattering angle, and λ (nm) is the wavelength of the X-ray. The small-angle scattering is a phenomenon of coherent scattering of incident light near the original beam, and the results can give the structure information of the nano-scale in the sample. When the electrode material has a highly ordered layered or mesoporous structure, SAXS can be used to study the structural transformation of the material during the cell cycle. The changes in the pore diameter and the total volume of electrode materials during charging and discharging will be reflected in the SAXS mode. To understand the structure change of the Fe@Fe2O3/PCNFs composite cathode during the charging and discharging process, the changed information was monitored via SAXS during the ex situ charging and discharging process of composite materials. The scattering patterns of composite materials with different potentials are shown in the figure (Fig. 5b–i). The blue dot in the center of the scattered light spot image and the vertical blue strip shadow extending downward from the center dot of the connecting light spot image are the beam occluder center and rod. Several horizontal and vertical blue stripe shadows in the image are caused by the detector sense itself. The scattering pattern needs to use special software to integrate the image information into data information, and finally obtain the relationship between scattering intensity (i) and scattering vector (q).
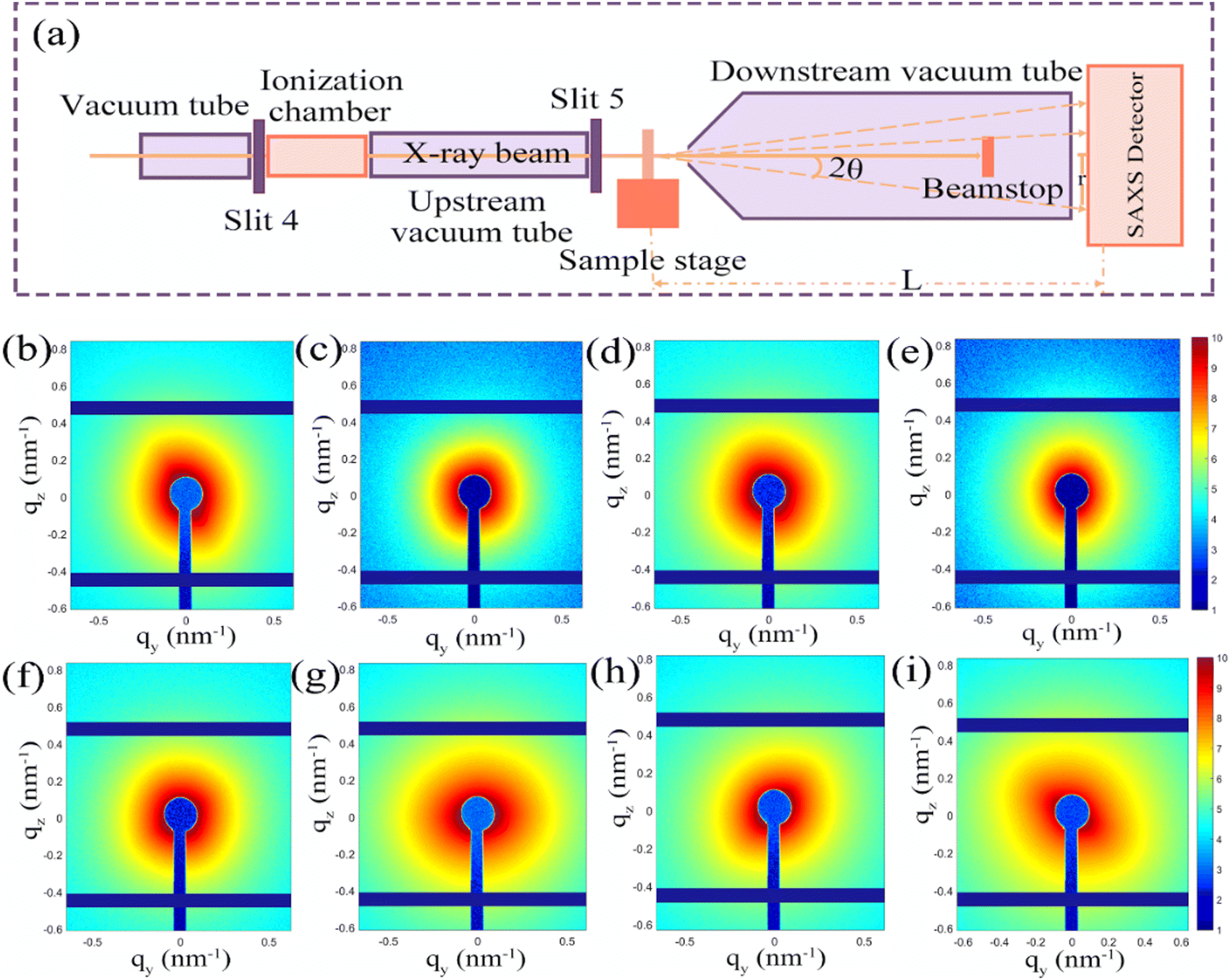 | ||
| Fig. 5 (a) Schematic diagram of beam line terminal device. The SAXS patterns of the composites with different potentials: (b) initial, (c) discharge to 1.5 V, (d) discharge to 1.0 V, (e) discharge to 0.5 V, (f) discharge to 0.01 V, (g) charge to 1.0 V, (h) charge to 1.5 V, and (i) charge to 3.0 V. | ||
Different levels of structure are represented in the scattering pattern. In some cases, the scattering system contains not only small particles but also large particles. When their sizes differ greatly, the scattering characteristics produced by larger scale structures generally occupy the lower q region; the information of the smaller scale structure will occupy the higher q region. Specific analysis of structural information at different organizational levels is required to arrive at more complete conclusions. Under different potentials, the SAXS curve of the Fe@Fe2O3/PCNFs composite is shown in Fig. 5b–i. q is the scattering vector, and its value is related to the diffraction angle (2θ) and light source wavelength (λ). The anode material produces two SAXS characteristics: one is in the low q zone, which corresponds to large-sized materials, and the other is in the high q region, which corresponds to small-sized materials. The former may be Fe@Fe2O3 particles, and the latter may correspond to pore information. The original sample is a composite material electrode sheet that has not been circulated by the battery, and the other electrode sheets are taken out after disassembling the battery. By observing Fig. 6a, a changing trend of the SAXS curve of Fe@Fe2O3/PCNFs composites can be found. It is found that the high q signal gradually weakens in the early stage of the discharge reaction, which may be due to the gradual infiltration of electrolytes, resulting in the reduction of the electron density inhomogeneity. After the weakening trend continues to the state of 1.0 V discharge, the signal starts to gradually increase to the state of 1.0 V charge, which relates to how the SEI film forms and how the potassium reaction is represented by the CV curve. Therefore, the signal enhancement at this stage can be contributed to the development of the SEI film and the volume expansion of Fe@Fe2O3/PCNFs particles caused by potassium reaction. The depotassiation reaction occurs mainly after charging to 1.0 V, which leads to the shrinkage of the particles and the rupturing of the SEI membrane. This phenomenon is consistent with the variation in the signal in the figure, with a decrease in the signal observed from 1.0 V continuous charge to full charge. The characteristics of the low q region change little during the discharge, which may be due to the filling of the pores by potassium. SAXS technology can also characterize the fractal structure of materials. For pinhole collimated SAXS, when the change of electron density on the system surface is discontinuous, the scattering intensity I(q) corresponding to each q value shows the following relationship:
| I(q) = I(0)q−α | (9) |
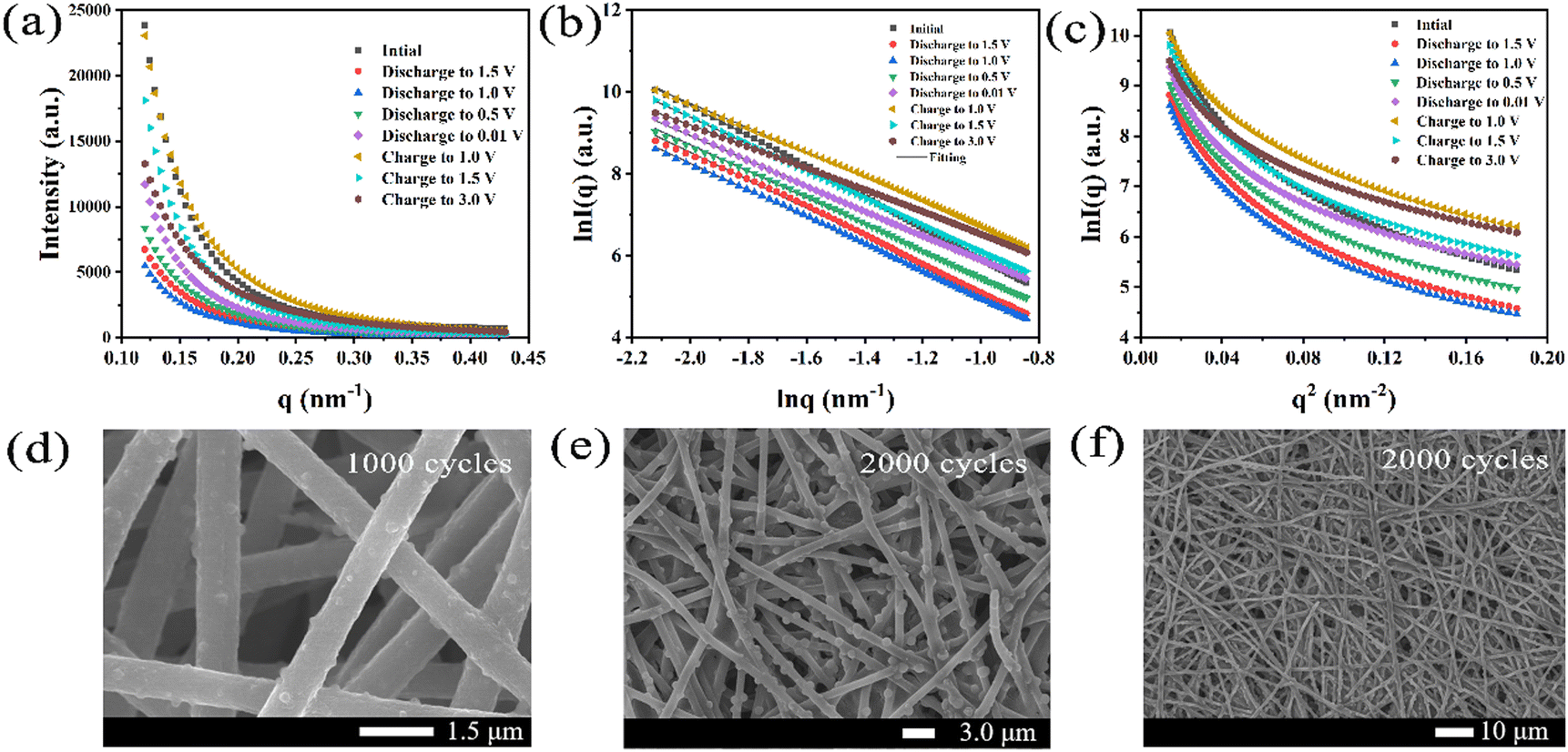 | ||
| Fig. 6 (a) Curves of intensity (I) and scattering vector (q) under different charge–discharge potentials. (b) Curves of lnI and lnq under different charge–discharge potentials. (c) Curves of lnI and q2 under different charge–discharge potentials. SEM images of the electrode plate after cycling: (d) 1000 cycles, (e) 2000 cycles, and (f) 2000 cycles. | ||
The following relationships can be derived from simple variations:
| lnI(q) = lnI(0) − αlnq | (10) |
I
0 in the formula is a constant and α is a parameter that determines the fractal dimension. Only when 3 < α < 4, the material has surface fractal (Ds), and the fractal dimension Ds = 6 − α. Ds can characterize the roughness of the material surface. In addition, the closer Ds are to 3, the more complex the surface is. When 0 < α < 3, the material is a mass fractal (Dm). In this case, the fractal dimension Dm = α. The lnI(q)–lnq diagrams of Fe@Fe2O3/PCNFs composites at different potentials were made after understanding the above-mentioned relations (Fig. 6b). The data were fitted according to the above-mentioned relations, and the relevant information obtained are presented in Table 1.
| Sample states | Slope of linear fitting | α | Fractal dimension | Fractal type |
|---|---|---|---|---|
| Initial | −3.7 | 3.7 | 2.3 | D s |
| Discharge to 1.5 V | −3.4 | 3.4 | 2.6 | D s |
| Discharge to 1.0 V | −3.3 | 3.3 | 2.7 | D s |
| Discharge to 0.5 V | −3.2 | 3.2 | 2.8 | D s |
| Discharge to 0.01 V | −3.0 | 3.0 | 3.0 | D s |
| Charge to1.0 V | −3.0 | 3.0 | 3.0 | D s |
| Charge to 1.5 V | −3.3 | 3.3 | 2.7 | D s |
| Charge to 3.0 V | −2.6 | 2.6 | 2.6 | D m |
Comparing the fractal dimensions of materials at different potentials, it can be found that the fractal dimension Ds is getting closer to 3 with the discharge reaction progress, indicating that the surface fractal is increasingly complex in this process. If the fractal type belongs to surface fractal, it can be explained to some extent that the material belongs to loose status and has a relatively rough surface. When fully charged, the composite material changes from surface fractal to mass fractal, indicating that the material changes into a relatively dense type and the surface becomes relatively smooth, which may be related to the structural change brought by the potassiation and depotassiation reaction, such as the filling of pore structure, the full infiltration of electrolytes, and the generation of SEI films.
The presence of numerous small-scale pores was found in the composites, and the scattering intensity in the high q region is directly influenced by the small-angle scattering signal induced by these pore structures. In order to study how the pore structure is dynamically evolving during charging and discharging, the scattering signal is analyzed according to the classical SAXS analysis theory to obtain the qualitative or quantitative structure information of the pores. The scattering intensity I(q) can be used to obtain the average radius of gyration (Rg) of the scatterer projected on the scattering system and its related parameter information. The radius of rotation Rg was calculated using the root mean square distance between the center of gravity and the scattering entity's electrons, which is usually calculated using the following Guinier approximation:
| I(q) = I(0)exp(−1/3Rg2q2) | (11) |
Take natural logarithms on both sides of the Guinier approximation to obtain the following relationship:
| lnI(q) = lnI(0) − 1/3(Rg2q2) | (12) |
As shown in Fig. 6c, the figure of the Fe@Fe2O3/PCNFs composite lnI(q)–q2 was used to calculate the average radius of gyration of pores through the slope. Because the particle size of Fe@Fe2O3 is larger than that of the pore size, the information of the higher q region can be analyzed to study the pore structure change. In order to compare, two parts of the data were selected for analysis. Table 2 shows the parameters obtained by linear fitting in the higher q region of the Fe@Fe2O3/PCNFs composites at different potentials. In the calculation of the hole radius, the hole shape is assumed to be circular.
| Sample states | Middle q zone | High q zone | ||||
|---|---|---|---|---|---|---|
| k | R g | r (nm) | k | R g | r (nm) | |
| Initial | −23.8 | 8.4 | 10.9 | −10.7 | 5.6 | 7.3 |
| Discharge to 1.5 V | −22.5 | 8.2 | 10.6 | −9.6 | 5.4 | 7.3 |
| Discharge to 1.0 V | −21.3 | 8.0 | 10.3 | −8.8 | 5.1 | 6.3 |
| Discharge to 0.5 V | −21.0 | 7.9 | 10.2 | −8.9 | 5.2 | 6.7 |
| Discharge to 0.01 V | −18.6 | 7.5 | 9.6 | −8.5 | 5.0 | 6.5 |
| Charge to 1.0 V | −19.0 | 7.6 | 9.7 | −9.5 | 5.3 | 6.9 |
| Charge to 1.5 V | −20.5 | 7.8 | 10.1 | −9.1 | 5.2 | 6.7 |
| Charge to 3.0 V | −16.8 | 7.1 | 9.2 | −8.4 | 5.0 | 6.5 |
By comparing the pore radius of the composite materials under different potentials, it was found that the pore radius shows a decreasing trend in the discharge stage, whether in the middle q zone or in the high q zone. The main reasons may include that the scattering signal of the pore structure is weakened due to the electrolyte infiltration, and part of the pores are filled due to the disordered carbon participating in the potassium reaction. In the charging stage of the middle q zone, the hole radius increases continuously to 1.5 V, and after full charging, the hole radius decreases. It may be due to the occurrence of the depotassiation reaction. The pore structure of the composite becomes larger due to the potassium exudation from the composite. After exudation, the electrolyte further soaks and the unevenness in electron density decreases. The changing trend of the pore structure in the charging stage of the high q zone is generally consistent with that in the middle q zone. The only difference is that the pore radius decreases from charging to 1.0 V, which indicates that smaller pore structures are more sensitive to reaction changes. The Fe@Fe2O3/PCNFs composite cells with different cycle numbers are disassembled and the anode electrode materials are observed by SEM. Interestingly, comparing the SEM images of the electrodes with different cycle numbers (Fig. 6d and e), the size of Fe@Fe2O3 particles changed obviously with the increase in cycle numbers, while the pore structure of the fibers was almost invisible. This phenomenon is consistent with the results obtained by the SAXS technique, which increases the reliability of the results. As shown in Fig. 6f, after cycles, the fiber morphology of the electrode sheet maintained integrity, which confirmed that the fiber structure had a moderating effect on the fragmentation of the material during the potassiation/depotassiation process.
4. Conclusions
In this study, carbon-coated Fe@Fe2O3/PCNFs composites were fabricated by electrospinning and used for potassium-ion batteries. They showed good electrochemical properties. The ex situ dynamic characterization of pore structure was performed using synchrotron radiation technique.In terms of electrochemical properties, in the potassium ion battery system, a discharge capacity of 273 mAh g−1 was maintained after 150 cycles at 100 mA g−1 current density, and at 1000 mA g−1 current density, after 1500 discharge/charge cycles, a specific capacity of 144.8 mAh g−1 can be obtained. The sodium ion battery system displayed a stable discharge capacity of 596.5 mAh g−1 by cycling 150 times under 100 mA g−1, and a specific capacity of 157.8 mAh g−1 was obtained after 1500 discharge/charge cycles at 1000 mA g−1.
In the mechanism characterization, SAXS was used to analyze the potassium reaction kinetics of Fe@Fe2O3/PCNFs composites during the charge/discharge process, and the changes of particle size and pore structure in the reaction process were analyzed. It confirmed that the potassiation reaction of Fe@Fe2O3/PCNFs composites can be divided into four stages: (1) at the initial stage of discharge, the electrolyte is constantly saturated, and the pores are filled with electrolytes, resulting in a reduction in the scattering system unevenness. (2) In the process of potassiation reaction, part of the potassium fills the pore structure, and the formation of the SEI membrane further reduces the pore size. This stage is accompanied by an increase in particle size. (3) During the potassium removal reaction, potassium is removed from the pores, and the SEI membrane is broken, which temporarily increases the pore structure. (4) The pores after stripping are further infiltrated by the electrolyte. The pore structure of the electrode after cycling is much smaller than that of the initial state, and the SEM image of the electrode sheet after cycling also proves this conclusion. Therefore, the pore structure design of electrode materials for potassium ion batteries should follow easy electrolyte penetration, high pore density, and large pore radius (or reduce the proportion of small pore size pore structure). This work provides a new research direction for exploring the storage of nano-pore structures and designing porous composite materials with appropriate nano-pore sizes to achieve higher capacity and higher stability of alkali metal ion batteries.
Conflicts of interest
The authors declare no competing conflicts of interest.Acknowledgements
The work was funded by the National Natural Science Foundation of China (U2032133 and 12105202). We would like to thank the Analytical & Testing Center of Tiangong University for the rewarded machine usage time work, including the Transmission Electron Microscope, X-Ray diffractometer, transmission electron microscope, transmission electron microscope, transmission electron microscope, etc. The support of the x-ray small angle scattering beamline station (BL16B1) at the Shanghai Synchrotron Light Facility (SSRF) in China is gratefully acknowledged.Notes and references
- D. Deng, Energy Sci. Eng., 2015, 3, 385–418 CrossRef.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- Y. Z. Pan, L. Yin and M. Q. Li, Ceram. Int., 2019, 45, 12072–12079 CrossRef CAS.
- M. Z. Chen, Q. N. Liu, Z. Hu, Y. Y. Zhang, G. C. Xing, Y. X. Tang and S. L. Chou, Adv. Energy Mater., 2020, 10, 2002244 CrossRef CAS.
- Z. Y. Sang, D. Su, J. S. Wang, Y. Liu and H. M. Ji, Chem. Eng. J., 2020, 381, 122677 CrossRef CAS.
- I. Sultana, M. M. Rahman, T. Ramireddy, Y. Chen and A. M. Glushenkov, J. Mat. Chem. A, 2017, 5, 23506–23512 RSC.
- M. Wang and Y. B. Tang, Adv. Energy Mater., 2018, 8, 1703320 CrossRef.
- R. Rajagopalan, Y. G. Tang, X. B. Ji, C. K. Jia and H. Y. Wang, Adv. Funct. Mater., 2020, 30, 1909486 CrossRef CAS.
- J. Shi, H. Kang, N. Li, K. Teng, W. Sun, Z. Xu, X. Qian and Q. Liu, Appl. Surf. Sci., 2019, 478, 38–48 CrossRef CAS.
- X. Min, J. Xiao, M. H. Fang, W. Wang, Y. J. Zhao, Y. G. Liu, A. M. Abdelkader, K. Xi, R. V. Kumar and Z. H. Huang, Energy Environ. Sci., 2021, 14, 2186–2243 RSC.
- K.-Y. Zhang, Z.-Y. Gu, E. H. Ang, J.-Z. Guo, X.-T. Wang, Y. Wang and X.-L. Wu, Mater. Today, 2022, 54, 189–201 CrossRef CAS.
- M. Wang, B. Qin, F. Xu, W. Yang, Z. Liu, Y. Zhang and H. Fan, J. Colloid Interface Sci., 2023, 650, 446–455 CrossRef CAS PubMed.
- Y. Liu, Z.-Y. Gu, Y.-L. Heng, J.-Z. Guo, M. Du, H.-J. Liang, J.-L. Yang, K.-Y. Zhang, K. Li and X.-L. Wu, Green Energy Environ., 2023 DOI:10.1016/j.gee.2023.10.004.
- Q. M. Yin, Z. Y. Gu, Y. Liu, H. Y. Lü, Y. T. Liu, Y. N. Liu, M. Y. Su, J. Z. Guo and X. L. Wu, Adv. Funct. Mater., 2023, 33, 2304046 CrossRef CAS.
- H. B. Huang, S. H. Luo, C. L. Liu, Y. Yang, Y. C. Zhai, L. J. Chang and M. Q. Li, Appl. Surf. Sci., 2019, 487, 1159–1166 CrossRef CAS.
- A. Eftekhari, Z. L. Jian and X. L. Ji, ACS Appl. Mater. Interfaces, 2017, 9, 4404–4419 CrossRef CAS PubMed.
- K. M. Song, C. T. Liu, L. W. Mi, S. L. Chou, W. H. Chen and C. Y. Shen, Small, 2021, 17, 1903194 CrossRef CAS PubMed.
- L. B. Ma, Y. H. Lv, J. X. Wu, C. Xia, Q. Kang, Y. Z. Zhang, H. F. Liang and Z. Jin, Nano Res., 2021, 14, 4442–4470 CrossRef CAS.
- X. Wu, Y. L. Chen, Z. Xing, C. W. K. Lam, S. S. Pang, W. Zhang and Z. C. Ju, Adv. Energy Mater., 2019, 9, 1900343 CrossRef.
- K. Z. Cao, H. Q. Liu, W. Y. Li, Q. Q. Han, Z. Zhang, K. J. Huang, Q. S. Jing and L. F. Jiao, Small, 2019, 15, 1901775 CrossRef PubMed.
- Z. Q. Gu, G. J. Li, N. Hussain, B. B. Tian and Y. M. Shi, Appl. Surf. Sci., 2022, 592, 153323 CrossRef CAS.
- J. W. Wen, L. Xu, J. X. Wang, Y. Xiong, J. J. Ma, C. R. Jiang, L. H. Cao, J. Li and M. Zeng, J. Power Sources, 2020, 474, 228491 CrossRef CAS.
- H. Y. Jiang, Y. L. An, Y. Tian, J. K. Feng and X. L. Tian, Mater. Technol., 2020, 35, 594–599 CrossRef CAS.
- D. Adekoya, H. Chen, H. Y. Hoh, T. Gould, M. Balogun, C. Lai, H. J. Zhao and S. Q. Zhang, ACS Nano, 2020, 14, 5027–5035 CrossRef CAS PubMed.
- J. F. Zheng, Y. J. Wu, Y. J. Sun, J. H. Rong, H. Y. Li and L. Niu, Nano-Micro Lett., 2021, 13, 1–37 CrossRef CAS PubMed.
- W. L. Zhang, J. Yin, W. X. Wang, Z. Bayhan and H. N. Alshareef, Nano Energy, 2021, 83, 105792 CrossRef CAS.
- J.-M. Cao, I. V. Zatovsky, Z.-Y. Gu, J.-L. Yang, X.-X. Zhao, J.-Z. Guo, H. Xu and X.-L. Wu, Prog. Mater. Sci., 2023, 135, 101105 CrossRef CAS.
- S. Wu, F. Xu, Y. Li, C. Liu, Y. Zhang and H. Fan, J. Colloid Interface Sci., 2023, 649, 741–749 CrossRef CAS PubMed.
- H. Wang, N. Li, J. Niu, K. Teng, Z. Xu, M. Jing, F. Li, W. Wang and X. Zhang, Composites, Part A, 2019, 121, 321–329 CrossRef CAS.
- D. P. Li, Q. Sun, Y. M. Zhang, X. Y. Dai, F. J. Ji, K. K. Li, Q. H. Yuan, X. J. Liu and L. J. Ci, Nano Res., 2021, 14, 4502–4511 CrossRef CAS.
- H. L. Deng, L. Wang, S. Y. Li, M. Zhang, T. Wang, J. Zhou, M. X. Chen, S. Chen, J. H. Cao, Q. S. Zhang, J. Zhu and B. A. Lu, Adv. Funct. Mater., 2021, 31, 2107246 CrossRef CAS.
- J. Liu and L. B. Kong, Electrochim. Acta, 2021, 378, 138141 CrossRef CAS.
- R. Guo, X. Liu, B. Wen, F. Liu, J. Meng, P. Wu, J. Wu, Q. Li and L. Mai, Nano-Micro Lett., 2020, 12, 148 CrossRef CAS PubMed.
- R. Xu, Y. Yao, H. Wang, Y. Yuan, J. Wang, H. Yang, Y. Jiang, P. Shi, X. Wu, Z. Peng, Z.-S. Wu, J. Lu and Y. Yu, Adv. Mater., 2020, 32, 2003879 CrossRef PubMed.
- K. Lei, C. Wang, L. Liu, Y. Luo, C. Mu, F. Li and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 4687–4691 CrossRef CAS PubMed.
- Y. Qian, S. Jiang, Y. Li, Z. Yi, J. Zhou, J. Tian, N. Lin and Y. T. Qian, Energy Storage Mater., 2020, 29, 341–349 CrossRef.
- F. Yuan, Y. Li, D. Zhang, Z. Li, H. Wang, B. Wang, Y. Wu and Y. A. Wu, Inorg. Chem. Front., 2023, 10, 2547–2573 RSC.
- J. Du, S. Gao, P. Shi, J. Fan, Q. Xu and Y. Min, J. Power Sources, 2020, 451, 227727 CrossRef CAS.
- J. Liu and L.-B. Kong, Electrochim. Acta, 2021, 378, 138141 CrossRef CAS.
- Y. An, Y. Tian, L. Ci, S. Xiong, J. Feng and Y. Qian, ACS Nano, 2018, 12, 12932–12940 CrossRef CAS PubMed.
- K. Zhang, Q. He, F. Y. Xiong, J. P. Zhou, Y. Zhao, L. Q. Mai and L. N. Zhang, Nano Energy, 2020, 77, 105018 CrossRef CAS.
- H. Y. Li, Z. Cheng, Q. Zhang, A. Natan, Y. Yang, D. X. Cao and H. L. Zhu, Nano Lett., 2018, 18, 7407–7413 CrossRef CAS PubMed.
- C. J. Chen, Z. G. Wang, B. Zhang, L. Miao, J. Cai, L. F. Peng, Y. Y. Huang, J. J. Jiang, Y. H. Huang, L. N. Zhang and J. Xie, Energy Storage Mater., 2017, 8, 161–168 CrossRef.
- F. Tian, X. H. Li, Y. Z. Wang, C. M. Yang, P. Zhou, J. Y. Lin, J. R. Zeng, C. X. Hong, W. Q. Hua, X. Y. Li, X. R. Miao, F. G. Bian and J. Wang, Nucl. Sci. Tech., 2015, 26, 1–6 Search PubMed.
- S. Liang, J. Miao, H. Shi, M. Zeng, H. An, T. Ma, T. Li and Z. Xu, ACS Nano, 2022, 16, 19584–19593 CrossRef CAS PubMed.
- B. Zhu, R. Shao, N. Li, C. Guo, P. Liu, J. Shi, C. Min, S. Liu, X. Qian, L. Wang and Z. Xu, J. Membr. Sci., 2023, 667, 121187 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp05994d |
| This journal is © the Owner Societies 2024 |